
Figure 1.
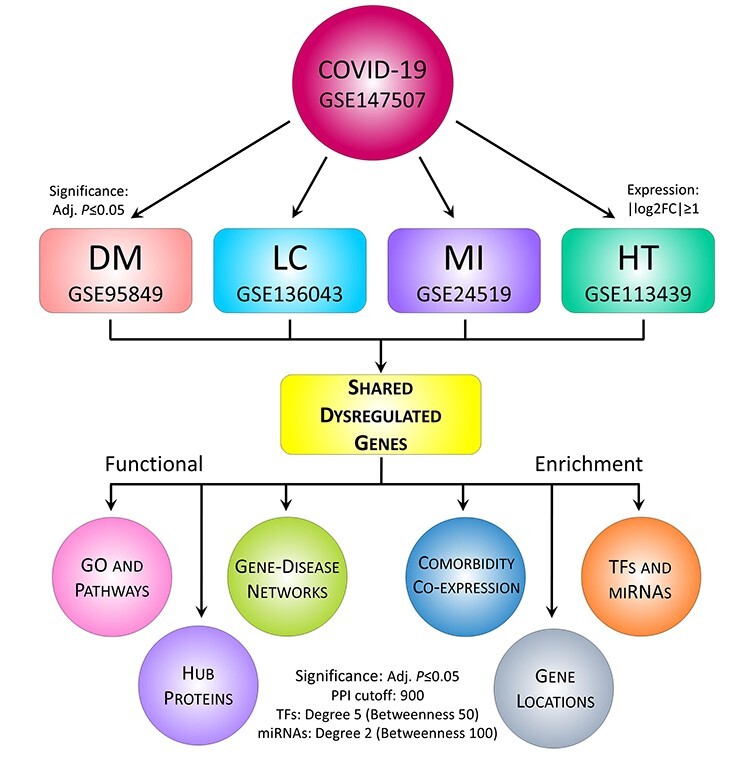
The proposed workflow for deciphering genetic determinants in COVID-19 pathophysiology. The RNA-Seq and microarray datasets of COVID-19 and four other diseases (i.e. DM, MI, LC and HT) were collected and analyzed for comparative gene expression analysis. In doing so, R package DSeq2 and Limma were applied for RNA-Seq normalization and differential expression, respectively. Herein, log2FC ≥ |1| was used to identify DEGs, and statistical significance was set to adjusted P-value ≤ 0.05. Next, the DEGs were subjected to functional enrichment in terms of distributions, GO and pathways, comorbidities, PPI, regulatory biomarkers, PDI and survival analysis. For PPI, the network was constructed with STRING confidence cutoff of 900 and potential hubs were identified using five different methods, while regulatory factors (e.g. TFs and miRNAs) were determined with degree centrality of 5 and 2, respectively. To detect the top-ranked factors, additional filtering was done using betweenness centrality of 50 and 100, respectively. For pathways, we considered Reactome (2016), KEGG (human; 2019) and WikiPathways (human; 2019) databases, while GO terms were determined with Biological Process (2018), Cellular Component (2018) and Molecular Function (2018) databases. In both cases, the adjusted P-value ≤ 0.05 was considered statistically significant.