

Abstract
Non-small cell lung cancer (NSCLC) represents one of the primary causes of cancer-related mortality all over the world. Following our initial finding of the upregulated expression of E2F transcription factor-1 (E2F1) in the NSCLC-related microarray, this study aimed to explore the regulatory role of E2F1 and underlying mechanism in NSCLC development. NSCLC cell viability, migration, and invasion were evaluated utilizing Cell Counting Kit 8 (CCK-8), 5-ethynyl-2′-deoxyuridine (EdU), wound-healing, and Transwell assays. Loss- and gain-function assays were performed to determine the effects of the fat mass and obesity-associated protein (FTO)/E2F1/neural epidermal growth factor-like 2 (NELL2) axis on NSCLC cell behaviors in vitro and NSCLC tumor growth in vivo. E2F1 was highly expressed in both NSCLC tissues and cells. E2F1 augmented the viability, migration, and invasion of NSCLC cells, which was attributable to E2F1 transcriptionally activating NELL2. FTO upregulated the expression of E2F1 by inhibiting the m6A modification of E2F1. The FTO/E2F1/NELL2 axis modulated NSCLC cell viability, migration, and invasion in vitro as well as affected NSCLC tumor growth and metastasis in vivo. The FTO/E2F1/NELL2 axis may impart pro-tumorigenic effects on the cell behavior of NSCLC cells and thus accelerate NSCLC progression.
Keywords: non-small cell lung cancer, demethylase, fat mass and obesity-associated protein, E2F transcription factor-1, neural epidermal growth factor-like 2, m6A modification, tumor growth, tumor metastasis
Graphical abstract
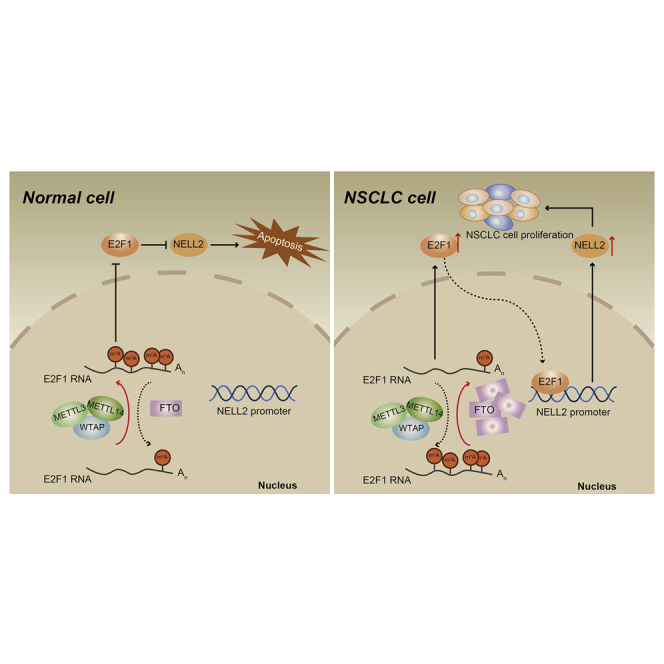
Non-small cell lung cancer (NSCLC) represents one of the primary causes of cancer-related mortality all over the world. This study may elucidate that the FTO/E2F1/NELL2 axis could augment the NSCLC cell behaviors and tumor formation, thus providing novel therapeutic targets for the development of targeted therapy for NSCLC.
Introduction
According to Global Cancer Statistics 2020, lung cancer remained the leading cause of cancer-related death, accounting for 18 percent of the total cancer deaths.1 Among lung cancer subtypes, non-small cell lung cancer (NSCLC) accounts for more than 80 percent of all lung malignancy.2 E2F transcription factor-1 (E2F1) is a key transcriptional factor that has been highlighted for participating in a series of biological processes including cell proliferation and apoptosis,3 oxidative metabolism,4 and cellular senescence.5 Notably, recent studies have pointed out the involvement of E2F1 activation in hepatocarcinogenesis, the progression of hepatocellular carcinoma, and also the brain metastasis of lung adenocarcinoma.6,7 Although there also exist reports on the correlation between E2F1 and NSCLC, such as E2F1 activating IRF5 transcription in NSCLC8 and LINC00461 promoting NSCLC cell proliferation through modulating E2F1,9 the specific regulation mechanism of E2F1 on NSCLC progression remains unclear. In this sense, the present study was aimed to explore the role of E2F1 in the pathogenesis of NSCLC.
Furthermore, a previous case has elucidated that activation of E2F1 could promote the expression of neural epidermal growth factor-like 2 (NELL2) in cancer cells.10 NELL2 represents an extracellular glycoprotein predominantly expressed in the nervous system.11 Interestingly, NELL2 has previously been highlighted for its regulatory effects on the cell behavior of renal cell carcinoma12 and breast cancer.13 Yu et al.14 also indicated that a long noncoding RNA (lncRNA) may promote NSCLC cell proliferation by association with the E2F1/NELL2 axis. Besides, m6A modification disorders may be related to the prognosis of NSCLC,15 and it has been established that fat mass and obesity-associated protein (FTO) could impede the m6A modification of E2F1.16 FTO, well-recognized for its involvement in adipogenesis and obesity in humans, has further been correlated with mitochondrial biogenesis and oxidative stress based on the capability to post-transcriptionally modify relevant genes.17 Due to its role in cellular metabolism, FTO has thus been considered as a promising factor involved in the pathogenesis of various cancers through inducing tumorigenesis and chemoresistance.18,19 Although the expression of FTO has been found to be upregulated in NSCLC,20 there is limited knowledge regarding the specific mechanism of FTO in NSCLC. On the basis of aforementioned evidence, we proposed a hypothesis that the FTO/E2F1/NELL2 axis may confer tumor-supporting roles in the cell behavior of NSCLC cells and thus accelerate NSCLC progression.
Results
E2F1 is highly expressed in both NSCLC tissues and cells
Through bioinformatics analysis, we identified 257 highly expressed genes in NSCLC-related microarray GEO: GSE74706 (Figure 1A), 318 human transcription factors from Cistrome, and 500 candidate NSCLC-related genes from GeneCards, the intersection of which consisted of two key transcription factors correlated with NSCLC, namely EZH2 and E2F1 (Figure 1B). Protein-protein interaction (PPI) analysis then revealed that E2F1 may interacted with 86 proteins. whereas EZH2 with 73 ones (Figure 1C). Although both E2F1 and EZH2 played a key role in the malignant process of NSCLC, we focused on E2F1 in the present study because EZH2 is an oncogene more related with histone-related pathways, whereas E2F1 is a transcription factor with unclear regulation mechanism. Moreover, the expression of E2F1 was found to be obviously upregulated in a NSCLC-related GEO: GSE74706 microarray (Figure 1D), as well as lung adenocarcinoma samples from The Cancer Genome Atlas (TCGA) database (Figure 1E). Results of by quantitative reverse transcription polymerase chain reaction (qRT-PCR) and western blot further revealed elevated levels of E2F1 mRNA and protein in NSCLC cell lines (A549, NCI-H1299, NCI-H460, and NCI-H23) as compared with those in the human normal lung epithelial cell line 16HBE, and A549 cell line was selected for subsequent experiments for presenting with the highest expression of E2F1 (Figures 1F and 1G).
Figure 1.

E2F1 is highly expressed in both NSCLC tissues and cells
(A) Heatmap of the expression of highly expressed genes in the NSCLC-related microarray GEO: GSE74706. (B) Venn map of the intersection of upregulated genes of in the GEO: GSE74706 microarray, human transcription factors from Cistrome, and disease-related genes from GeneCards. (C) The interaction genes of EZH2 and E2F1 predicted by GeneMANIA. (D) Boxplot of E2F1 expression in NSCLC and normal tissues in the GEO: GSE74706 microarray. (E) Boxplot of E2F1 expression in lung adenocarcinoma tissues and adjacent normal tissues retrieved from TCGA database. (F) qRT-PCR measurement of the expression of E2F1 in NSCLC cell lines (A549, NCI-H1299, NCI-H23, NCI-H460) and human lung epithelial cell line 16HBE. (G) Western blot measurement of the expression of E2F1 in NSCLC cell lines (A549, NCI-H1299, NCI-H23, NCI-H460) and human lung epithelial cell line 16HBE. ∗p < 0.05 versus data regarding 16HBE cells. One-way ANOVA with Tukey’s post hoc test was applied for comparison among multiple datasets. Each experiment was conducted in triplicates.
E2F1 augments the viability, migration, and invasion of NSCLC cells
After identifying the upregulation in E2F1 expression in NSCLC cells and tissues, we then explored the specific effects of E2F1 on the NSCLC cell by constructing A549 cells overexpressing/silencing E2F1. Among three short hairpin (sh)RNA targeting E2F1, shRNA targeting (shE2F1-2 was selected for E2F1 knockdown due to its optimal silencing efficiency, as shown by western blot results (Figure 2A). Similarly, overexpression (oe) plasmid E2F1-3 stood out from three E2F1-containing sequences for its optimal overexpression efficiency (Figure 2B) and was thus selected for subsequent experiments.
Figure 2.

E2F1 augments the viability, migration, and invasion of NSCLC cells
(A) Western blot to determine the silencing effect of plasmids carrying different E2F1-targeting shRNAs in NSCLC cells. (B) Western blot to determine the overexpression effect of different E2F1 overexpression plasmids in NSCLC cells. (C) CCK-8 assay to detect A549 cell viability in response to overexpression or silencing of E2F1. (D) EdU staining to detect A549 cell viability in response to overexpression or silencing of E2F1. (E) Wound-healing assay to detect the migration of A549 cells in response to overexpression or silencing of E2F1. (F) Transwell assay to evaluate A549 cell invasion in response to overexpression or silencing of E2F1. ∗p < 0.05. One-way ANOVA was applied for comparison among multiple datasets and two-way ANOVA for comparison among data of multiple datasets at different time points. Each experiment was conducted in triplicates.
According to results of Cell Counting Kit 8 (CCK-8) and 5-ethynyl-2′-deoxyuridine (EdU) staining, E2F1 overexpression enhanced the viability of A549 cells, and E2F1 interference repressed it (Figures 2C, 2D, and S1A). Further, E2F1 overexpression led to promoted migratory and invasive abilities of A549 cells, and E2F1 knockdown led to the opposite, which was reflected by wound-healing and Transwell assays (Figures 2E, 2F, S1B, and S1C). Taken together, E2F1 could trigger the viability, migration, and invasion of NSCLC cells.
E2F1 augments NSCLC cell viability, migration, and invasion through transcriptionally activating NELL2
Subsequently, we explored the downstream mechanism of E2F1 modulating NSCLC cell bioactivity. According to our bioinformatics analysis, E2F1 was predicted to bind to the promoter of NELL2 (Table S1), the expression of which was upregulated in lung adenocarcinoma (Figure 3A) and positively correlated with E2F1 expression (Figure 3B). Further, results of qRT-PCR showed upregulated expression of NELL2 in the four NSCLC cell lines, among which A549 cells presented with the most substantial upregulation (Figure 3C). Then, we validated the binding affinity between E2F1 and NELL2 through the chromatin immunoprecipitation (IP; ChIP) assay (Figure 3D) as well as the interaction between the NELL2 promoter and E2F1 through the dual-luciferase reporter gene assay (Figure 3E). In addition, E2F1 overexpression led to elevated expression of NELL2 in A549 cells, whereas silencing E2F1 led to the opposite, as reflected by qRT-PCR and western blot results (Figures 3F and 3G). Altogether, these results suggested that E2F1 may upregulate the expression of NELL2 in NSCLC cells by activating the NELL2 promoter.
Figure 3.

E2F1 augments NSCLC cell viability, migration, and invasion through transcriptionally activating NELL2
(A) Boxplot of NELL2 expression in lung adenocarcinoma samples from TCGA database. (B) Correlation between the expression of E2F1 and NELL2 in lung adenocarcinoma samples. (C) qRT-PCR to determine NELL2 expression in NSCLC cell lines (A549, NCI-H1299, NCI-H23, NCI-H460) and human lung epithelial cell line 16HBE. (D) ChIP assay to verify the interaction between the E2F1 and NELL2 promoter. (E) Dual-luciferase reporter gene assay to verify the binding between NELL2 and E2F1. (F) qRT-PCR to determine NELL2 expression in the presence of E2F1 overexpression/silencing in A549 cells. (G) Western blot to determine NELL2 expression in the presence of E2F1 overexpression/silencing in A549 cells. (H) qRT-PCR to determine the expression of E2F1 and NELL2 in A549 cells of each group. (I) Western blot to determine the expression of E2F1 and NELL2 in A549 cells of each group. (J) CCK-8 assay to detect the viability of A549 cells of each group. (K) EdU staining to detect the viability of A549 cells of each group. (L) Wound-healing assay to detect the migration of A549 cells of each group. (M) Transwell to assess the invasion of A549 cells of each group. ∗p < 0.05. One-way ANOVA was applied for comparison among multiple datasets and two-way ANOVA for comparison among data of multiple datasets at different time points. Each experiment was conducted in triplicates.
To further evaluate effects of the E2F1/NELL2 axis on NSCLC cells, we constructed A549 cells with altered expression of them. Consequently, the expression of both E2F1 and NELL2 in A549 cells was repressed in response to E2F1 knockdown alone, and additional NELL2 overexpression showed obvious effects on E2F1 expression (Figures 3H and 3I). Moreover, the proliferative potential of A549 cells was attenuated in the presence of E2F1 interference alone, whereas this attenuation was reversed when E2F1 interference was combined with NELL2 overexpression (Figures 3J, 3K, and S1D). Similarly, E2F1 interference also repressed the invasion and migration of the cells, whereas additional NELL2 overexpression could abrogate the inhibiting effects of E2F1 knockdown (Figures 3L, 3M, S1E, and S1F). Collectively, our data indicated that E2F1 may transcriptionally activate NELL2 and thereby enhance the proliferative, migratory, and invasive abilities of NSCLC cells.
FTO upregulates the expression of E2F1 by inhibiting the m6A modification of E2F1
After the identification of the E2F1/NELL2 axis, we then explored the upstream modulator of E2F1. Among 10 potential genes targeting E2F1 m6A modification (METTL3, METL16, IGF2BP3, ALKBH5, ZC3H13, WTAP, VIRMA, FTO, ZCCHC4, and METL14) obtained from bioinformatics analysis, only FTO has been previously reported for impeding the m6A modification of E2F1.13 On this basis, we then determined the expression of FTO in the four NSCLC cell lines, and all of them exhibited upregulation in FTO expression, which was the highest in A549 cells (Figure 4A). Furthermore, silencing/overexpressing FTO in A549 cells resulted in the same variation tendency for E2F1 expression (Figures 4B and 4C). Results of the methylated RNA IP (MeRIP)-PCR assay further revealed the negative correlation between FTO expression and the level of E2F1 m6A modification (Figure 4D). As such, FTO overexpression/interference led to an increased/decreased level of E2F1 pulldown in the photoactivatable ribonucleoside-enhanced crosslinking and IP (PAR-CLIP) assay (Figure 4E). Taken together, these results suggested that FTO could promote the expression of E2F1 by repressing the m6A modification of E2F1.
Figure 4.

FTO upregulates the expression of E2F1 by inhibiting the m6A modification of E2F1
(A) qRT-PCR to measure FTO expression in NSCLC cell lines (A549, NCI-H1299, NCI-H23, NCI-H460) and human lung epithelial cell line 16HBE. (B) qRT-PCR to determine E2F1 expression in A549 cells in the presence of FTO overexpression/knockdown. (C) Western blot to determine E2F1 expression in A549 cells in the presence of FTO overexpression/knockdown. (D) MeRIP-PCR assay to detect the E2F1 m6A modification level in A549 cells in the presence of FTO overexpression/knockdown. (E) PAR-CLIP assay to detect FTO enrichment on E2F1 in the presence of FTO overexpression/knockdown. ∗p < 0.05. One-way ANOVA was applied for comparison among multiple datasets and two-way ANOVA for comparison among data of multiple datasets at different time points. Each experiment was conducted in triplicates.
The FTO/E2F1/NELL2 axis modulates NSCLC cell viability, migration, and invasion
Since the aforementioned results have established the FTO/E2F1/NELL2 axis, we further managed to confirm the effects of the axis on NSCLC cell bioactivities. Through a series of gain- and loss-of-function experiments based on A549 cells, we uncovered that FTO overexpression alone upregulated the expression of FTO, E2F1, and NELL2, and additional E2F1 interference could reverse the upregulation in NELL2, whereas additional NELL2 interference could upregulate FTO and E2F1 expression (Figure 5A). These results further substantiated the FTO/E2F1/NELL2 cascade in NSCLC cells. Subsequently, the viability, migration, and invasion of the cells, as reflected by CCK-8, EdU staining, wound-healing, and Transwell assays, were augmented in the presence of FTO overexpression, and such stimulative effects of FTO overexpression could be abrogated by silencing either E2F1 or NELL2 (Figures 5B−5E). In summary, our data support the view that the FTO/E2F1/NELL2 axis could trigger the viability, migration, and invasion of NSCLC cells.
Figure 5.

The FTO/E2F1/NELL2 axis modulates NSCLC cell viability, migration, and invasion
(A) qRT-PCR to determine the expression of FTO, E2F1, and NELL2 in A549 cells in response to FTO overexpression alone or its combination with E2F1/NELL2 knockdown. (B) CCK-8 assay to detect A549 cell viability in response to FTO overexpression alone or its combination with E2F1/NELL2 knockdown. (C) EdU staining to detect A549 cell viability in response to FTO overexpression alone or its combination with E2F1/NELL2 knockdown. (D) Wound-healing assay to detect A549 cell migration in response to FTO overexpression alone or its combination with E2F1/NELL2 knockdown. (E) Transwell assay to detect A549 cell invasion in response to FTO overexpression alone or its combination with E2F1/NELL2 knockdown. ∗p < 0.05. One-way ANOVA was applied for comparison among multiple datasets and two-way ANOVA for comparison among data of multiple datasets at different time points. Each experiment was conducted in triplicates.
The FTO/E2F1/NELL2 axis affects NSCLC tumor growth and metastasis in vivo
Following the aforementioned in vitro experiments, we then constructed a NSCLC mouse model to substantiate the effects of the FTO/E2F1/NELL2 axis on the NSCLC tumor in vivo. According to the results, FTO overexpression led to increased size and weight of xenografted lung tumors, which could be reversed by additional knockdown of either E2F1 or NELL2 (Figures 6A and 6B). Further, the levels of FTO, E2F1, and NELL2 in the lung tumor tissues were all upregulated following FTO overexpression, whereas the increase in NELL2 expression could be abrogated by E2F1 or NELL2 interference (Figure 6C). We then employed immunohistochemistry to assess the proliferation marker Ki-67, the positive rate of which was shown to be elevated in the lung tissues of nude mice overexpressing FTO and reduced in response to E2F1 or NELL2 knockdown (Figures 6D and S2A). Moreover, the metastatic ability of NSCLC cells, shown by the number of extrapulmonary metastases and fluorescence signal intensity in intravital fluorescence imaging, was enhanced in response to FTO overexpression, and the effects of FTO overexpression were repressed when it was combined with E2F1/NELL2 knockdown (Figures 6E and S2B). Moreover, the expression levels of FTO, E2F1, and NELL2 in tumor tissues in the lung, as reflected by western blot and immunohistochemistry, were upregulated in response to FTO overexpression, and the increase in NELL2 level induced by FTO overexpression alone was reversed when it was combined with knockdown of either E2F1 or NELL2 (Figures 6F, 6G, and S2C). Taken together, the FTO/E2F1/NELL2 axis may stimulate the NSCLC tumor growth and cell metastasis in vivo.
Figure 6.

The FTO/E2F1/NELL2 axis affects NSCLC tumor growth and metastasis in vivo
(A) Volume of lung-xenografted tumors in nude mice in response to FTO overexpression alone or its combination with E2F1/NELL2 knockdown. (B) Weight of lung-xenografted tumors in nude mice in response to FTO overexpression alone or its combination with E2F1/NELL2 knockdown. (C) qRT-PCR determination of FTO-, E2F1-, and NELL2 mRNA-level tumor tissues of nude mice in response to FTO overexpression alone or its combination with E2F1/NELL2 knockdown. (D) Immunohistochemical staining to measure the expression of proliferation marker Ki-67 in the lung tumor tissues of nude mice in response to FTO overexpression alone or its combination with E2F1/NELL2 knockdown. (E) The number and distribution of extrahepatic metastases in nude mice observed by fluorescence imaging. (F) Western blot determination of FTO, E2F1, and NELL2 protein expression in xenografted tumor tissues. (G) Immunohistochemical detection of FTO, E2F1, and NELL2 protein-positive rates in xenografted tumor tissues. n = 6. One-way ANOVA was applied for comparison among multiple datasets and repeated-measurement ANOVA for comparison among data of multiple datasets at different time points. Each experiment was conducted in triplicates.
Discussion
Approximately 85% of patients with lung cancer suffer from a group of histological subtypes collectively known as NSCLC.21 The past decade has witnessed the growing interest in the investigation of molecularly targeted agents for the treatment of NSCLC, especially that of early-stage NSCLC.22 In the present study, we illuminated that the FTO/E2F1/NELL2 axis could promote NSCLC development via triggering the viability, migration, and invasion of NSCLC cells.
Our initial finding of the upregulated expression of transcriptional factor E2F1 in NSCLC tissues from NSCLC-related microarray indicated the potential role of E2F1 in NSCLC development. In relation to this, accumulating evidence also revealed the involvement of E2F1 in the progression of NSCLC.8,23 We then validated the upregulation of E2F1 expression in A549 cells, a NSCLC cell line, and uncovered that E2F1 could augment the viability, migration, and invasion of NSCLC cells. Our findings corroborate several previous studies where E2F1 was found to promote the proliferation and migration of NSCLC cells through activating transforming growth factor (TGF)-beta and also other pathways.14,24 Besides, it has been elucidated that downregulation of serine-arginine protein kinase exerted promoting effects on cell-cycle arrest in NSCLC though modulating E2F1,25 and the CDK4-pRB-E2F1 pathway regulated cell metastasis in NSCLC,26 which further supports our finding that E2F1 could trigger NSCLC cell viability, migration, and invasion.
By continuing to explore the regulation mechanism with E2F1 as the center, we confirmed NELL2 as the downstream target gene of E2F1, and FTO, a m6A demethylase, as the upstream modulator of E2F1 m6A modification. We observed that FTO upregulated the expression of E2F1 by inhibiting the m6A modification of E2F1. In agreement with our finding, activation of E2F1 has previously been reported for inducing the expression of NELL2 in cancer cells,10 and FTO has been highlighted for repressing the m6A modification of E2F1.16 Afterward, we substantiated through a series of gain- and loss-of-function experiments that E2F1 overexpression accelerated NSCLC cell behaviors through transcriptionally activating NELL2, and the FTO/E2F1/NELL2 axis positively modulates NSCLC cell viability, migration, and invasion. Our findings are consistent with quite a few previous reports. For instance, Yu et al.14 have indicated that a lncRNA may trigger NSCLC cell viability through the E2F1/NELL2 axis; the upregulated expression of FTO has been identified in NSCLC, responsible for the accelerated growth of lung cancer cells through affecting the m6A level of mRNAs.20,27 Furthermore, our in vivo experiments validated that the FTO/E2F1/NELL2 axis may stimulate the NSCLC tumor growth and cell metastasis.
Taken together, the data acquired in the present study may lead to a conclusion that the FTO/E2F1/NELL2 axis could augment NSCLC cell viability, migration, and invasion in vitro and also promote NSCLC tumor formation and metastasis in vivo (Figure 7). Herein, our findings deepened our understanding of the pathogenesis of NSCLC and more importantly, provided novel insights into the development of targeted therapy for NSCLC treatment based on the newly discovered therapeutic targets.
Figure 7.

The regulation mechanisms of the FTO/E2F1/NELL2 axis in NELL2
FTO upregulates E2F1 through inhibiting E2F1 m6A modification, enhances the expression of NELL2, and thus promotes the growth and metastasis of NSCLC.
Materials and methods
Ethics statement
Animal experiments were approved by the Animal Care and Use Committee of the First Affiliated Hospital of Jinzhou Medical University (approval number: JUH1-19043) and performed in accordance with Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Bioinformatics analysis
Differentially expressed genes were identified in NSCLC-related microarray GEO: GSE74706 retrieved from the GEO database using Limma package in R language. NSCLC-related transcription factors were obtained from Cistrome and GeneCards by taking the intersection and then validated with GeneMANIA and TCGA. Then, potential downstream targets of these factors were analyzed with University of California, Santa Cruz (UCSC) data and hTFtarget tools. Besides, upstream targets of these factors were predicted using the m6A2Target database.
Cell culture and grouping
Human NSCLC cell lines (A549, NCI-H1299, NCI-H460, and NCI-H23) and a human normal lung epithelial cell line (16HBE) were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences. NSCLC cells were cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA), and 16HBE cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen), both of which were supplemented with 10% fetal bovine serum (FBS; Invitrogen) and penicillin-streptomycin (Sigma, USA) and incubated under 37°C and 5% CO2. When cells grew adherent to the vessel walls, they were digested with 0.25% trypsin (Sigma, St. Louis, MO, USA), and the cells in the logarithmic growth phase were selected for subsequent experiments.
Overexpression and silencing plasmids used for cell transfection were synthesized by Gemma Gene (Shanghai, China). Upon cells seeded in a six-well plate (5 × 105 cells/well) reaching the confluence of 70%, the transfection was performed following the protocols of the Lipofectamine 2000 kit (Invitrogen). After 48 h of incubation, cells were harvested to detect the transfection efficiency. The cells were divided into 14 groups and respectively transfected with plasmids overexpressing FTO/E2F1/NELL2 or shuttling shRNA targeting FTO/E2F1/NELL2 or corresponding negative control (NC), as listed in Table S2.
RNA extraction and qRT-PCR
The mRNA expression of FTO, E2F1, and NELL2 in tissues and cells was determined with qRT-PCR. Total RNA was utilizing the TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA), and 1 μg of it was reversely transcribed into cDNA following the instructions of the PrimeScript RT Reagent Kit with genomic DNA (gDNA) Eraser (Takara, Shiga, Japan). Then, the qRT-PCR was performed using SYBR Premix Ex Taq II (Takara, Japan) and ABI PRISM 7500 RT-PCR system (ABI, Foster City, CA, USA), with β-glucuronidase (GUSB) as the internal control. The relative quantification (2−△△CT) was used to calculate the relative transcription level of target genes: △△Ct = △Ct experimental group − △Ct control group, △Ct = Ct (target gene) − Ct (internal control). Each sample was repeated in three wells. The primers (Invitrogen) involved were listed in Table S3.
Western blot assay
Tissues and cells were digested with radioimmunoprecipitation assay (RIPA) lysis buffer (R0010; Solarbio, Beijing, China), followed by the determination of protein concentration utilizing a bicinchoninic acid (BCA) detection kit (GBCBIO Technologies, Guangzhou, Guangdong, China). Then, the protein was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), electro-transferred to polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA), and blocked with 5% skim milk powder for 2 h to suppress non-specific binding. Afterward, the proteins were incubated overnight at 4°C with diluted primary antibodies, including anti-FTO (1:1,500, ab126605; Abcam, Cambridge, UK), anti-E2F1 (1:1,000, ab179445; Abcam), anti-NELL2 (1:1,000, ab182002; Abcam), and anti-β-actin (1:1,000; Sigma, St. Louis, MO, USA) antibodies. After washing, the membrane was further incubated for 2 h with goat anti-rabbit immunoglobulin G (IgG) secondary antibody (ab97051, 1:2,000; Abcam). The protein bands were then visualized by enhanced chemiluminescence reagent and exposed to UV light using Image Quant LAS 4000C (GE Healthcare, Waukesha, WI, USA). Further, the gray level of protein bands was quantified with the Quantity One version (v.)4.6.2 analysis software, and the protein level was normalized to β-actin. Each experiment was conducted in triplicates to take the average.
MeRIP-PCR
Total RNA was extracted from NSCLC cells of each group using TRIzol reagent, and mRNA was then isolated from it using PolyATtract mRNA Isolation Systems (A-Z5300; Aide Technology, Beijing, China). Next, IP buffer (20 mM Tris, pH 7.5, 140 mM NaCl, 1% NP-40, 2 mM EDTA), supplemented with anti-m6A antibody (1:500, ab151230; Abcam) or anti-IgG antibody (ab109489, 1:100; Abcam), was incubated with protein A/G magnetic beads for 1 h. The purified bead-antibody complex as well as mRNA was then added to the IP buffer, supplemented with ribonuclease inhibitor and protease inhibitor, allowed to stand overnight at 4°C. Subsequently, the RNA was eluted with elution buffer, followed by extraction and purification using phenol-chloroform. The mRNA expression of E2F1 was then analyzed by qRT-PCR.
PAR-CLIP
NSCLC cells were incubated with 200 μM 4-thiopyridine (4SU) (Sigma-Aldrich, USA) for 14 h and crosslinked under radiant exposure of 0.4 J/cm2 at 365 nm. After lysis, IP was performed with FTO antibody (ab124892; Abcam) or goat anti-mouse IgG (ab205718; Abcam), followed by overnight incubation at 4°C. Next, precipitated RNA was labeled with [γ-32-P]-ATP and observed by autoradiography. The precipitate was then treated with proteinase K to remove protein, and RNA was extracted for qRT-PCR determination of E2F1 expression, with IgG as the control.
CCK-8
The viability of NSCLC cells was detected with the CCK-8 assay following the protocols of the CCK-8 kit (Dojindo, Kumamoto, Japan). The optical density (OD) was measured with a microplate reader at 450 nm.
EdU fluorescent staining
Mitochondrial DNA of NSCLC cells was fluorescently labeled with the EdU kit (KeyGEN, Nanjing, Jiangsu, China) as previously described.28 The percentage of EdU-positive cells was then calculated under a fluorescence microscope to evaluate the viability of the cells.
Wound-healing assay
Cells were seeded at a density of 5 × 105 cells/well into a 6-well plate with horizon markings on the outer bottom, followed by 24 h incubation at 37°C with 5% CO2. Then, lines were etched on the bottom of the plate following the markings. After PBS washing, cells were incubated for another 24 h. Cell migration was then observed under a microscope and analyzed with ImageJ software.
Transwell assay
Transwell system with upper chamber pre-coated by Matrigel (356234; Becton Dickinson, Franklin Lakes, NJ, USA) was incubated at 37°C for 30 min to polymerize Matrigel. Cells were cultured in a serum-free medium for 12 h, harvested, and resuspended in the medium (1 × 105/mL). Then, 10% FBS was added to the lower chamber and 100 μL of cell suspension was added to the upper chamber, followed by 24 h incubation at 37°C. Invaded cells were fixed with 100% methanol and stained with 1% toluidine blue (Sigma), and the stained cells were counted in 5 randomly selected fields of view under an inverted optical microscope (Carl Zeiss, Jena, Germany).
Dual-luciferase reporter gene assay
The potential E2F1 binding-site sequence in the NELL2 promotor and a corresponding mutated sequence were cloned into the pGLO vector to construct pGLO-NELL2-wild-type (WT) and pGLO-NELL2-mutant (MUT) reporter plasmids. The two reporter plasmids were co-transfected into HEK293T cells (ATCC, Manassas, VA, USA) using the Lipofectamine 2000 kit (Invitrogen), and the Renilla luciferase plasmid pRL-SV40 was transfected as a control. After 24 h of transfection, the luciferase activity was measured using the dual-luciferase reporter gene-detection system (Promega, Madison, WI, USA). The relative luciferase activity was normalized by calculating the ratio of the firefly luciferase activity to that of Renilla luciferase.
ChIP assay
The ChIP assay was performed with the EZ-ChIP Kit (Millipore) following its protocols. The anti-E2F1 antibody (ab179445; Abcam) and normal rabbit IgG (ab172730; Abcam) were used to precipitate protein-DNA complexes, followed by qRT-PCR to measure NELL2 expression in the precipitated DNA.
Establishment of the NSCLC mouse model
A total of 24 specific pathogen-free (SPF) BALB/c male nude mice (aged 5 to 6 weeks) in good body condition were purchased from Hunan Slac Laboratory Animals (Changsha, China) and housed in separate cages (a rat per cage) in a SPF laboratory under 60%−65% humidity, 22°C−25°C. The nude mice were randomly divided into 4 groups (n = 6) and respectively subjected to subcutaneous injection (into the left thoracic cavity) of NSCLC cells (5 × 106) overexpressing FTO alone, overexpressing FTO and silencing E2F1, overexpressing FTO and silencing NELL2, and expressing NCs. On the 7th day after inoculation, the weight of nude mice was measured every 3 days, accompanied by observation and photography of tumor growth. Subsequently, the nude mice were sacrificed by cervical dislocation on the 25th day, whereupon the extrapulmonary metastases were observed with in vivo fluorescence imaging, and the metastases outside of the lung were counted. Pathological samples were then collected from lung in situ lesions and metastatic nodules. Xenografted tumors were weighed, and tumor tissues were extracted for qRT-PCR to analyze the mRNA expression of FTO, E2F1, and NELL2. The remaining tumor tissues were fixed in 10% neutral formalin for 24 h and prepared into paraffin-embedded sections for immunohistochemical staining to determine Ki-67 expression.
Immunohistochemistry
The paraffin-embedded sections were deparaffinized and treated with (pH 6.0) citric acid buffer for retrieval of antigens, followed by 20 min incubation in a pressure cooker at 104°C and 10 min cooling at room temperature. The sections were then blocked with a mouse IgG blocking agent (#MKB-2213; Vector Labs) for 60 min and quenched with 0.03% H2O2 and sodium azide for 5 min. Subsequently, the sections were incubated with primary mouse monoclonal anti-Ki-67 antibody (1:100, #VP-K452; Vector Labs) or rabbit polyclonal anti-FTO antibody (1;1,000, #PA5-84785; Thermo Fisher Scientific) for 60 min, followed by 20 min incubation with the anti-mouse horseradish peroxidase (HRP)-labeled secondary antibody (K4007, Dako Envision + System; Dako, Carpinteria, CA, USA). After visualization using DAB + chromogen (ab64238; Abcam) for 5 min, the sections were observed with microscopy, and the positive cells in each field of view were counted.
In vivo fluorescence imaging
Following the aforementioned establishment of NSCLC mouse models with altered expression of FTO/E2F1/NELL2, the growth site and range of xenografted tumors as well as the distribution of extrapulmonary metastases in mice were weekly detected with the intravital fluorescence imaging system Kodak IS2000MM, and images were photographed.
Statistical analysis
Data in this study were processed utilizing SPSS v.21.0 (IBM, Armonk, NY, USA) software. Measurement data were summarized as mean ± standard error of the mean (SEM). Unpaired t test was applied for comparison between data of two groups, and two-way analysis of variance (ANOVA) with Tukey’s post hoc test was performed for comparison among data of multiple groups at different time points. Moreover, p < 0.05 indicated a statistically significant difference.
Acknowledgments
The authors would like to acknowledge the helpful suggestions concerning this study received from their colleagues. The datasets generated/analyzed during the current study are available.
Author contributions
Y.W. and M.L. wrote the paper. L.Z. and Y.C. conceived the experiments and collected and provided the sample for this study. S.Z. and Y.W. analyzed the data. All authors have read and approved the final submitted manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.04.011.
Supplemental information
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021 doi: 10.3322/caac.21660. Published online February 4, 2021. [DOI] [PubMed] [Google Scholar]
- 2.Zhou X., He X., Shi K., Yuan L., Yang Y., Liu Q., Ming Y., Yi C., Qian Z. Injectable Thermosensitive Hydrogel Containing Erlotinib-Loaded Hollow Mesoporous Silica Nanoparticles as a Localized Drug Delivery System for NSCLC Therapy. Adv. Sci. (Weinh.) 2020;7:2001442. doi: 10.1002/advs.202001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimova D.K., Dyson N.J. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–2826. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- 4.Blanchet E., Annicotte J.S., Lagarrigue S., Aguilar V., Clapé C., Chavey C., Fritz V., Casas F., Apparailly F., Auwerx J., Fajas L. E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol. 2011;13:1146–1152. doi: 10.1038/ncb2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie Q., Peng S., Tao L., Ruan H., Yang Y., Li T.M., Adams U., Meng S., Bi X., Dong M.Q., Yuan Z. E2F transcription factor 1 regulates cellular and organismal senescence by inhibiting Forkhead box O transcription factors. J. Biol. Chem. 2014;289:34205–34213. doi: 10.1074/jbc.M114.587170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song H., Liu Y., Li X., Chen S., Xie R., Chen D., Gao H., Wang G., Cai B., Yang X. Long noncoding RNA CASC11 promotes hepatocarcinogenesis and HCC progression through EIF4A3-mediated E2F1 activation. Clin. Transl. Med. 2020;10:e220. doi: 10.1002/ctm2.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoj J.P., Mayro B., Pendergast A.M. The ABL2 kinase regulates an HSF1-dependent transcriptional program required for lung adenocarcinoma brain metastasis. Proc. Natl. Acad. Sci. USA. 2020;117:33486–33495. doi: 10.1073/pnas.2007991117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng D.D., Cao Q., Zhang D.Q., Wu X.L., Yang C.X., Chen Y.F., Yu T., Qi H.X., Zhou G.P. Transcription factor E2F1 positively regulates interferon regulatory factor 5 expression in non-small cell lung cancer. OncoTargets Ther. 2019;12:6907–6915. doi: 10.2147/OTT.S215701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meng Q., Liu M., Cheng R. LINC00461/miR-4478/E2F1 feedback loop promotes non-small cell lung cancer cell proliferation and migration. Biosci. Rep. 2020;40 doi: 10.1042/BSR20191345. BSR20191345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim D.H., Roh Y.G., Lee H.H., Lee S.Y., Kim S.I., Lee B.J., Leem S.H. The E2F1 oncogene transcriptionally regulates NELL2 in cancer cells. DNA Cell Biol. 2013;32:517–523. doi: 10.1089/dna.2013.1974. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Nakamoto C., Durward E., Horie M., Nakamoto M. Nell2 regulates the contralateral-versus-ipsilateral visual projection as a domain-specific positional cue. Development. 2019;146:dev170704. doi: 10.1242/dev.170704. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura R., Oyama T., Tajiri R., Mizokami A., Namiki M., Nakamoto M., Ooi A. Expression and regulatory effects on cancer cell behavior of NELL1 and NELL2 in human renal cell carcinoma. Cancer Sci. 2015;106:656–664. doi: 10.1111/cas.12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang W., Guo X., Niu L., Song D., Han B., Zhang H. Identification of key molecular targets that correlate with breast cancer through bioinformatic methods. J. Gene Med. 2020;22:e3141. doi: 10.1002/jgm.3141. [DOI] [PubMed] [Google Scholar]
- 14.Yu L., Fang F., Lu S., Li X., Yang Y., Wang Z. lncRNA-HIT promotes cell proliferation of non-small cell lung cancer by association with E2F1. Cancer Gene Ther. 2017;24:221–226. doi: 10.1038/cgt.2017.10. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y., Guo X., Zhao M., Ao H., Leng X., Liu M., Wu C., Ma J., Zhu J. Contributions and prognostic values of m6 A RNA methylation regulators in non-small-cell lung cancer. J. Cell. Physiol. 2020;235:6043–6057. doi: 10.1002/jcp.29531. [DOI] [PubMed] [Google Scholar]
- 16.Zou D., Dong L., Li C., Yin Z., Rao S., Zhou Q. Correction to: The m6A eraser FTO facilitates proliferation and migration of human cervical cancer cells. Cancer Cell Int. 2020;20:423. doi: 10.1186/s12935-020-01473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang X., Liu B., Nie Z., Duan L., Xiong Q., Jin Z., Yang C., Chen Y. The role of m6A modification in the biological functions and diseases. Signal Transduct. Target. Ther. 2021;6:74. doi: 10.1038/s41392-020-00450-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tao L., Mu X., Chen H., Jin D., Zhang R., Zhao Y., Fan J., Cao M., Zhou Z. FTO modifies the m6A level of MALAT and promotes bladder cancer progression. Clin. Transl. Med. 2021;11:e310. doi: 10.1002/ctm2.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qing Y., Dong L., Gao L., Li C., Li Y., Han L., Prince E., Tan B., Deng X., Wetzel C. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m6A/PFKP/LDHB axis. Mol. Cell. 2021;81:922–939.e9. doi: 10.1016/j.molcel.2020.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J., Han Y., Zhang H., Qian Z., Jia W., Gao Y., Zheng H., Li B. The m6A demethylase FTO promotes the growth of lung cancer cells by regulating the m6A level of USP7 mRNA. Biochem. Biophys. Res. Commun. 2019;512:479–485. doi: 10.1016/j.bbrc.2019.03.093. [DOI] [PubMed] [Google Scholar]
- 21.Herbst R.S., Morgensztern D., Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 22.Duma N., Santana-Davila R., Molina J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019;94:1623–1640. doi: 10.1016/j.mayocp.2019.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Yin J., Fu W., Dai L., Jiang Z., Liao H., Chen W., Pan L., Zhao J. ANKRD22 promotes progression of non-small cell lung cancer through transcriptional up-regulation of E2F1. Sci. Rep. 2017;7:4430. doi: 10.1038/s41598-017-04818-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi J., Li J., Yang S., Hu X., Chen J., Feng J., Shi T., He Y., Mei Z., He W. LncRNA SNHG3 is activated by E2F1 and promotes proliferation and migration of non-small-cell lung cancer cells through activating TGF-β pathway and IL-6/JAK2/STAT3 pathway. J. Cell. Physiol. 2020;235:2891–2900. doi: 10.1002/jcp.29194. [DOI] [PubMed] [Google Scholar]
- 25.Li X., Yang S., Zhang M., Xie S., Xie Z. Downregulation of SRPK2 promotes cell cycle arrest though E2F1 in non-small cell lung cancer. Eur. J. Histochem. 2019;63:3067. doi: 10.4081/ejh.2019.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu B., Liu R. PAQR4 promotes cell proliferation and metastasis through the CDK4-pRB-E2F1 pathway in non-small-cell lung cancer. OncoTargets Ther. 2019;12:3625–3633. doi: 10.2147/OTT.S181432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi H., Zhao J., Han L., Xu M., Wang K., Shi J., Dong Z. Retrospective study of gene signatures and prognostic value of m6A regulatory factor in non-small cell lung cancer using TCGA database and the verification of FTO. Aging (Albany NY) 2020;12:17022–17037. doi: 10.18632/aging.103622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong Z., Liang S., Sanchez-Lopez E., He F., Shalapour S., Lin X.J., Wong J., Ding S., Seki E., Schnabl B. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203. doi: 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.