

Abstract
Background:
Pucotenlimab is a humanized immunoglobulin G4 (IgG4) anti programmed cell death protein 1 (anti-PD-1) monoclonal antibody (mAb) with a S228P hinge mutation and an engineered Fc domain. Preclinical data suggests that pucotenlimab exerts antitumor effects. In this phase I study, which was prospectively registered on www.chinadrugtrials.org.cn (CTR20180125), the safety, maximum tolerated dose, preliminary antitumor activity, pharmacokinetics, and immunogenicity of pucotenlimab were evaluated in patients with advanced solid tumors.
Methods:
Patients with advanced solid tumors refractory to standard therapies were recruited. In a 3+3 dose escalation study, 13 patients received pucotenlimab intravenously every 3 weeks (Q3W) until disease progression or unacceptable toxicity occurred at doses of 1 mg/kg, 3 mg/kg, 10 mg/kg, and 200 mg. 17 additional patients were assigned in the expansion period.
Results:
A total of 30 patients were enrolled. No dose-limiting toxicity was observed. The maximum tolerated dose was not reached. The most common treatment-related adverse events of any grade were proteinuria (40%), fatigue (36.7%), weight loss (26.7%), fever (26.7%), increased aspartate aminotransferase (26.7%), rash (23.3%), and anorexia (20.0%). Partial responses occurred in five patients, with an objective response rate of 16.7%. Pharmacokinetics analysis showed rapid absorption followed by slow terminal elimination, with a mean half-life of 17.1–23.5 days across all dose groups.
Conclusions:
Pucotenlimab had an acceptable toxicity profile at doses up to 10 mg/kg and the maximum tolerated dose was not reached. Based on the pharmacokinetics, efficacy, and safety profile, 3 mg/kg Q3W or 200 mg Q3W are optimal for further drug development.
Keywords: anti-PD-1, pharmacokinetics, pucotenlimab, solid tumor, safety
Introduction
The programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1) pathway represents a critical immune control switch that may contribute to tumor cell suppression of T-cell immune surveillance.1–3 Preclinical experiments in vitro and in vivo have shown that PD-1/PD-L1 blockade via monoclonal antibodies (mAbs) enhances tumor cell-specific T-cell activation, cytokine production, anti-tumor effector mechanisms, and the clearance of tumor cells by the immune system.4,5 PD-1 and PD-L1 inhibitors have significantly changed the therapeutic landscape in a variety of malignancies with durable antitumor responses,6–10 including melanoma and cancers of the lung, kidney, head and neck, bladder, stomach, and breast.
Pucotenlimab is a humanized immunoglobulin G4 (IgG4) mAb against human PD-1 containing an Fc domain with S228P and S254T/V308P/N434A mutations, which has a similar PD-1 binding affinity to the approved nivolumab. 11 Pucotenlimab mainly recognizes glycosylated PD-1 through a unique epitope. It has no antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) activity by using the IgG4 Fc isotype to avoid killing of PD-1-expressing immune cells. According to preclinical data, pucotenlimab significantly inhibits tumor growth and shows an effective antitumor response, comparable to those of approved anti-PD-1 drugs, suggesting that it is a suitable drug candidate for cancer immunotherapy. 11
The objectives of this phase I study were to evaluate the safety, pharmacokinetics (PK), and pharmacodynamics of pucotenlimab in patients with advanced solid tumors. The tumor response to pucotenlimab was also evaluated as an exploratory objective.
Materials and methods
Patient population
This study enrolled patients aged ⩾18 years with a histologically- or cytologically-confirmed diagnosis of locally-advanced or metastatic solid tumors that progressed or were intolerant to standard treatment or had no standard treatments available. Additional key eligibility criteria were as follows: patients with at least one measurable lesion at baseline as assessed by the Response Evaluation Criteria in Advanced Solid Tumors version 1.1 (RECIST version 1.1), Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, a life expectancy ⩾3 months, and adequate organ function. The main exclusion criteria were as follows: patients with active or a history of autoimmune disease (such as systemic lupus erythematosus, rheumatoid arthritis, inflammatory bowel disease, autoimmune thyroid disease, multiple sclerosis, vasculitis, glomerulonephritis, etc), active central nervous system metastases, a history of or current interstitial lung disease or pulmonary fibrosis; prior treatment with an agent directed against PD-1/PD-L1, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), or another co-inhibitory T-cell receptor; a history of allogeneic hematopoietic stem cell transplantation; and adverse events (AEs) from previous therapy without recovery to grade ⩽1.
Study design
This was a single-center, single-arm, open-label, phase I study (CTR20180125) sponsored by Taizhou Hanzhong Biomedical Co., Ltd. The study protocol and all amendments were approved by the Ethics Committee of Fudan University Shanghai Cancer Center (approval number: 1711178-3) and conducted in accordance with the Declaration of Helsinki guidelines and international standards of good clinical practice. Informed consent was obtained from all patients.
The study consisted of a dose-escalation and expansion phase. Dose-escalation was conducted using a traditional 3+3 design. Thirteen patients were administered pucotenlimab at doses of 1, 3, and 10 mg/kg intravenously over 60 min every 3 weeks until disease progression or intolerable toxicity occurred. The first 21-day treatment cycle was designed for the observation of dose-limiting toxicity (DLT), which was defined as grade ⩾3 non-hematological toxicity, except for grade 3 rash, nausea, vomiting, or diarrhea lasting ⩽3 days after optimal supportive treatment; or treatment interruption for >14 days due to toxicity; grade 4 neutrophil count reduction lasting for ⩾5 days; febrile neutropenia; grade 4 thrombocytopenia; grade 3 thrombocytopenia with bleeding tendency; or other grade 4 hematological toxicity.
Dose escalation proceeded when all three patients completed the safety evaluation at a given dose level with DLTs occurring in less than one-third of patients. Intra-patient dose escalation was prohibited. During the subsequent expansion period, 17 patients were randomly assigned to cohorts administered 1, 3, 10 mg/kg, or a fixed dose of 200 mg. Clinically stable patients with first radiographic progressive disease (PD) might continue treatment at the investigator’s discretion until PD is confirmed.
Safety assessment
DLTs were assessed during the first 21 days of treatment. All AEs were recorded from the initiation of treatment to 90 days after the last dose or the start date of subsequent anti-tumor therapy following the last dose, whichever came first. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA, version 22.1) and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE, version 4.03). The causality between AEs and pucotenlimab was evaluated by investigators.
Assessment of antitumor effects
Efficacy was evaluated by the investigator according to RECIST version 1.1 and immune response evaluation criteria in solid tumor (iRECIST). Tumor assessments were performed by computed tomography or magnetic resonance imaging at baseline and every 6 weeks during the first 24 weeks and every 12 weeks thereafter.
Pharmacokinetics and immunogenicity assessments
The concentration of pucotenlimab was detected for PK studies. Plasma samples for pucotenlimab PK profiling were collected predose, postdose (+5 min), 2 (±1/6), 8 (±1/2), 24 (±1), 72 (±2), 168 (±4), and 336 (±12) h from the initiation of the drug in cycle 1 and cycle 6, as well as predose and postdose (+5 min) in cycles 2–5, 9, 13, 17, and every 8 cycles thereafter. Samples were stored at −80°C until measurement. Pucotenlimab serum concentrations were quantified using a validated electrochemiluminescence assay (lower limit of quantification, 8 ng/ml). The PK data were described using a noncompartmental approach.
Blood samples for anti-drug antibody analyses were collected predose in cycles 1–6, 9, 13, 17, and every 8 cycles thereafter, as well as 28 days after the last pucotenlimab dose. Antidrug antibodies to pucotenlimab were detected by an electrochemiluminescence immunoassay.
Biomarker assessments
For patients with available tumor samples, PD-L1 tumor expression was measured by immunohistochemistry with formalin-fixed paraffin-embedded tissue sections at a central laboratory. Evaluation of PD-L1 was performed using a mouse anti-PD-L1 monoclonal antibody (clone 22C3). The tumor cell percentage with membranous PD-L1 staining was recorded. Scores of <1% were categorized as “PD-L1 negative” [Figure S1(a)], those of ⩾1% were categorized as “PD-L1 positive”. Polymerase chain reaction-based microsatellite instability (MSI) testing and immunohistochemical detection of mismatch repair (MMR) proteins (MLH1, MSH2, MSH6, and PMS2) were also performed.
Statistical analysis
The objective response rate (ORR) and disease control rate (DCR) with 95% confidence interval (CI) were calculated using the Clopper–Pearson method based on the binomial distribution. Patients without tumor assessment data were considered non-responders. Data analyses were conducted using SAS version 9.4. PK parameters for pucotenlimab were calculated using non-compartmental approaches implemented in WinNonlin 5.3.
Results
Patients
Between February 2018 and December 2018, 44 patients were assessed and 30 eligible patients with advanced solid tumors were enrolled in the study. Patients had a variety of tumor types, including colorectal cancer (n = 6), breast cancer (n = 6), esophageal cancer (n = 3), lung cancer (n = 2), pancreatic cancer (n = 2), and so on. The median age was 54 years (range, 24–72 years). A total of 56.7% of patients were male, and 100% had an ECOG performance status of 1. The dose-limiting toxicity analysis set included 13 patients: 3 at 1 mg/kg, 4 at 3 mg/kg, 3 at 10 mg/kg, and 3 at 200 mg. One patient receiving a dose of 3 mg/kg had to be replaced as a result of rapid disease progression before the completion of the evaluation period. Owing to the requirement for PK sample collection (at least three subjects for each dose in cycle 6), another 17 patients were recruited, that is, 3 at 1 mg/kg, 4 at 3 mg/kg, 3 at 10 mg/kg, and 7 at 200 mg. The baseline characteristics of patients are summarized in Table 1. All 30 patients enrolled in the study were evaluable for toxicity and tumor response.
Table 1.
Baseline characteristics of treated patients (FAS).
| Characteristic | 1 mg/kg Q3W | 3 mg/kg Q3W | 10 mg/kg Q3W | 200 mg Q3W | All patients |
|---|---|---|---|---|---|
| Number of patients | 6 | 8 | 6 | 10 | 30 |
| Age, years | |||||
| Median | 64.5 | 49.5 | 47.5 | 55.5 | 54 |
| Range | 35–72 | 24–66 | 36–61 | 27–68 | 24–72 |
| Sex | |||||
| Male | 1 (16.7) | 7 (87.5) | 3 (50.0) | 6 (60.0) | 17 (56.7) |
| Female | 5 (83.3) | 1 (12.5) | 3 (50.0) | 4 (40.0) | 13 (43.3) |
| ECOG performance status | |||||
| 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 6 (100.0) | 8 (100.0) | 6 (100.0) | 10 (100.0) | 30 (100.0) |
| Prior surgery, n (%) | 4 (66.7) | 8 (100.0) | 5 (83.3) | 7 (70.0) | 24 (80.0) |
| Tumor type, n (%) | |||||
| Colorectal cancer | 1 (16.6) | 1 (12.5) | 2 (33.3) | 2 (20.0) | 6 (20.0) |
| Breast cancer | 3 (50.0) | 0 | 1 (16.6) | 2 (20.0) | 6 (20.0) |
| Esophageal cancer | 1 (16.6) | 2 (25.0) | 0 | 0 | 3 (10.0) |
| Pancreatic cancer | 0 | 0 | 0 | 2 (20.0) | 2 (6.7) |
| Soft tissue sarcoma | 0 | 2 (25.0) | 0 | 0 | 2 (6.7) |
| Neuroendocrine tumor | 0 | 0 | 1 (16.6) | 1 (10.0) | 2 (6.7) |
| Lung cancer | 0 | 0 | 2 (33.3) | 0 | 2 (6.7) |
| Gastric cancer | 1 (16.6) | 0 | 0 | 0 | 1 (3.3) |
| Cecum cancer | 0 | 0 | 0 | 1 (10.0) | 1 (3.3) |
| Thymic cancer | 0 | 0 | 1 (16.6) | 0 | 1 (3.3) |
| Kidney cancer | 0 | 1 (12.5) | 0 | 0 | 1 (3.3) |
| Ovarian cancer | 0 | 0 | 1 (16.6) | 0 | 1 (3.3) |
| Cholangiocarcinoma | 0 | 1 (12.5) | 0 | 0 | 1 (3.3) |
| Melanoma | 0 | 1 (12.5) | 0 | 0 | 1 (3.3) |
ECOG,; Eastern Cooperative Oncology Group; FAS, full analysis set; n, number of patients; Q3W, every 3 weeks.
The data cutoff date was March 24, 2020, with a median follow-up duration of 13.0 months (range 1.4–22.5 months).
Safety and tolerability
During dose escalation, no DLTs were observed. Therefore, the maximum tolerated dose (MTD) was not reached. Table 2 summarizes treatment-related AEs in the safety analysis set of 30 patients, while Table 3 presents an overview of the safety profile for pucotenlimab. Nearly all patients (28/30) experienced treatment-related adverse events (TRAEs). There were no obvious correlations between the type and frequency of AEs and the dosage of the test drug. Across all dose levels, the most frequent (⩾10% of patients) TRAEs of any grade were proteinuria (40%), fatigue (36.7%), weight loss (26.7%), fever (26.7%), increased aspartate aminotransferase (26.7%), rash (23.3%), anorexia (20%), increased alanine aminotransferase (16.7%), decreased white blood cell count (16.7%), increased blood bilirubin (16.7%), decreased neutrophil count (13.3%), hematuria (13.3%), hypothyroidism (13.3%), sinus tachycardia (13.3%), pneumonitis (10.0%), dyspnea (10.0%), hypokalemia (10.0%), nausea (10.0%), gastrointestinal bleeding (10.0%), and anemia (10.0%).
Table 2.
Treatment-related adverse effects occurring in ⩾10% of patients, by treatment group (SS).
| Preferred term, n (%) | 1 mg/kg Q3W | 3 mg/kg Q3W | 10 mg/kg Q3W | 200 mg Q3W | All patients | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| n = 6 | n = 8 | n = 6 | n = 10 | n = 30 | ||||||
| Any grade | Grade ⩾3 | Any grade | Grade ⩾3 | Any grade | Grade ⩾3 | Any grade | Grade ⩾3 | Any grade | Grade ⩾3 | |
| Total | 6 (100.0) | 1 (16.7) | 8 (100.0) | 3 (37.5) | 5 (83.3) | 2(33.3) | 9 (90.0) | 4 (40.0) | 28 (93.9) | 10 (33.3) |
| Proteinuria | 2 (33.3) | 0 | 3 (37.5) | 0 | 4 (66.7) | 0 | 3 (30.0) | 0 | 12 (40.0) | 0 |
| Fatigue | 0 | 0 | 5 (62.5) | 0 | 4 (66.7) | 0 | 2 (20.0) | 0 | 11 (36.7) | 0 |
| Fever | 4 (66.7) | 0 | 3 (37.5) | 0 | 1 (16.7) | 0 | 0 | 0 | 8 (26.7) | 0 |
| Weight loss | 1 (16.7) | 0 | 1 (12.5) | 0S | 3 (50.0) | 0 | 3 (30.0) | 0 | 8 (26.7) | 0 |
| Aspartate aminotransferase increased | 0 | 0 | 2 (25.0) | 0 | 3 (50.0) | 0 | 3 (30.0) | 0 | 8 (26.7) | 0 |
| Rash | 2 (33.3) | 0 | 4 (50.0) | 0 | 0 | 0 | 1 (10.0) | 0 | 7 (23.3) | 0 |
| Anorexia | 2 (33.3) | 1 (16.7) | 1 (12.5) | 0 | 1 (16.7) | 0 | 2 (20.0) | 1 (10.0) | 6 (20.0) | 2 (6.7) |
| White blood cell decreased | 0 | 0 | 1 (12.5) | 0 | 1 (16.7) | 0 | 3 (30.0) | 0 | 5 (16.7) | 0 |
| Alanine aminotransferase increased | 0 | 0 | 2 (25.0) | 0 | 2 (33.3) | 0 | 1 (10.0) | 0 | 5 (16.7) | 0 |
| Blood bilirubin increased | 0 | 0 | 2 (25.0) | 0 | 1 (16.7) | 0 | 2 (20.0) | 0 | 5 (16.7) | 0 |
| Sinus tachycardia | 0 | 0 | 1 (12.5) | 0 | 2 (33.3) | 0 | 1 (10.0) | 0 | 4 (13.3) | 0 |
| Hematuria | 0 | 0 | 1 (12.5) | 0 | 2 (33.3) | 0 | 1 (10.0) | 0 | 4 (13.3) | 0 |
| Free triiodothyronine decreased | 1 (16.7) | 0 | 1 (12.5) | 0 | 2 (33.3) | 0 | 0 | 0 | 4 (13.3) | 0 |
| Neutrophil count decreased | 0 | 0 | 1 (12.5) | 0 | 1 (16.7) | 1 (16.7) | 2 (20.0) | 0 | 4 (13.3) | 1 (3.3) |
| Hypokalemia | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 2 (20.0) | 1 (10.0) | 3 (10.0) | 1 (3.3) |
| Nausea | 0 | 0 | 0 | 0 | 2 (33.3) | 0 | 1 (10.0) | 0 | 3 (10.0) | 0 |
| Dyspnea | 0 | 0 | 1 (12.5) | 1 (12.5) | 1 (16.7) | 0 | 1 (10.0) | 1 (10.0) | 3 (10.0) | 2 (6.7) |
| Anemia | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 | 2 (20.0) | 0 | 3 (10.0) | 1 (3.3) |
| Gastrointestinal bleeding | 1 (16.7) | 0 | 0 | 0 | 1 (16.7) | 1 (16.7) | 1 (10.0) | 1 (10.0) | 3 (10.0) | 2 (6.7) |
| Blood thyroid stimulating hormone increased | 1 (16.7) | 0 | 0 | 0 | 1 (16.7) | 0 | 1 (10.0) | 0 | 3 (10.0) | 0 |
| Free thyroxine decreased | 0 | 0 | 1 (12.5) | 0 | 1 (16.7) | 0 | 1 (10.0) | 0 | 3 (10.0) | 0 |
SS, safety set; n, number of patients; Q3W, every three weeks; SS.
Table 3.
Overview of safety.
| 1 mg/kg (n = 6) | 3 mg/kg (n = 8) | 10 mg/kg (n = 6) | 200 mg (n = 10) | |
|---|---|---|---|---|
| Any treatment-related adverse event | 6 (100.0%) | 8 (100.0%) | 5 (83.3%) | 9 (90.0%) |
| Grade⩾3 treatment-related adverse event | 1 (16.7%) | 2 (25.0%) | 2 (33.3%) | 5 (50.0%) |
| Treatment-related adverse event leading to treatment discontinuation | 1 (16.7%) | 0 (0.0%) | 1 (16.7%) | 4 (40.0%) |
| Serious treatment-related adverse event | 1 (16.7%) | 1 (12.5%) | 2 (33.3%) | 3 (30.0%) |
| Immune-related treatment-related adverse event | 0 (0.0%) | 1 (12.5%) | 1 (16.7%) | 3 (30.0%) |
n, number of patients.
Grade ⩾3 TRAEs occurred in 10 of 30 patients (33.3%), including bronchitis and anemia in one patient (1 mg/kg), hypoglycemia and epilepsy in one patient (3 mg/kg), gastrointestinal bleeding and thromboembolism in one patient (10 mg/kg), dyspnea, ventricular arrhythmias, hypokalemia, and alkalosis in the same patient (200 mg), and one patient each for dyspnea, anorexia, decreased platelet count, decreased neutrophil count, gastrointestinal bleeding, and hepatitis. Among them, no grade 5 TRAEs occurred, only two patients developed a grade 4 TRAE (one patient in 3 mg/kg developed grade 4 hypoglycemia, the other patient in 10 mg/kg developed grade 4 thromboembolism). Notably, this was not the same as it was reported before: “HX008 was well tolerated at 10 mg/kg with no reported grade 4 or 5 drug-related adverse events”, 11 due to the different cutoff date of data.
Serious AEs related to treatment were observed in seven patients. Six patients (20%) discontinued therapy as a result of TRAEs (one patient each for gastrointestinal hemorrhage, thromboembolism, gastrointestinal bleeding, immune induced hepatitis, immune-mediated pneumonitis, and arrhythmia). One patient died due to respiratory failure, and investigators determined that the death may be unrelated to the treatment.
Pharmacokinetic and immunogenicity evaluations
PK parameters are shown in Table 4 and concentration–time profiles by dose are shown in Figure 1. Pucotenlimab showed a dose-proportional increase in the area under the curve (AUC) and maximum serum concentration between doses of 1 mg/kg and 10 mg/kg. For all doses, the median time to the maximum concentration was within 3.10 h from the end of infusion. The observed mean half-life for pucotenlimab ranged from 17.1 to 23.5 days. After about 21 days of the single administration of pucotenlimab, nearly half of the drug remained, and the continued administration of pucotenlimab could produce pharmacodynamic effects; accordingly, the pucotenlimab administration cycle was set to every 3 weeks. After multiple administrations, a steady state was reached at the 6th cycle for each dose group.
Table 4.
Pharmacokinetic parameters for pucotenlimab.
| 1 mg/kg (n = 6) | 3 mg/kg (n = 8) | 10 mg/kg (n = 6) | 200 mg (n = 10) | |
|---|---|---|---|---|
| Cycle 1 | ||||
| AUC0-t (h*µg/ml) | 3146.93 (483.72) | 11303.14 (4151.55) | 33912.21 (3867.77) | 11792.82 (2447.67) |
| Cmax (µg/ml) | 14.49 (3.00) | 59.49 (17.48) | 169.77 (24.30) | 58.41 (11.72) |
| Tmax (h) | 2.97 (1.03,8.52) | 2.86 (0.98,2.97) | 2.94 (0.98, 9.18) | 2.88 (1.02, 8.83) |
| T1/2 (h) | 553.81 (376.79) | 411.52 (228.83) | 564.21 (129.22) | 522.30 (335.11) |
| Cycle 6 | ||||
| AUC0-t (h*µg/ml) | 8142.02 (3823.98) | 24307.45 (4527.51) | 87646.46 (29017.73) | 31700.03 (4960.76) |
| Cmax (µg/ml) | 27.74 (10.37) | 82.04 (13.20) | 345.72 (84.76) | 102.22 (20.50) |
| Tmax (h) | 2.95 (2.95,3.05) | 3.10 (2.87,3.13) | 2.82 (2.80,8.75) | 2.87 (2.85, 9.12) |
| T1/2 (h) | 441.77 (111.90) | 865.48 (445.15) | 518.86 (112.35) | 915.94 (541.89) |
Mean values (SD) provided, except for Tmax, which is reported as medians (range).
AUC0-t, area under the curve from zero up to a definite time t; Cmax, maximum serum concentration; SD, standard deviation; T1/2, half-life; Tmax, time at Cmax
Figure 1.
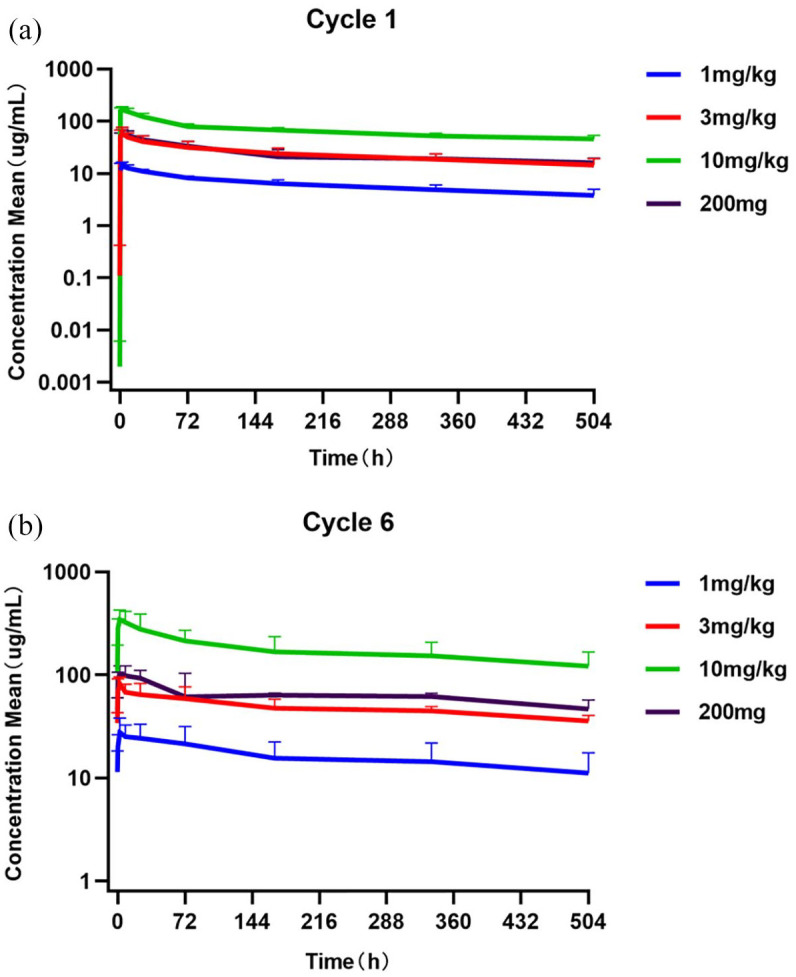
Average pucotenlimab concentration-time curve in each dose group in the first and sixth cycle (semi-logarithmic). (a) Cycle 1. (b) Cycle 6.
Of the 30 patients included in the safety set, the anti-pucotenlimab antibody test results for one patient were positive in cycle 13 (within 60 min before dosing) and 28 days after the last dose in cycle 13 (within 60 min before dosing), which has been described previously. 11 Compared with patients who did not develop antibodies, this patient showed no clinically substantial differences in PK, safety, and effectiveness.
Antitumor activity
The median treatment duration was 2.1 months (range, 0–20.5 months). A total of 17 patients (56.7%) received treatment for ⩾2 months, and 3 patients (10%) received treatment for longer than 12 months. In addition, 29 patients (96.7%) were evaluable for the maximum percentage change in the sum of target lesion diameters from baseline (Figure 2). A waterfall plot of this parameter for each individual patient is shown in [Figure 3(a)]. One patient with pancreatic cancer was not evaluable owing to PD, and was regarded as a non-responder.
Figure 2.

Percentage change from baseline in target lesions over time by dose group.
Figure 3.

Tumor response evaluation. (a) Waterfall plot of the maximum percentage change in the sum of target lesion diameters from baseline in evaluable patients (n = 29). Most patients underwent MMR and MSI assessments (18 for MSI and 15 for MMR); dMMR and MSI-H are indicated by triangles and pentagrams, respectively. (b, c) One PR with tumor shrinkage at 1 mg/kg after 23 weeks of treatment (c) with pucotenlimab compared with baseline (b). (d, e) One PR with tumor shrinkage at 10 mg/kg after 23 weeks of treatment (e) with pucotenlimab compared with baseline (d).
dMMR, deficient MMR; MMR, mismatch repair; MSI, microsatellite instability; MSI-H; MSI high; PR, partial response.
According to RECIST version 1.1, a partial response (PR) was observed in five patients and stable disease (SD) was observed in six patients, resulting in an ORR of 16.7% and DCR of 36.7% (Table 5). Among those showing a response, one patient (1 mg/kg) with a diagnosis of gastric cancer with liver, splenic, and lymph node metastasis, showed a duration of response of approximately 15.2 months, and the best percentage decrease from baseline in the sum of target lesion diameters was 88.5% [Figure 3(b) and (c)]. The response was ongoing until the patient withdrew from the study owing to gastrointestinal bleeding. In one patient (10 mg/kg) with a diagnosis of ovarian endometrioid adenocarcinoma, the duration of response was approximately 4.2 months, and the best percentage decrease from baseline in the sum of target lesion diameters was 78.9% [Figure 3(d) and (e)]. The response was ongoing until the patient withdrew from the study owing to thromboembolism. The other responding patients had a diagnosis of mediastinal thymus tumor (10 mg/kg), non-small cell lung cancer (200 mg), and breast cancer (200 mg). The response durations were 2.9 months, 7 months, and 5.6 months, respectively, after which the patients developed PD. The best percentage decreases from baseline in the sum of target lesion diameters were 27.2%, 48.6%, and 45.5%, respectively.
Table 5.
Best overall response, by treatment group (investigator assessed according to RECIST V1.1 criteria).
| 1 mg/kg | 3 mg/kg | 10 mg/kg | 200 mg | All patients | |
|---|---|---|---|---|---|
| (n = 6) | (n = 8) | (n = 6) | (n = 10) | (n = 30) | |
| CR, n (%) | 0 | 0 | 0 | 0 | 0 |
| PR, n (%) | 1 (16.7) | 0 | 2 (33.3) | 2 (20.0) | 5 (16.7) |
| SD, n (%) | 1 (16.7) | 3 (37.5) | 0 | 2 (20.0) | 6 (20.0) |
| PD, n (%) | 4 (66.7) | 5 (62.5) | 4 (66.7) | 5 (50.0) | 18 (60.0) |
| NE, n (%) | 0 | 0 | 0 | 1 (10.0) | 1 (3.3) |
| ORR (95% CI) | 16.7 (0.4–64.1) | 0 | 33.3 (4.3–77.7) | 20.0 (2.5–55.6) | 16.7 (5.6–34.7) |
| DCR (95% CI) | 33.3 (4.3–77.7) | 37.5 (8.5–75.5) | 33.3 (4.3–77.7) | 40.0 (12.2–73.7) | 36.7 (19.9–56.1) |
CI, confidence interval; CR, complete response; DCR, disease control rate; PR, partial response; SD, stable disease; PD, progressive disease; NE, not evaluable; ORR, overall response rate.
Per investigator review as assessed by iRECIST, a PR was observed in six patients and SD in seven patients. The ORR was 20.0% (95% CI 7.71–38.57) and DCR was 43.3% (95% CI 25.46–62.57).
Biomarker analyses
For 20 patients, tumor samples were available for biomarker assessment. Tumor tissues from 10 patients were used for a PD-L1 expression assay, 18 samples were used for microsatellite instability (MSI) detection, and 15 were used for mismatch repair (MMR) assessment. PD-L1 expression was negative in tumor samples from all 10 patients. Among these 10 patients with PD-L1-negative tumors, the ORR was 30.0% (95% CI 6.67–65.25). These results provide initial evidence for an effect of the drug in PD-L1 negative patients. However, it is not yet possible to confirm the exact relationship between PD-L1 expression and drug efficacy owing to the limited sample size.
Only 1 of 18 samples was classified as MSI high (MSI-H); however, this patient obtained PD/unconfirmed PD with a treatment duration of 0.7 months. Due to the small sample size, it was not possible to evaluate the correlation between MSI type and efficacy. In addition, 2 out of 15 patients had deficient mismatch repair (dMMR) tumors, one of them obtained PR. The correlation between MMR and therapeutic effect could not be confirmed.
Discussion
The primary objective of this study was to investigate the safety, tolerability, and PK profiles of pucotenlimab in 30 patients with advanced solid tumors. No DLT was observed at any dose tested up to 10 mg/kg, and the MTD could not be determined. TRAEs were reported in almost all patients, most of which have been described previous in patients treated with other PD-1/PD-L1 antibodies.8,12–14 Grade ⩾3 TRAEs were observed in 10 patients in our study. We found no major differences in the incidences or severities of TRAEs among the four dose groups.
Notably, proteinuria and hematuria were more frequent than most previously reported immune checkpoint inhibitors. Fortunately, these AEs were low in degree, short in duration, and did not need intervention. In this study, there was no proteinuria or hematuria of grade 3 or above. All patients with proteinuria or hematuria recovered within 47 days without treatment.
It is worth mentioning that grade 3 hypoglycemia and epilepsy occurred in one patient with renal cancer after 3 cycles of 3 mg/kg for three weeks (Q3W) pucotenlimab, and three patients developed gastrointestinal bleeding. One patient with a shrunken tumor discontinued therapy because of thromboembolism, which was judged as ‘possibly related’, and then grade 3 gastrointestinal bleeding occurred during anticoagulation. Another patient with unresected gastric cancer developed positive fecal occult blood (grade 2 gastrointestinal bleeding) after receiving HX008. Another patient with colon cancer had hematochezia (grade 3 gastrointestinal bleeding); digital subtraction angiography (DSA) showed staining in ileocecal mass and there was no bleeding after intravascular embolization. These gastrointestinal bleeding incidents might be related to the disease itself and the use of anticoagulation drugs, but as we still could not rule out the possibility of coagulation disorders caused by PD-1 inhibitor, these bleeding were strictly judged as ‘possibly related’.
The half-life of pucotenlimab (17–24 days) is relatively long compared with those of other anti-PD-1 monoclonal antibodies, including nivolumab (12–20 days) 14 and pembrolizumab (14–22 days). 8 S254T/V308P/N434A mutations results in increased Fc Rn binding that extends the half-life of pucotenlimab. Compared with competitors, pucotenlimab is the only anti-PD1 antibody employing extended half-life design in clinics. Based on the half-life data for pucotenlimab, the expansion of the dosing cycle to once every 4 weeks can be explored. At 3 mg/kg Q3W, the targeted Cycle 6 AUC0–336h levels of over 1000 day*μg/ml were reached, comparable to marketed PD-1 inhibitors. 8 Based on the PK and safety analyses, 3 mg/kg Q3W was selected as the recommended phase II dose, with an alternative dosing schedule of 200 mg Q3W expected to achieve similar results.
Despite the heterogeneous and heavily pretreated patient population, our results provide initial evidence for the antitumor activity of pucotenlimab. According to the RECIST version 1.1 standard, reviewed by the researchers, the overall ORR was 16.7% and the overall DCR was 36.7%. We also observed durable responses. In one case, SD was maintained for longer than 2 years.
There is growing evidence that PD-L1 expression, MSI-H, high tumor mutation burden, and immune gene expression signatures are correlated with the high efficacy of PD-1/PD-L1 inhibitors,15,16 indicating the importance of predictive biomarkers for the identification of patients suitable for anti-PD-1 and anti-PD-L1 therapy. However, no predictive biomarkers have been established to date. In our study, objective responses were observed in 3/10 patients (30.0%) with negative PD-L1 expression and in 5/15 patients (33.3%) with microsatellite-stable tumors. This could be explained by the small sizes of patients and tumor samples, making it difficult to detect a relationship between the tumor response and PD-L1 expression and/or MSI-H/dMMR. Further biomarker analyses are on-going.
In conclusion, pucotenlimab at dosages of 1 mg/kg–10 mg/kg Q3W was well tolerated in patients with advanced solid tumors and showed encouraging antitumor activity. Based on the PK and safety characteristics of pucotenlimab, the recommended dosage for phase II/III clinical trials is 3 mg/kg or a fixed dose of 200 mg given every 3 weeks. The efficacy and safety of pucotenlimab at a 200 mg Q3W is being examined in several phase II/III trials, including single-agent (ClinicalTrial.gov identifiers: NCT03704246 and NCT04574817) and combination (ClinicalTrial.gov identifiers: NCT04508803 and NCT04486651) studies. These trials will provide detailed information about the therapeutic potential of pucotenlimab in a range of tumor types.
Supplemental Material
Supplemental material, sj-pdf-1-tam-10.1177_17588359211020528 for Phase I study of pucotenlimab (HX008), an anti-PD-1 antibody, for patients with advanced solid tumors by Rujiao Liu, Wenhua Li, Yanchun Meng, Shuiping Gao, Jian Zhang and Xichun Hu in Therapeutic Advances in Medical Oncology
Acknowledgments
The authors sincerely thank all of the patients, families, nurses, and staff members who participated in our study.
Footnotes
Conflict of interest statement: The authors declare that there is no conflict of interest.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Science and Technology Major Project (2020ZX09201-013); National Natural Science Foundation of China (grant no. 82072915); the Shanghai Municipal Science and Technology Commission Guidance Project, P.R. China (contract no. 18411967800); Shanghai Hospital Development Center (grant no. SHDC12018X03); and the CSCO-ROCHE Cancer Research Fund 2019 (grant no. Y-2019Roche-171).
Supplemental material: Supplemental material for this article is available online.
Contributor Information
Rujiao Liu, Department of Medical Oncology, Fudan University Shanghai Cancer Center, Shanghai, P.R. China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, P.R., China.
Wenhua Li, Department of Medical Oncology, Fudan University Shanghai Cancer Center, Shanghai, P.R. China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, P.R., China.
Yanchun Meng, Department of Medical Oncology, Fudan University Shanghai Cancer Center, Shanghai, P.R. China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, P.R., China.
Shuiping Gao, Department of Medical Oncology, Fudan University Shanghai Cancer Center, Shanghai, P.R. China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, P.R., China.
Jian Zhang, Department of Medical Oncology, Fudan University Shanghai Cancer Center, Shanghai, 200032, P.R. China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, P.R. China.
Xichun Hu, Department of Medical Oncology, Fudan University Shanghai Cancer Center, Shanghai, 200032, P.R. China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, P.R. China.
References
- 1. Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother 2005; 54: 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Disis ML. Immune regulation of cancer. J Clin Oncol 2010; 28: 4531–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jiang X, Wang J, Deng X, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer 2019; 18: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li HY, McSharry M, Bullock B, et al. The tumor microenvironment regulates sensitivity of murine lung tumors to PD-1/PD-L1 antibody blockade. Cancer Immunol Res 2017; 5: 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res 2020; 10: 727–742. [PMC free article] [PubMed] [Google Scholar]
- 6. Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366: 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol 2015; 33: 1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti–PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015; 21: 4286–4293. [DOI] [PubMed] [Google Scholar]
- 9. Shimizu T, Seto T, Hirai F, et al. Phase 1 study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in Japanese patients with advanced solid tumors. Invest New Drugs 2016; 34: 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Constantinidou A, Alifieris C, Trafalis DT. Targeting programmed cell death-1 (PD-1) and ligand (PD-L1): a new era in cancer active immunotherapy. Pharmacol Ther 2019; 194: 84–106. [DOI] [PubMed] [Google Scholar]
- 11. Zhang J, Huang Y, Xi G, et al. HX008: a humanized PD-1 blocking antibody with potent antitumor activity and superior pharmacologic properties. InMabs 2020; 12: 1724751–1724762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khoja L, Butler MO, Kang SP, et al. Pembrolizumab. J Immunother Cancer 2015; 3: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brahmer JR, Hammers H, Lipson EJ. Nivolumab: targeting PD-1 to bolster antitumor immunity. Future Oncol 2015; 11: 1307–1326. [DOI] [PubMed] [Google Scholar]
- 14. Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010; 28: 3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Le DT, Uram JN, Wang H, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372: 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology: mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348: 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-tam-10.1177_17588359211020528 for Phase I study of pucotenlimab (HX008), an anti-PD-1 antibody, for patients with advanced solid tumors by Rujiao Liu, Wenhua Li, Yanchun Meng, Shuiping Gao, Jian Zhang and Xichun Hu in Therapeutic Advances in Medical Oncology