
Figure 1: High-throughput screening of solid drug nanoparticles and machine learning model development.
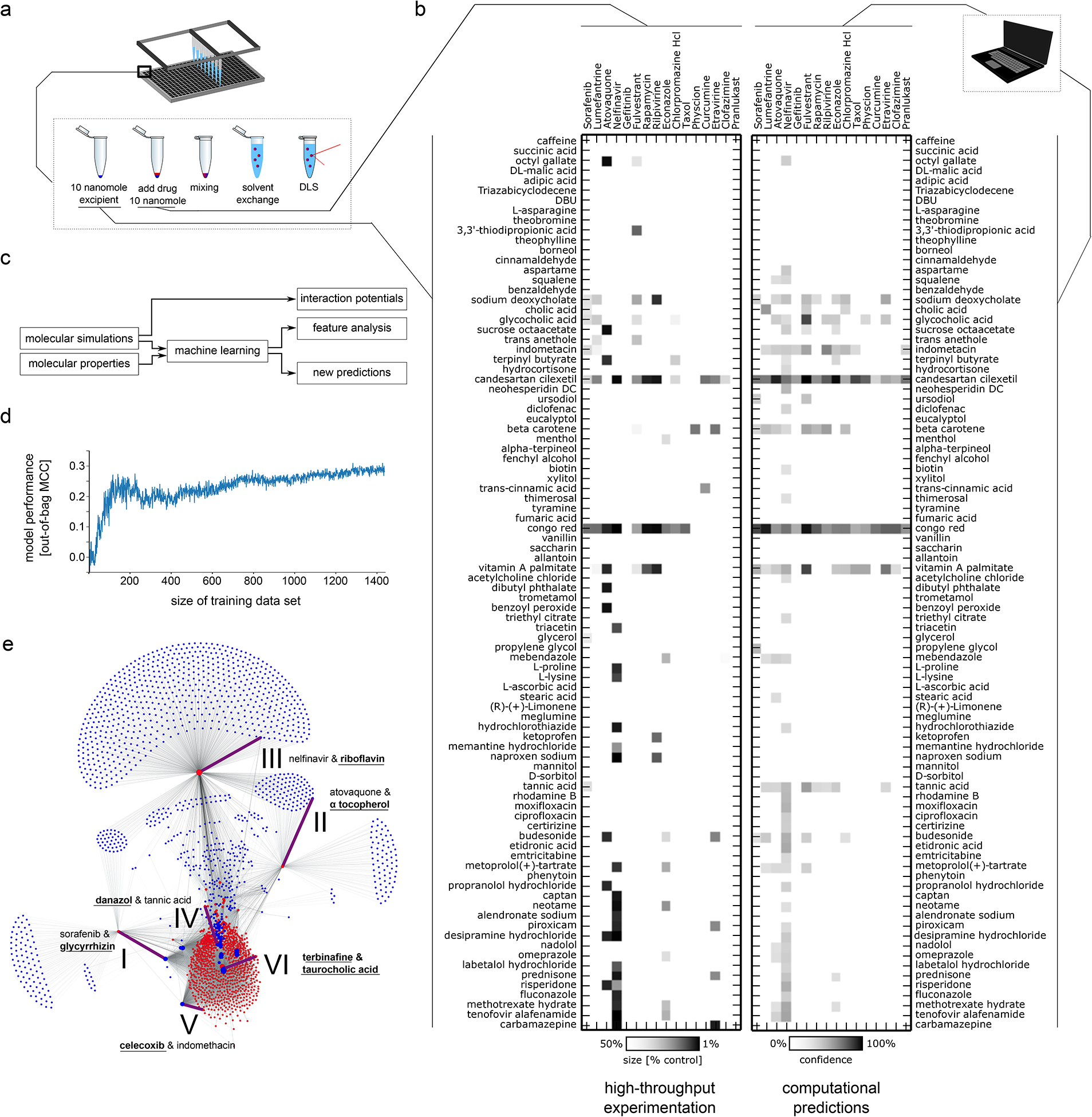
a Schematic of high-throughput experimental workflow to create nanoparticles using nanoprecipitation technique and rapid assessment using dynamic light scattering (DLS). b Left: High-throughput testing of all 1440 combinations of 16 drugs and 90 excipients (inactive ingredients, generally-recognized-as-safe food and drug additives, and other FDA-approved approved compounds). Colour gradient indicates size reduction of nanoparticle compared to the unformulated drug, where white corresponds to less than a 50% reduction in size and black correspond to a 90% reduction in size with linear interpolation (cf colour bar at bottom). Right: Machine learning-based assessment of nanoparticle-forming potential of drug-excipient pairs according to their chemical structures, physicochemical properties, and pairwise interaction potential determined from short molecular dynamic simulations. Gradient indicates predictive confidence from ten-fold cross validation (white 0% confidence, black 100% confidence, linear interpolation, cf colour bar at bottom). We found good agreement between the computational assessments (right) and the real-world experimentation (left), with 91% of the experiments correctly predicted. c Schematic explaining relationship of molecular dynamic simulations and machine learning. The molecular dynamic simulations of drug-excipient systems are computationally analysed to quantify non-covalent interaction potentials. These potentials serve as input for our machine learning model. The machine learning model uses both these interaction potentials and the molecular properties of drugs and excipients to predict which drug-excipient pairs will most likely lead to nanoparticle formation. Furthermore, the analysis of the predictive architecture of the machine learning model enables us to determine the most important variables that govern co-aggregation prediction (Supplementary Table 4). In a separate analysis, molecular dynamics trajectories were analysed to identify the most relevant non-covalent interactions for different drug-excipient pairs (Supplementary Table 8). d Performance analysis of machine learning model trained on different training dataset sizes. Shown is the mean performance of 20 independent models trained on random data subsets. e Using the developed computational prediction model, we predicted 2.1 million pairs constituting all exhaustive combinations of 788 drugs each paired with one of 2686 excipients. The machine learning model predicted that a majority of the drugs and excipients would not co-aggregate, resulting in colloidal self-aggregation of the drug and precipitation. For 38,464 combinations (1.8%), the machine learning model predicted a potential interaction between the drug and the excipient, leading to a stabilized, co-assembled nanoparticle formation. Some excipients (blue dots) are predicted to enable creation of nanoparticles with many different drugs (red dots) while other excipients were predicted to be able to create nanoparticles with only a single drug. Node size corresponds to the number of predicted excipients/drugs enabling formation of nanoparticles with this compound. Combinations further characterized in this paper are highlighted with purple edges, are numbered (cf. Figure 2) and labelled with the corresponding drug and excipient constituting this pair. The novel component that was not previously used in the screen is highlighted in bold and underlined.