

Abstract
Fusion transcription factors generated by genomic translocations are common drivers of several types of cancers including sarcomas and leukemias. Oncofusions of the FET (FUS, EWSR1, and TAF15) family proteins result from the fusion of the prion‐like domain (PLD) of FET proteins to the DNA‐binding domain (DBD) of certain transcription regulators and are implicated in aberrant transcriptional programs through interactions with chromatin remodelers. Here, we show that FUS‐DDIT3, a FET oncofusion protein, undergoes PLD‐mediated phase separation into liquid‐like condensates. Nuclear FUS‐DDIT3 condensates can recruit essential components of the global transcriptional machinery such as the chromatin remodeler SWI/SNF. The recruitment of mammalian SWI/SNF (mSWI/SNF) is driven by heterotypic PLD‐PLD interactions between FUS‐DDIT3 and core subunits of SWI/SNF, such as the catalytic component BRG1. Further experiments with single‐molecule correlative force‐fluorescence microscopy support a model wherein the fusion protein forms condensates on DNA surface and enrich BRG1 to activate transcription by ectopic chromatin remodeling. Similar PLD‐driven co‐condensation of mSWI/SNF with transcription factors can be employed by other oncogenic fusion proteins with a generic PLD‐DBD domain architecture for global transcriptional reprogramming.
Keywords: intrinsically disordered proteins, liquid‐liquid phase separation, optical tweezers, protein‐protein interactions, single molecule imaging, transcription factor phase separation, transcriptional condensates
1. INTRODUCTION
FET fusion proteins are key drivers of several types of cancers including sarcomas and leukemias. 1 , 2 These chimeric proteins are created by the fusion of two non‐homologous genes. 3 , 4 , 5 In the case of FET (FUS, EWSR1, and TAF15) family fusion proteins, their N‐terminal disordered domain fuse to the DNA‐binding domain of the transcription factor family ETS (E‐twenty‐six). The N‐terminal domain of FET proteins features a low complexity (LC) sequence enriched in aromatic and polar amino acids (Q/N/Y/S/G) and is classified as “prion‐like.” 6 Prion‐like domains (PLDs) are present in nearly 1.2% of the human proteome, predominantly in ribonucleoproteins (RNPs) including the FET proteins, TDP43, TIA1, and hnRNPA1. 7 , 8 PLD‐containing proteins are highly enriched in various biomolecular condensates such as stress granules and transcriptional hubs. 7 , 9 , 10 , 11 At the molecular level, PLDs enable the formation of dynamic protein condensates through a physical process known as liquid–liquid phase separation (LLPS), which is mediated by multivalent self‐interactions among PLD chains involving distributed aromatic residues. 12
Although the phase separation of PLDs has been well characterized in the context of RNA binding proteins, 13 the impact of PLDs fused to DNA‐binding proteins such as transcription factors is relatively less explored. Transcription factors typically utilize their DNA‐binding domain (DBD) to bind to specific gene loci and use their activation domains to recruit additional regulatory coactivators and RNA Polymerase II. 14 , 15 , 16 , 17 For FET oncofusion transcription factors, PLDs can act as the activation domain and provide additional functionalities to the fusion transcription factors including the capacity to form phase‐separated transcriptional hubs. 18 , 19 Moreover, these fused PLDs may establish new interactions or modulate interactions with existing partners of the parent transcription factor.
Recent studies have provided evidence that FET fusion oncoproteins aberrantly interact with the mammalian SWI/SNF (mSWI/SNF) chromatin remodeling complex. 2 , 20 , 21 , 22 , 23 These abnormal engagements between FET oncoproteins and mSWI/SNF can drive ectopic chromatin remodeling and oncogenic transcriptional events. The mSWI/SNF or BAF is an evolutionarily conserved multiprotein complex that uses ATP hydrolysis to reposition nucleosomes and remodel chromatin landscape. 24 , 25 , 26 The mSWI/SNF represents a wide variety of complexes with varying subunit compositions that are expressed in a developmental stage and tissue‐specific manner. 27 , 28 Predominantly localized at enhancers and promoters, mSWI/SNF plays key roles in regulating transcription. 29 Mutations in subunits of this complex are documented in ~20% of all cancers implying a high propensity for oncogenesis in the event of their deregulation and/ or mislocalization. 20 , 24 , 30 , 31 Since the subunits of mSWI/SNF lack DNA‐recognition motifs, their recruitment to specific genomic locations is typically mediated via their interactions with transcription factors and coactivators. 29 , 32 , 33
In this study, we use the FET oncofusion FUS‐DDIT3/FUS‐CHOP as a model system to investigate the molecular behavior of such fusion oncoproteins and the mechanism of their engagement with the mSWI/SNF chromatin remodeler. FUS‐DDIT3 (Type II fusion 34 ; Figure 1a) is composed of the PLD of FUS fused to the ETS family transcription factor, DDIT3/CHOP (see Table B: Materials & Methods for protein sequence [Appendix S1]), and is detected in more than 90% of the myxoid/round cell liposarcoma. 35 , 36 Our results suggest that the PLD drives phase separation of FUS‐DDIT3 in the mammalian cell nucleus, which is unlike the behavior of the parent protein FUS that remains predominantly soluble due to its interactions with nuclear RNAs. We further uncover the existence of PLDs in multiple subunits of the mSWI/SNF complex that can enable a synergistic engagement with the FET fusion oncoproteins through heterotypic PLD‐PLD interactions. Such dynamic and multivalent interactions, mediated by prion‐like LC domains, provide a molecular mechanism for the recruitment of the mSWI/SNF to FET‐oncoprotein condensates and rewiring of transcriptional programs via altered chromatin dynamics.
FIGURE 1.
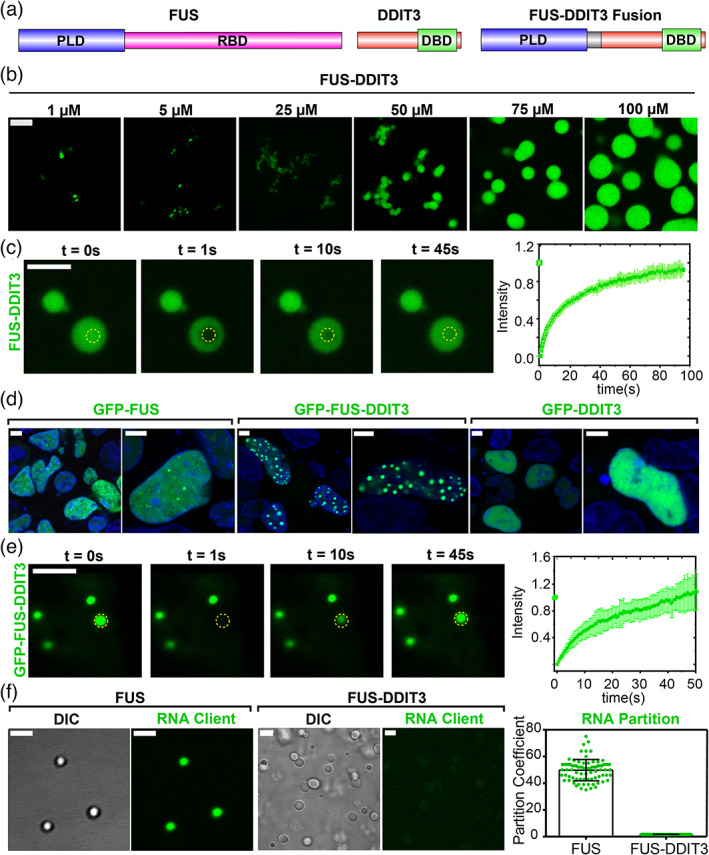
Oncofusion protein FUS‐DDIT3 undergoes liquid–liquid phase separation in vitro and in mammalian cells. (a) Schematics of FUS, DDIT3, and FUS‐DDIT3 domain architectures. DBD, DNA‐binding domain; PLD, prion‐like domain; RBD, RNA‐binding domain. (b) Fluorescence microscopy images of the assemblies formed by recombinantly purified FUS‐DDIT3 (mixed with 250 nM AlexaFluor488‐labeled FUS‐DDIT3) at varying protein concentrations. (c) Representative fluorescence recovery after photobleaching (FRAP) images of recombinant FUS‐DDIT3 condensates at 50 μM (t = 0 s: pre‐bleach; t = 1 s: bleach; t > 1 s: recovery). Right: The FRAP curve shows the average intensity and standard deviation of the intensity profiles over time (n = 3). (d) Fluorescence microscopy images of HEK293T cells expressing GFP‐tagged proteins (FUS, FUS‐DDIT3, or DDIT3), as indicated. Hoechst was used to stain the nucleus and is shown in blue. (e) Representative FRAP images of GFP‐FUS‐DDIT3 condensates expressed in HEK293T cells (t = 0 s: pre‐bleach; t = 1 s: bleach; t > 1 s: recovery). Right: The FRAP curve shows the average intensity and standard deviation of the intensity profiles over time (n = 3). (f) Partitioning of FAM‐labeled RNA client into condensates of recombinant FUS‐DDIT3 (50 μM) and recombinant FUS (6 μM). DIC, differential interference contrast; FAM, 6‐Carboxyfluorescein. Right: Enrichment is calculated as partition coefficients. Mean and standard deviation are shown. The scale bar is 5 μm for all images
2. RESULTS
2.1. FUS‐DDIT3 undergoes LLPS in vitro and in mammalian cells
Prior studies have established a key role for the PLD in driving the phase separation of FUS into liquid‐like condensates. 13 , 37 , 38 We, therefore, wanted to test if the same PLD enables FUS‐DDIT3 fusion protein to undergo a similar liquid phase condensation. Utilizing recombinant FUS‐DDIT3, we found that the fusion oncoprotein forms condensates in vitro in a concentration‐dependent manner (Figure 1b). FUS‐DDIT3 condensates were stable across a broad range of NaCl concentrations (10–300 mM, Figure S1a), and the presence of polymer crowders facilitated their formation (Figure S1b). Fluorescence recovery after photobleaching (FRAP) experiments showed that FUS‐DDIT3 molecules within these condensates are dynamic, implying a liquid‐like behavior (Figure 1c). Interestingly, we observed that FUS‐DDIT3 assemblies appear as a mixture of “irregular” bodies and spherical condensates at lower protein and crowder concentrations but form predominantly large spherical condensates at higher concentrations (CFUS‐DDIT3 > 50 μM and Ficoll PM70 ≥ 10%; Figure 1b, Figure S1b). This is analogous to a previous report on SPOP‐DAXX condensates where competition between inter‐chain interactions of variable strengths determine the material state of the resultant assemblies. 39 , 40 When compared with the PLD of FUS alone, we observed that the FUS‐DDIT3 fusion protein formed condensates at a significantly lower protein concentration (Figure 1b, Figure S1c). These data suggest that DDIT3 may directly contribute to the homotypic interactions between FUS‐DDIT3 molecules. Indeed, recombinantly purified DDIT3 is known to undergo homo‐oligomerization 41 that can increase the valency of the fusion protein and promote phase separation. Consistent with this report, we observed that DDIT3 can form micron‐scale assemblies in vitro that appear gel‐like and are less dynamic than FUS‐DDIT3 assemblies (Figure S1d,e). Therefore, intermolecular PLD‐PLD interactions along with DDIT3‐DDIT3 interactions synergistically facilitate the formation of FUS‐DDIT3 condensates.
To investigate the phase behavior of FUS‐DDIT3 in cell culture models, we employed transgenic expression of GFP‐tagged FUS‐DDIT3 in HEK293T cells and observed the formation of FUS‐DDIT3 enriched spherical assemblies in the nucleus (Figure 1d, center panel). These FUS‐DDIT3 foci were dynamic as evidenced by their near‐complete fluorescence recovery after photobleaching (Figure 1e). We also observed that FUS‐DDIT3 droplet/foci formation was concentration‐dependent as cells with low concentrations of FUS‐DDIT3 lacked these foci (Figure S1f), suggesting that the oncofusion protein's condensation is mediated via an LLPS mechanism. On the contrary, both GFP‐DDIT3 and GFP‐FUS showed a diffused distribution in the nucleus, with small punctate structures observed for GFP‐FUS (Figure 1d). Taken together, these results demonstrate that the PLD of FUS when fused to the transcription factor DDIT3 can drive phase separation of the fusion protein both in vitro and in the cell nucleus.
2.2. FUS‐DDIT3 forms stable condensates within the RNA‐rich nuclear environment
To understand the distinct behavior of FUS‐DDIT3 and the parent FUS protein in the nucleus, we turned to the role of RNA in regulating protein phase separation. FUS is an RNA binding protein, and RNA can regulate the phase behavior and physicochemical properties of FUS condensates. 42 , 43 Although recombinant FUS can undergo phase separation at low protein concentrations (~2 μM) in vitro, which is lower than FUS's nuclear concentration, the high level of RNA in the nucleus suppresses phase separation of FUS. 44 Our previous studies have established that RNA has a dual role in regulating the LLPS of FUS protein. At low RNA concentrations, RNA promotes FUS phase separation via the formation of “sticky” complexes, whereas at higher concentrations, it inhibits FUS condensation due to the formation of negatively charged FUS‐RNA complexes. 43 Since FUS‐DDIT3 lacks an RNA‐binding domain, we hypothesized that FUS‐DDIT3 may have a lower affinity for RNA and therefore, FUS‐DDIT3 condensates may be refractory to the high RNA concentrations within the nucleus. This idea is directly supported by our observations that nuclear FUS‐DDIT3 forms stable condensates whereas nuclear FUS remains predominantly diffused (Figure 1d). To test this model further, we examined the partitioning of a short RNA client ([6FAM]UGAAGGAC) into recombinant FUS and FUS‐DDIT3 condensates in vitro. While FUS condensates could readily interact with and enrich RNA (partition coefficient: 50 ± 8; Figure 1f), FUS‐DDIT3 condensates did not show any significant enrichment (partition coefficient: 1.60 ± 0.03; Figure 1f). We also note a subset of GFP‐FUS‐DDIT3 condensates exhibited hollow spherical morphologies in the nucleus (Figure S2) similar to what was observed previously for the condensates formed by TDP43 mutants with impaired RNA binding ability. 45 Overall, these results indicate that RNA does not significantly interact with FUS‐DDIT3 and provide an explanation for FUS‐DDIT3's ability to form condensates within the RNA‐rich environment of the cell nucleus where wild‐type FUS primarily remains soluble.
2.3. FUS‐DDIT3 condensates enrich BRG1, a catalytic subunit of the mSWI/SNF complex
Nuclear condensates can compartmentalize machineries responsible for many biochemical processes such as transcription, splicing, and chromatin organization. 46 Recent studies have indicated that transcription factor condensates can activate genes by creating phase‐separated transcriptional hubs at super‐enhancer sites that enrich coactivators and RNA polymerase II. 47 , 48 , 49 Based on our observations that FUS‐DDIT3 forms stable nuclear condensates, we aimed to investigate if these condensates can enrich transcriptional activators/coactivators. Previous studies have established that FET fusion protein interactomes are enriched in the components of mSWI/SNF complex and that BRG1, a key ATPase subunit that drives chromatin remodeling by mSWI/SNF, co‐immunoprecipitates with the FUS‐DDIT3 fusion protein. 2 , 22 Therefore, we hypothesized that nuclear FUS‐DDIT3 condensates may compartmentalize BRG1. To test this idea, we expressed GFP‐tagged BRG1 along with mCherry‐tagged FUS‐DDIT3 in HEK293T cells. We observed that BRG1 remained diffused in the nucleus in absence of FUS‐DDIT3, but was readily recruited within FUS‐DDIT3 nuclear condensates when co‐expressed (Figure 2a,b, Figures S3 and S4). Independently, using purified proteins in vitro, we observed that recombinant BRG1 protein is enriched within the FUS‐DDIT3 condensates (partition coefficient: 27 ± 7 [Figure 2c]). These data suggest that phase separation of the FUS‐DDIT3 leads to the enrichment and ectopic compartmentalization of the chromatin remodeler BRG1 into nuclear oncoprotein condensates.
FIGURE 2.
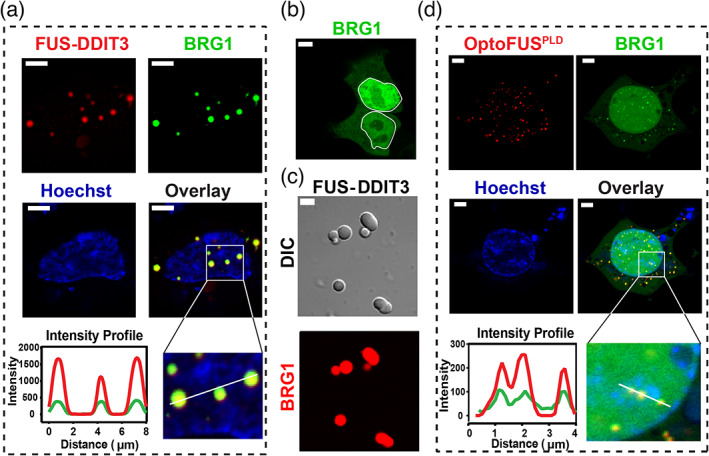
FUS‐DDIT3 condensates enrich mSWI/SNF subunit BRG1 via the FUSprion‐like domain. (a) HEK293T cells co‐expressing GFP‐BRG1 and Cry2‐mCherry‐FUS‐DDIT3. FUS‐DDIT3 condensates were spontaneously formed via protein overexpression (without blue light activation) and co‐localized with GFP‐BRG1 (see Figure S3 top panel for additional data on mCherry‐FUS‐DDIT3 condensates without the Cry2 domain). Hoechst (blue) was used to stain the nucleus. The region demarcated in the white square is magnified and the fluorescence intensity profiles are shown across the linear section (white line). Green represents the intensity profile of GFP‐BRG1 and red represents the profile for Cry2‐mCherry‐FUS‐DDIT3. (b) HEK293T cells expressing the GFP‐BRG1 protein show diffused distribution in both nucleus (demarcated by the white line) and the cytoplasm. (c) Partitioning of RED‐tris‐NTA‐labeled BRG1 (0.5 μM) into recombinant FUS‐DDIT3 condensates (50 μM). (d) HEK293T cells co‐expressing GFP‐BRG1 and Cry2‐mCherry‐FUSPLD (OptoFUSPLD). OptoFUSPLD droplets were formed by activating with blue light for 60 sec and then enrichment of GFP‐BRG1 was analyzed within these light‐induced condensates. Hoechst (blue) was used to stain the nucleus. The region demarcated in the white square is magnified and the fluorescence intensity profiles are shown across the linear section (white line). Green represents the intensity profile of GFP‐BRG1 and red represents the profile for Cry2‐mCherry‐FUSPLD. The scale bar is 5 μm for all images
What is the molecular mechanism of BRG1 recruitment within FUS‐DDIT3 condensates? Linden et al. reported that the N‐terminal prion‐like domains of FET proteins can co‐immunoprecipitate with multiple subunits of the mSWI/SNF complex. 2 In addition to the PLD‐containing FET fusion oncoproteins, PLD‐harboring transcription activators such as EBF1 and MN1 have also been suggested to interact with BRG1. 10 , 50 We, therefore, hypothesized that the PLD of FUS‐DDIT3 is responsible for recruiting BRG1 within FUS‐DDIT3 condensates. To test this, we used a previously characterized OptoFUSPLD construct containing a Cry2 tag that homo‐oligomerizes on exposure to blue light (488 nm) and nucleates FUSPLD condensates. 51 We observed that when co‐expressed with GFP‐BRG1 protein, OptoFUSPLD droplets enriched GFP‐BRG1 (Figure 2d, Figure S4). These results suggest that the PLD of FUS is sufficient to recruit and compartmentalize the chromatin remodeler BRG1 in FUS‐DDIT3 condensates.
2.4. Prion‐like domains can act as scaffolds to recruit mSWI/SNF proteins in FUS‐DDIT3 condensates
PLDs are known to self‐assemble into a variety of assemblages including phase‐separated condensates, amorphous aggregates, and fibrillar solids. 13 , 37 , 38 Furthermore, distinct PLDs are also known to cooperate and co‐condense through heterotypic PLD‐PLD interactions in transcription factor condensates. 11 Given the PLD of FUS is sufficient to recruit BRG1 into FUS‐DDIT3 condensates, we enquired if BRG1 also carries a prion‐like domain. Sequence analysis using the PLAAC algorithm 52 revealed that the N‐terminus of BRG1 (aa 1–340) is disordered and has a significant prion‐like amino acid composition (Figure S5). Therefore, it is conceivable that heterotypic PLDFUS‐PLDBRG1 interactions drive the recruitment of BRG1 into FUS‐DDIT3 condensates. We first characterized the phase separation capability of BRG1PLD and observed that recombinant BRG1PLD has a low intrinsic capacity to phase separate in vitro, only forming condensates at relatively high protein and crowder concentrations (Figure S6a). Furthermore, when expressed in HEK293T cells, OptoBRG1PLD proteins did not form condensates even at concentrations that are ~4‐fold higher than that required for the formation of OptoFUSPLD condensates (Figure 3a, Figure S6b). Thus, the N‐terminal prion‐like domain of BRG1 has a weak capacity to undergo self‐condensation both in vitro and in cells.
FIGURE 3.
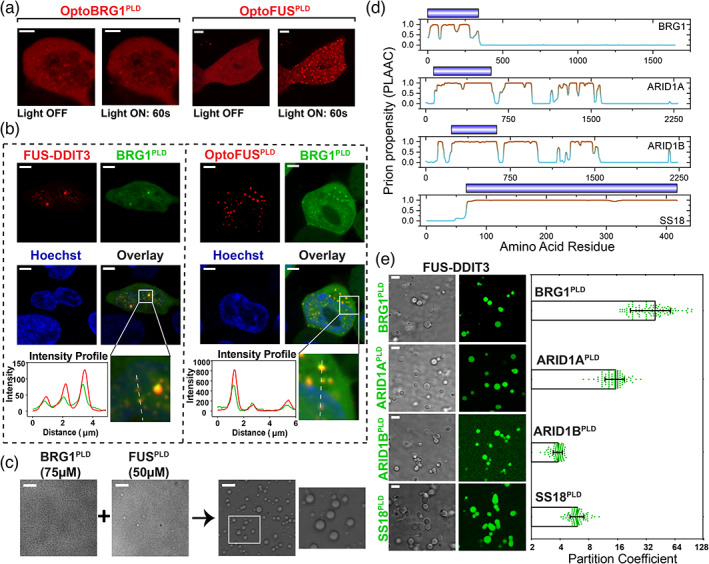
Many mSWI/SNF complex subunits contain PLDs and enrich into FUS‐DDIT3 condensates via heterotypic PLD‐PLD interactions. (a) HEK293T cells expressing OptoBRG1PLD (left) or OptoFUSPLD (right) before and after activation with blue light for 60 sec. Scale bar is 5 μm. (b) HEK293T cells co‐expressing GFP‐BRG1PLD and either Cry2‐mCherry‐FUS‐DDIT3 (left) or OptoFUSPLD (right). OptoFUSPLD droplets were formed by blue light stimulation for 60 sec and then enrichment of GFP‐BRG1PLD was analyzed within the condensates. Cry2‐mCherry‐FUS‐DDIT3 condensates were spontaneously formed via protein overexpression (see Figure S3 middle panel for additional data on mCherry‐FUS‐DDIT3 condensates without the Cry2 domain). Hoechst (blue) was used to stain the nucleus. The region demarcated in the white square is magnified and the fluorescence intensity profile is shown across the linear section (white line). Green represents the intensity profile of GFP‐BRG1PLD and red represents the profile for either Cry2‐mCherry‐FUS‐DDIT3 or OptoFUSPLD. The scale bar is 5 μm. (c) Co‐condensation of purified BRG1PLD and FUSPLD. The region demarcated in the white square is magnified and shown for better clarity. The scale bar is 10 μm. (d) PLAAC analysis showing multiple regions with high prion‐propensity for the four selected subunits of mSWI/SNF complex (see Figure S5 for PLAAC profiles for all PLD‐containing mSWI/SNF complex subunits). Domains corresponding to royal blue bars were recombinantly expressed and purified and used in our experiments (panel e). (e) Partitioning of recombinant PLDs from panel d (fluorescently‐labeled with AlexaFluor488) into FUS‐DDIT3 condensates (50 μM). Enrichment is calculated as partition coefficient. Mean and standard deviation are shown. (Partition coefficients: BRG1PLD = 40 ± 20; ARID1APLD = 15 ± 4; ARID1BPLD = 3.8 ± 0.4; SS18PLD = 6 ± 1). The scale bar is 10 μm
Although the PLD of BRG1 did not undergo phase separation in cells, we tested whether it could engage with the PLD of FUS through heterotypic PLD‐PLD interactions. We expressed GFP‐tagged BRG1PLD in cells expressing OptoFUSPLD. Upon blue light activation, cells expressing OptoFUSPLD formed condensates of FUSPLD that enriched GFP‐BRG1PLD (Figure 3b right panel). Using independent experiments, we further observed that GFP‐BRG1PLD is enriched within the nuclear FUS‐DDIT3 condensates (Figure 3b left panel, Figure S3). Consistent with these results, we also found that when recombinant BRG1PLD and FUSPLD were mixed at a concentration below their individual phase separation thresholds in vitro, they form co‐condensates (Figure 3c, Figure S7). This is likely due to heterotypic PLD‐PLD interactions leading to the lowering of phase separation saturation concentration of the PLD mixture. 53 Thus, BRG1PLD can interact and undergo co‐condensation with the prion‐like domain of FUS.
A broader analysis using PLAAC revealed that in addition to BRG1, multiple subunits of the mSWI/SNF complex contain bona fide PLDs (Figure 3d, Figure S5). These include SMARCC1 and SMARCC2, which along with BRG1, are three of the four core subunits of the complex that orchestrate chromatin remodeling. 54 , 55 In addition, accessory (and/or signature) subunits such as ARID1A and ARID1B, mutations of which are highly correlated with oncogenic transformation, 24 , 56 also have long tracts of prion‐like LC sequences (Figure 3d, Figure S5). To test if these PLDs could also mediate interactions with FET fusion oncoproteins, we reconstituted FUS‐DDIT3 condensates in vitro and probed for the enrichment of PLDs from three SWI/SNF accessory factors, namely, ARID1A, ARID1B, and SS18. All of the tested PLDs were enriched within FUS‐DDIT3 condensates (Figure 3e), although with varied partition coefficients (partition coefficient ranges from ~4 to 40), suggesting differential strengths of their interactions. This can be attributed to distinct sequence compositions, chain lengths, and charge patterns of the tested PLDs. Together, these data suggest that the prion‐like domain of FET fusion oncoproteins can engage with prion‐like domains of multiple mSWI/SNF proteins and recruit them to ectopic nuclear condensates.
2.5. FUS‐DDIT3 forms condensates on dsDNA and recruits chromatin remodeler BRG1
Our results discussed above suggest that the PLD of FUS enables phase separation of the oncogenic FUS‐DDIT3 transcription factor. Transcription factors can interact with the surface of DNA and such interactions can contribute to their phase separation at specific genomic loci. 19 , 57 , 58 To test if FUS‐DDIT3 can form condensates on DNA, we tethered a single double‐stranded (ds) λ‐phage genomic DNA between two optically‐trapped polystyrene beads using laminar‐flow in a microfluidic glass chamber (see Materials & Methods for further details and Figure 4a, Figure S8). Upon transferring this tethered DNA molecule into a channel containing 250 nM AlexaFluor488‐labeled FUS‐DDIT3, we detected the formation of distinct FUS‐DDIT3 clusters on the dsDNA (Figure 4b, top left panel). The FUS‐DDIT3 clusters formed at multiple loci on a single DNA chain. This is likely because FUS‐DDIT3 interacts promiscuously with the λ‐phage genomic DNA. 19 Next, to determine if these FUS‐DDIT3 condensates can recruit BRG1, the single‐molecule DNA tether with FUS‐DDIT3 condensates was transferred to a microfluidic channel containing 10 nM of RED‐tris‐NTA‐labeled BRG1 (Figure S8). In line with our above‐mentioned results, we observed that the full‐length BRG1 co‐localizes with the FUS‐DDIT3 condensates on the surface of dsDNA (Figure 4b, top right panel). The co‐localization of BRG1 within FUS‐DDIT3 condensates was confirmed using the confocal images as well as by analyzing the intensity profiles which revealed a clear overlap of FUS‐DDIT3 and BRG1 peaks (Figure 4b bottom panel). We also noted a few isolated BRG1 peaks that are present across the dsDNA without any detectable spatial overlap with FUS‐DDIT3 condensates and the occurrence of such peaks increases with increasing bulk concentration of BRG1 from 10 to 50 nM (Figure S9). Such foci formation indicates the presence of a possible interaction between BRG1 and the λ‐phage genomic DNA that is independent of FUS‐DDIT3.
FIGURE 4.
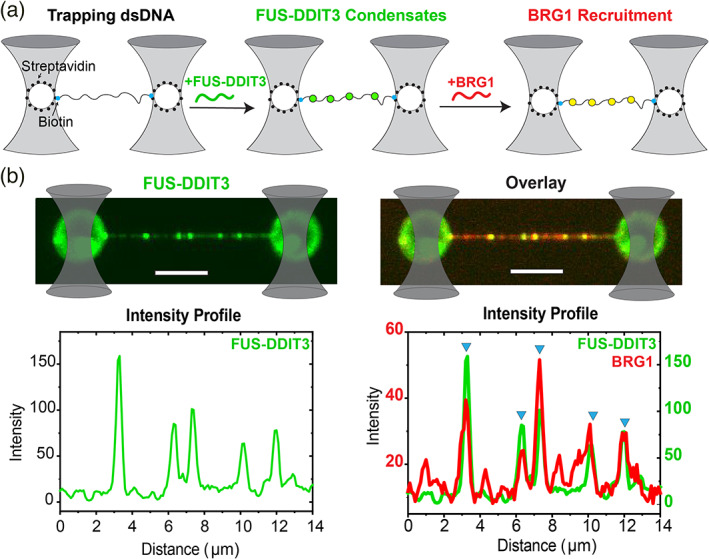
FUS‐DDIT3 condensates on dsDNA recruit BRG1. (a) A schematic representation of the single‐molecule DNA tethering assay. A single molecule of dsDNA (with ends biotinylated; represented by solid cyan circles) is tethered between two optically‐trapped polystyrene beads (traps are shown as grey cones; beads are shown as white circles) coated with streptavidin (solid black circles). The transfer of DNA molecule into a microfluidic channel containing FUS‐DDIT3 leads to the formation of FUS‐DDIT3 condensates on the dsDNA surface (solid green circles). Subsequent transfer of the DNA chain decorated with FUS‐DDIT3 condensates to a separate microfluidic channel containing BRG1 leads to the recruitment of BRG1 into the FUS‐DDIT3 condensates (solid yellow circles). (b) Experimental data: multicolor confocal fluorescence micrographs and intensity profiles showing the formation of FUS‐DDIT3 puncta/condensates (green) on a single DNA molecule followed by the recruitment of BRG1 (red) into the FUS‐DDIT3 condensates. Blue triangles show the position of the overlapping intensity peaks of FUS‐DDIT3 and BRG1 representing the recruitment of BRG1 into FUS‐DDIT3 puncta/condensates. [FUS‐DDIT3] = 250 nM and [BRG1] = 10 nM. The scale bar is 5 μm for all images
2.6. Many recurrent oncogenic translocations result in the fusion of prion‐like domains with transcriptional regulators
Our results, presented so far, indicate that both partners in the fusion pair (i.e., FUSPLD and DDIT3) are likely to contribute to the neomorphic activity of the FET oncofusion protein – the DDIT3DBD can recruit the fusion transcription factor to specific genomic loci, whereas the FUSPLD can drive their condensation and the subsequent recruitment of the chromatin remodeler mSWI/SNF. Since many transcription factors are involved in oncogenic fusions, 59 we asked whether fusion between the DNA localization domain of transcriptional regulators and PLD of prion‐containing proteins represents a generic category of oncogenic transcription regulators. To answer this, we focused on chromosomal translocations involving transcription regulators. Specifically, we retrieved sequences of transcription regulator fusions that are recurrently present in cancer patients using the following selection criteria: (i) present in at least 25 curated patient samples in the COSMIC database, 60 and (ii) present in >10% of tumor samples in a given malignancy (the list of recurrent translocations involving transcriptional regulators was obtained from the Reference 61). We further narrowed down our list to the oncofusions that harbor at least one prion‐like domain and identified a total of 16 different recurrent oncofusions involving 10 fusion pairs (Table 1). These proteins represent in‐frame fusions between a prion‐like domain and a specific DNA localization domain. The DNA localization domains were either a specific DNA‐binding domain (e.g., ERGDBD in FUS‐ERG fusion) or a protein–protein interaction motif that specifically interacts with transcription regulators (e.g., SSXSSXRD in SS18‐SSX family fusions). In all these oncofusions, prion‐like domains and DNA‐localization domains originate from the two separate genes involved in respective in‐frame genomic translocations. Not surprisingly, we find that many of these recurrent oncofusions have already been implicated in chromatin reorganization (Table 1). Therefore, as observed for FUS‐DDIT3 fusion protein in our experiments, other PLD‐containing oncogenic transcription regulators may similarly engage with mSWI/SNF chromatin remodelers to drive aberrant transcriptional outcomes via protein co‐condensation.
TABLE 1.
Recurrent fusions of transcriptional regulators with prion‐like domains
| Fusion pair | Head gene | Head junction | Tail gene | Tail junction | Domain organization | Implicated in chromatin reorganization |
|---|---|---|---|---|---|---|

|

|

|
||||
| (FET protein) – (transcription factor) oncofusions | Yes [2, 20] | |||||
| FUS‐ERG | FUS | 31198157 | ERG | 39755845 |

|
|
| FUS‐ERG | FUS | 31198157 | ERG | 39763637 |

|
|
| EWSR1‐ATF1 | EWSR1 | 29683123 | ATF1 | 51207793 |

|
|
| EWSR1‐ATF1 | EWSR1 | 29683123 | ATF1 | 51208063 |

|
|
| EWSR1‐ATF1 | EWSR1 | 29684775 | ATF1 | 51203238 |

|
|
| EWSR1‐FLI1 | EWSR1 | 29683123 | FLI1 | 128675261 |

|
|
| EWSR1‐FLI1 | EWSR1 | 29683123 | FLI1 | 128679052 |

|
|
| EWSR1‐FLI1 | EWSR1 | 29683125 | FLI1 | 128651853 |

|
|
| EWSR1‐NR4A3 | EWSR1 | 29688595 | NR4A3 | 102591275 |

|
|
| TAF15‐NR4A3 | TAF15 | 34149837 | NR4A3 | 102590321 |

|
|
| (Nuclear receptor) – (transcription factor) oncofusion | N/A | |||||
| HEY1‐NCOA2 | HEY1 | 80678885 | NCOA2 | 71057083 |

|
|
| Fusion between transcription factors | Yes [71, 72] | |||||
| PAX3‐FOXO1 | PAX3 | 223084858 | FOXO1 | 41134997 |

|
|
| PAX7‐FOXO1 | PAX7 | 19029790 | FOXO1 | 41134997 |

|
|
| Fusion between transcription co‐factors | Yes [69, 73] | |||||
| SS18‐SSX1 | SS18 | 23615029 | SSX1 | 48123278 |

|
|
| SS18‐SSX1 | SS18 | 23612362 | SSX1 | 48123216 |

|
|
| SS18‐SSX2 | SS18 | 23612362 | SSX2 | 52729628 |

|
|
Abbreviations: DBD, DNA‐binding domain; PLD, prion‐like domain; PPI motif, protein–protein interaction motif.
3. DISCUSSION
A major consequence of FET oncofusions is the transfer of the prion‐like domain from an RNA‐binding protein to a DNA‐binding protein. Our results described here along with two recent reports suggest that FET‐fusion oncoproteins can form ectopic nuclear condensates. 18 , 19 These reports also suggest that FET‐fusion protein condensates can activate transcription at DNA enhancer sites by engaging with essential transcriptional co‐activators such as BRD4 and by recruiting RNA polymerase II. 18 , 19 In our study, we focus on characterizing the interactions between FET‐fusion condensates with the ATP‐dependent chromatin remodeler complex mSWI/SNF. Chromatin remodeling is one of the primary and essential steps in the regulation of gene expression and the aberrant targeting of mSWI/SNF complexes can be detrimental to physiologic cellular processes. 62 Our results reveal that FUS‐DDIT3 condensates can compartmentalize BRG1, a key catalytic subunit (ATPase) of mSWI/SNF that is responsible for ATP‐dependent remodeling of DNA‐histone interactions. Since a continuous activity of the mSWI/SNF is often required to maintain the appropriate chromatin state at the target genomic locus, 63 , 64 the selective enrichment of mSWI/SNF components by FUS‐DDIT3 condensates provides two possible routes to transcriptional reprograming (Figure 5). First, condensates of FET‐fusion proteins formed at ectopic genomic regions, as specified by binding sites of the DNA‐binding domain, can recruit BRG1 and modify local chromatin dynamics to activate transcription (Figure 5 left panel). Previous studies with FET oncoproteins, such as EWS‐FLI1 and FUS‐DDIT3 showed retargeting of chromatin remodelers to microsatellites and enhancers, respectively, 20 , 65 lending support to this model. Congruently, condensates formed by both FET proteins and FET fusion‐oncoproteins are capable of enhancing gene transcription. 19 , 66 Alternatively, we propose a model where sequestration of mSWI/SNF subunits away from their natural targets and trapping them in ectopic condensates can contribute to a loss‐of‐function phenotype (Figure 5 right panel). This model is consistent with the recently reported observations that mSWI/SNF complex suppresses the H3K27Me3 histone modification under normal conditions, but when FET oncoproteins are expressed, the H3K27Me3 levels within the cells are upregulated likely due to the sequestration of mSWI/SNF away from its physiological target sites. 2
FIGURE 5.
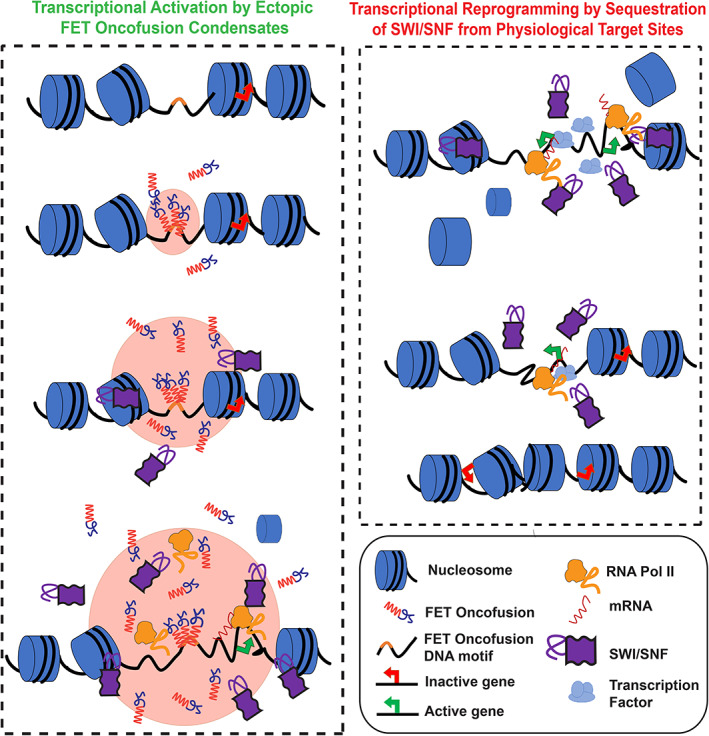
Proposed model for transcriptional reprogramming by FET‐fusion oncoproteins. Left: FET fusion proteins can bind to specific DNA motifs defined by the DNA‐binding domain at physiologically inactive genes to form phase‐separated condensates. The process of LLPS is facilitated by the prion‐like domain. FET oncofusion protein condensates can recruit the ATP‐dependent chromatin remodeler SWI/SNF leading to remodeling of the closed chromatin to an open chromatin state, which subsequently provides access to transcriptional coactivators and RNA polymerase II to mediate transcription of otherwise silenced genes. Right: The sequestration of chromatin remodeler SWI/SNF into FUS‐DDIT3 condensates, as shown in the left panel, can lead to diminished chromatin remodeling activity at physiologically active genes where continuous SWI/SNF activity is required. This sequestration of SWI/SNF away from the physiological target genes may trigger a switch to a closed chromatin state, thereby decreasing their transcriptional output
At the molecular level, the presence of PLDs in multiple subunits of the mSWI/SNF (Figure S5) suggests that PLD‐mediated dynamic interactions could be a generic mechanism for the recruitment of these complexes to their target sites. This is based on our observations that the PLD of FUS is sufficient to target the mSWI/SNF ATPase, BRG1, to FUS‐DDIT3 condensates (Figures 2 and 3). Moreover, the PLDs of different mSWI/SNF proteins can synergistically engage with FETPLD and reduce the phase separation threshold as observed in mixtures of FUSPLD and BRG1PLD (Figure S7). These data also indicate that heterotypic PLD‐mediated co‐condensation may play a fundamental role in the functional assembly of the mSWI/SNF complex itself.
We envision that hijacking of mSWI/SNF complex by FET oncofusions through heterotypic PLD‐PLD interactions is a generic strategy employed by many aberrant transcription regulators. This is supported by our analysis that many recurrent oncogenic translocations involve the fusion of a prion‐like domain to a DNA recruitment domain and result in a widespread reorganization of the chromatin landscape (Table 1). In addition to the FET oncofusions, SS18‐SSX fusions represent an interesting category of oncogenic translocations. In SS18‐SSX fusions, the prion‐like domain of SS18 is fused to the C‐terminal segments of SSX family proteins containing the SSXRD domain. 67 , 68 SSXRD domain is a protein–protein interaction module that directly interacts with the polycomb complex DNA‐binding protein, KDM2B. 69 Not surprisingly, SS18‐SSX fusions recruit mSWI/SNF complex to KDM2B‐binding genomic loci and result in the aberrant activation of numerous otherwise repressed genes. 69 , 70 Therefore, similar to the FUSPLD, it is likely that SS18PLD is capable of recruiting mSWI/SNF chromatin remodelers via SS18PLD‐SWI/SNFPLD interactions. However, unlike FET fusions where the neomorphic transcriptional activator is recruited to specific genomic locations via DNA‐binding domains fused to the FETPLD, SS18‐SSX fusions may utilize specific protein–protein interaction module from SSX family members (i.e, the SSXRD domain) to recruit oncofusion proteins to targeted genomic loci.
In summary, our results provide a molecular mechanism with regards to how FET oncofusions can synergistically engage with the ATP‐dependent chromatin remodeler mSWI/SNF. Our bioinformatics analysis reveals that PLD fusions to DNA‐binding domains go beyond FET fusion oncoproteins and the proposed role of heterotypic PLD‐PLD interactions in recruiting mSWI/SNF complex at non‐native genomic loci may play a central role in all such cases. Future studies can test the generality of this model and subsequently target the phase separation and/or mSWI/SNF complex engagement capacities of PLD‐containing oncofusion proteins for potential cancer therapeutics.
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Richoo Davis: Conceptualization; data curation; formal analysis; methodology; writing‐original draft. Taranpreet Kaur: Data curation; formal analysis; writing‐review & editing. Mahdi Moosa: Conceptualization; methodology; writing‐review & editing. Priya Banerjee: Conceptualization; funding acquisition; investigation; writing‐original draft.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENT
The authors acknowledge Drs. M. C. Yu, J. Lee, and S. Lee of the University at Buffalo for helping with the cell culture experiments. The authors would also like to thank Dr. S. Walker and Mr. A. Sahoo of the University at Buffalo for valuable help with protein purification. Priya R. Banerjee acknowledges the College of Arts and Sciences at the University at Buffalo, SUNY, and the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (R35 GM138186) for financial support.
Davis RB, Kaur T, Moosa MM, Banerjee PR. FUS oncofusion protein condensates recruit mSWI/SNF chromatin remodeler via heterotypic interactions between prion‐like domains. Protein Science. 2021;30:1454–1466. 10.1002/pro.4127
Funding information National Institute of General Medical Sciences, Grant/Award Number: R35 GM138186
DATA AVAILABILITY STATEMENT
All data relevant to the findings of this manuscript are included in the manuscript and the supplementary appendix. Additional data are available from the authors upon reasonable request.
REFERENCES
- 1. Panagopoulos I, Höglund M, Mertens F, Mandahl N, Mitelman F, Aman P. Fusion of the EWS and CHOP genes in myxoid liposarcoma. Oncogene. 1996;12:489–494. [PubMed] [Google Scholar]
- 2. Lindén M, Thomsen C, Grundevik P, et al. FET family fusion oncoproteins target the SWI/SNF chromatin remodeling complex. EMBO Rep. 2019;20:e45766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bunting SF, Nussenzweig A. End‐joining, translocations and cancer. Nat Rev Cancer. 2013;13:443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perry JA, Seong BKA, Stegmaier K. Biology and therapy of dominant fusion oncoproteins involving transcription factor and chromatin regulators in sarcomas. Ann Rev Cancer Biol. 2019;3:299–321. [Google Scholar]
- 5. Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–245. [DOI] [PubMed] [Google Scholar]
- 6. Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. March ZM, King OD, Shorter J. Prion‐like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 1647;2016:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gotor NL, Armaos A, Calloni G, et al. RNA‐binding and prion domains: The yin and Yang of phase separation. Nucleic Acids Res. 2020;48:9491–9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Franzmann TM, Jahnel M, Pozniakovsky A, et al. Phase separation of a yeast prion protein promotes cellular fitness. Science. 2018;359:eaao5654. [DOI] [PubMed] [Google Scholar]
- 10. Wang Y, Zolotarev N, Yang CY, Rambold A, Mittler G, Grosschedl R. A prion‐like domain in transcription factor EBF1 promotes phase separation and enables B cell programming of progenitor chromatin. Immunity. 2020;53:1151–1167. [DOI] [PubMed] [Google Scholar]
- 11. Frazer C, Staples MI, Kim Y, et al. Epigenetic cell fate in Candida albicans is controlled by transcription factor condensates acting at super‐enhancer‐like elements. Nat Microbiol. 2020;5:1374–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martin EW, Holehouse AS, Peran I, et al. Valence and patterning of aromatic residues determine the phase behavior of prion‐like domains. Science. 2020;367:694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang J, Choi JM, Holehouse AS, et al. A molecular grammar governing the driving forces for phase separation of prion‐like RNA binding proteins. Cell. 2018;174:688–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keegan L, Gill G, Ptashne M. Separation of DNA binding from the transcription‐activating function of a eukaryotic regulatory protein. Science. 1986;231:699–704. [DOI] [PubMed] [Google Scholar]
- 15. Triezenberg SJ. Structure and function of transcriptional activation domains. Curr Opin Genet Develop. 1995;5:190–196. [DOI] [PubMed] [Google Scholar]
- 16. Liou S‐H, Singh SK, Singer Robert H, Coleman Robert A, & Liu W‐L. (2021). Structure of the p53/RNA polymerase II assembly. Communications Biology, 4(1). 10.1038/s42003-021-01934-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Keaveney M, Struhl K. Activator‐mediated recruitment of the RNA polymerase II machinery is the predominant mechanism for transcriptional activation in yeast. Mol Cell. 1998;1:917–924. [DOI] [PubMed] [Google Scholar]
- 18. Owen I, Yee D, Wyne H, et al. The oncogenic transcription factor FUS‐CHOP can undergo nuclear liquid‐liquid phase separation. bioRxiv. 2021. 10.1101/2021.02.24.432743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zuo L, Zhang G, Massett M, et al. Loci‐specific phase separation of FET fusion oncoproteins promotes gene transcription. Nat Commun. 2021;12:1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boulay G, Sandoval GJ, Riggi N, et al. Cancer‐specific retargeting of BAF complexes by a prion‐like domain. Cell. 2017;171:163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Riggi N, Knoechel B, Gillespie SM, et al. EWS‐FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell. 2014;26:668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jamie SE, Colborne S, Hughes CS, Morin GB, & Nielsen TO. The FUS‐DDIT3 interactome in myxoid liposarcoma. Neoplasia. 2019;21:740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shorter J. Prion‐like domains program Ewing's sarcoma. Cell. 2017;171:30–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv. 2015;1:e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mashtalir N, D'Avino AR, Michel BC, et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell. 2018;175:1272–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tolstorukov MY, Sansam CG, Lu P, et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc Natl Acad Sci U S A. 2013;110:10165–10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alver BH, Kim KH, Lu P, et al. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun. 2017;8:14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu F, Flowers S, Moran E. Essential role of ARID2 protein‐containing SWI/SNF complex in tissue‐specific gene expression. J Biol Chem. 2012;287:5033–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yudkovsky N, Logie C, Hahn S, Peterson CL. Recruitment of the SWI/SNF chromatin remodeling complex by transcriptional activators. Genes Dev. 1999;13:2369–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kadoch C, Hargreaves DC, Hodges C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PloS One. 2013;8:e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Neely KE, Hassan AH, Wallberg AE, et al. Activation domain–mediated targeting of the SWI/SNF complex to promoters stimulates transcription from nucleosome arrays. Mol Cell. 1999;4:649–655. [DOI] [PubMed] [Google Scholar]
- 33. Vierbuchen T, Ling E, Cowley CJ, et al. AP‐1 transcription factors and the BAF complex mediate signal‐dependent enhancer selection. Mol Cell. 2017;68:1067–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Panagopoulos I, Mandahl N, Ron D, et al. Characterization of the CHOP breakpoints and fusion transcripts in myxoid liposarcomas with the 12;16 translocation. Cancer Res. 1994;54:6500–6503. [PubMed] [Google Scholar]
- 35. Crozat A, Åman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA‐binding protein in human myxoid liposarcoma. Nature. 1993;363:640–644. [DOI] [PubMed] [Google Scholar]
- 36. Rabbitts TH, Forster A, Larson R, & Nathan P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t (12; 16) in malignant liposarcoma. Nat Genet. 1993;4:175–180. [DOI] [PubMed] [Google Scholar]
- 37. Patel A, Lee HO, Jawerth L, et al. A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066–1077. [DOI] [PubMed] [Google Scholar]
- 38. Kato M, Han TW, Xie S, et al. Cell‐free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schmit JD, Bouchard JJ, Martin EW, & Mittag T. Protein network structure enables switching between liquid and gel states. J Am Chem Soc. 2019;142:874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bouchard JJ, Otero JH, Scott DC, et al. Cancer mutations of the tumor suppressor SPOP disrupt the formation of active, phase‐separated compartments. Mol Cell. 2018;72:19–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Singh VK, Pacheco I, Uversky VN, Smith SP, MacLeod RJ, Jia Z. Intrinsically disordered human C/EBP homologous protein regulates biological activity of colon cancer cells during calcium stress. J Mol Biol. 2008;380:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alshareedah I, Kaur T, Ngo J, et al. Interplay between short‐range attraction and long‐range repulsion controls reentrant liquid condensation of ribonucleoprotein–RNA complexes. J Am Chem Soc. 2019;141:14593–14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Banerjee PR, Milin AN, Moosa MM, Onuchic PL, Deniz AA. Reentrant phase transition drives dynamic substructure formation in ribonucleoprotein droplets. Angew Chem. 2017;129:11512–11517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maharana S, Wang J, Papadopoulos DK, et al. RNA buffers the phase separation behavior of prion‐like RNA binding proteins. Science. 2018;360:918–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu H, Lu S, Gasior K, et al. HSP70 chaperones RNA‐free TDP‐43 into anisotropic intranuclear liquid spherical shells. Science. 2021;371:eabb4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sabari BR, Dall'Agnese A, Young RA. Biomolecular condensates in the nucleus. Trends Biochem Sci. 2020;45:961–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boija A, Klein IA, Sabari BR, et al. Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell. 2018;175:1842–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sabari BR, Dall’Agnese A, Boija A, et al. Coactivator condensation at super‐enhancers links phase separation and gene control. Science. 2018;361:eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cho W‐K, Spille JH, Hecht M, et al. Mediator and RNA polymerase II clusters associate in transcription‐dependent condensates. Science. 2018;361:412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Riedel SS, Lu C, & Xie HM, (2021). Intrinsically disordered Meningioma‐1 stabilizes the BAF complex to cause AML. Molecular Cell, 10.1016/j.molcel.2021.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shin Y, Berry J, Pannucci N, Haataja MP, Toettcher JE, Brangwynne CP. Spatiotemporal control of intracellular phase transitions using light‐activated optoDroplets. Cell. 2017;168:159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lancaster AK, Nutter‐Upham A, Lindquist S, King OD. PLAAC: A web and command‐line application to identify proteins with prion‐like amino acid composition. Bioinformatics. 2014;30:2501–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ruff KM, Dar F, Pappu RV. Ligand effects on phase separation of multivalent macromolecules. Proc Natl Acad Sci U S A. 2021;118:e2017184118. 10.1073/pnas.2017184118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mani U, S. AS, Goutham R. N. A, Mohan S. S. SWI/SNF Infobase‐an exclusive information portal for SWI/SNF remodeling complex subunits. PLoS One. 2017;12:e0184445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Phelan ML, Sif S, Narlikar GJ, Kingston RE. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell. 1999;3:247–253. [DOI] [PubMed] [Google Scholar]
- 56. Davoli T, Xu AW, Mengwasser KE, et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Morin JA, Wittmann S, Choubey S, et al. Surface condensation of a pioneer transcription factor on DNA. bioRxiv. 2020. 10.1101/2020.09.24.311712. [DOI] [Google Scholar]
- 58. Renger R, Morin JA, Lemaitre R, et al. Co‐condensation of proteins with single‐and double‐stranded DNA. bioRxiv. 2021. 10.1101/2021.03.17.435834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim P, Ballester LY, Zhao Z. Domain retention in transcription factor fusion genes and its biological and clinical implications: A pan‐cancer study. Oncotarget. 2017;8:110103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tate JG, Bamford S, Jubb HC, et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brien GL, Stegmaier K, Armstrong SA. Targeting chromatin complexes in fusion protein‐driven malignancies. Nat Rev Cancer. 2019;19:255–269. [DOI] [PubMed] [Google Scholar]
- 62. McBride MJ, Kadoch C. Disruption of mammalian SWI/SNF and polycomb complexes in human sarcomas: Mechanisms and therapeutic opportunities. J Pathol. 2018;244:638–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schick S, Grosche S, Kohl KE, et al. Acute BAF perturbation causes immediate changes in chromatin accessibility. Nat Genet. 2021;53:269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Iurlaro M, Stadler MB, Masoni F, Jagani Z, Galli GG, Schübeler D. Mammalian SWI/SNF continuously restores local accessibility to chromatin. Nat Genet. 2021;53:279–287. [DOI] [PubMed] [Google Scholar]
- 65. Chen M, Foster JP, Lock IC, et al. Disruption of oncogenic targeting by ISWI via phosphorylation of a prion‐like domain. bioRxiv. 2020. 10.1101/2020.03.11.987750. [DOI] [Google Scholar]
- 66. Wei M‐T, Chang YC, Shimobayashi SF, Shin Y, Strom AR, Brangwynne CP. Nucleated transcriptional condensates amplify gene expression. Nat Cell Biol. 2020;22:1187–1196. [DOI] [PubMed] [Google Scholar]
- 67. Lim FL, Soulez M, Koczan D, Thiesen HJ, Knight JC. A KRAB‐related domain and a novel transcription repression domain in proteins encoded by SSX genes that are disrupted in human sarcomas. Oncogene. 1998;17:2013–2018. [DOI] [PubMed] [Google Scholar]
- 68. Skytting B, Nilsson G, Brodin B, et al. A novel fusion gene, SYT‐SSX4, in synovial sarcoma. J Natl Cancer Inst. 1999;91:974–975. [DOI] [PubMed] [Google Scholar]
- 69. Banito A, Li X, Laporte AN, et al. The SS18‐SSX oncoprotein hijacks KDM2B‐PRC1.1 to drive synovial sarcoma. Cancer Cell. 2018;33:527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Boulay G, Cironi L, Garcia SP, et al. The chromatin landscape of primary synovial sarcoma organoids is linked to specific epigenetic mechanisms and dependencies. Life Sci Alliance. 2021;4:e202000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gryder BE, Yohe ME, Chou HC, et al. PAX3–FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017;7:884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sunkel B, Wang M, LaHaye S, et al. Pioneer activity of an oncogenic fusion transcription factor at inaccessible chromatin. bioRxiv. 2020. 10.1101/2020.12.11.420232. [DOI] [Google Scholar]
- 73. Nagai M, Tanaka S, Tsuda M, et al. Analysis of transforming activity of human synovial sarcoma‐associated chimeric protein SYT‐SSX1 bound to chromatin remodeling factor hBRM/hSNF2 alpha. Proc Natl Acad Sci U S A. 2001;98:3843–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
All data relevant to the findings of this manuscript are included in the manuscript and the supplementary appendix. Additional data are available from the authors upon reasonable request.