

Abstract
Silicosis is a scarring lung disease caused by inhaling fine particles of crystalline silica in the workplace of many industries. Due to the lack of effective treatment and management, the continued high incidence of silicosis remains a major public health concern worldwide, especially in the developing countries. Till now, related molecular mechanisms underlying silicosis are still not completely understood. Multiple pathways have been reported to be participated in the pathological process of silicosis, and more complex signaling pathways are receiving attention. The activated extracellular signal-regulated kinase (ERK) signaling pathway has been recognized to control some functions in the cell. Recent studies have identified that the ERK signaling pathway contributes to the formation and development of silicosis through regulating the processes of oxidative stress, inflammatory response, proliferation and activation of fibroblasts, epithelial–mesenchymal transformation, autophagy, and apoptosis of cells. In this review article, we summarize the latest findings on the role of ERK signaling pathway in silica-induced experimental models of silicosis, as well as clinical perspectives.
Keywords: silica, silicosis, extracellular signal-regulated kinase
Introduction
Crystalline silica, as an environmental pollutant, exists in various natural phenomenon, such as sandstorms and volcanic eruptions. More importantly, as an occupational hazard, crystalline silica was also generated in the process of quarrying, mining, tunneling, construction, pottery making, and sandblasting. It has been well recognized that long-term inhalation of crystalline silica dust may cause silicosis, which is one of the most serious occupational diseases worldwide [1–3]. Although prevention efforts have been implemented for many decades, the prevalence rate of silicosis is still increasing in the recent decades, and the burden of disease caused by silicosis remained high worldwide, especially in developing countries including China [4–6]. In addition, outbreaks of silicosis occurred in developed countries such as the USA, Australia, and Spain [1, 7–9]. The large scale of silica exposed workers maybe one of the reasons for high incidence of silicosis. Published reports have indicated that there are approximately 11.5 million workers and 23 million workers exposed to crystalline silica in India and China, respectively [10, 11]. Apart from this reason, the complex and unclear pathogenesis of silicosis also makes it difficult to prevent and manage silicosis.
Silicosis is a diffuse pulmonary interstitial disease characterized by a fibrotic response in lung parenchyma [12, 13]. In addition, silicosis is a progressive condition, meaning it gets worse overtime, even if subjects were out of exposure to silica dust [13]. Normally, symptoms of silicosis may start out as an intense cough, shortness of breath, or weakness, and followed by present chest pain, weight loss, difficulty breathing. Different theories have been put forward to explain how silicosis is triggered, but all of those cannot comprehensively summarize it. The generally accepted process including that (1) respirable crystalline silica particles are inhaled through respiratory tract and deposit in the small bronchi and alveolar areas. Then, silica particles could be engulfed by alveolar macrophages and damage cells by their direct cytotoxicity, as well as make more immune cells gather and uptake them; (2) uncleared silica particles could induce the release of reactive oxygen species (ROS) and cause the activation of inflammasomes. (3) Macrophages are damaged and then release silica particles and cytokines, causing persistent inflammation and stimulating the proliferation and differentiation of fibroblasts. (4) The above 1–3 can be circulated, leading to the formation and deposition of extracellular matrix (ECM). Besides, silica-induced macrophages recruitment and their transformation into epithelioid histiocytes lead to a delayed hypersensitivity reaction, resulting in the silicotic granuloma [13–16].
Mitogen-activated protein kinases (MAPK) pathways are evolutionarily conserved signaling pathways that participate in many cellular processes. Extracellular signal-related kinases (ERK), Jun amino-terminal kinases, and p38 proteins are three main subfamilies of MAPKs [17]. Of these, ERK signaling pathway has been thoroughly studied, which is involved in diverse cellular events, including cell growth, proliferation, differentiation, migration, survival, and apoptosis [18–20]. The core members of the ERK signaling pathway include Ras (a small GTPase), Raf (MAPK kinase kinase), MEK (MAPK kinase), and ERK (MAPK) [17, 21]. Classically, growth factor, cytokines, and other extracellular substances bind to specific receptors on the cell membrane, causing the combination of growth factor receptor bound protein-2 (Grb-2) and son of sevenless (SOS) to switch the inactive GDP-bound Ras to active GTP-bound Ras. Activated Ras triggers a cascade of Raf–MEK–ERK phosphorylation. Ras recruits Raf (serine/threonine kinase) from the cytoplasm to the cell membrane and phosphorylates it, and activated Raf phosphorylates MEK (MEK1 and MEK2), which in turn phosphorylates ERK (ERK1 and ERK2) [20]. Activated ERK1/2 can not only regulate the target substrates in the cytoplasm, but also translocate into the nucleus and phosphorylate various transcription factors to regulate the expression of genes (Fig. 1). In addition, the activation of ERK substrates will result in the formation of feedback loops, which can control the ERK signaling pathway in a positive or negative way according to the substrates [22].
Figure 1.
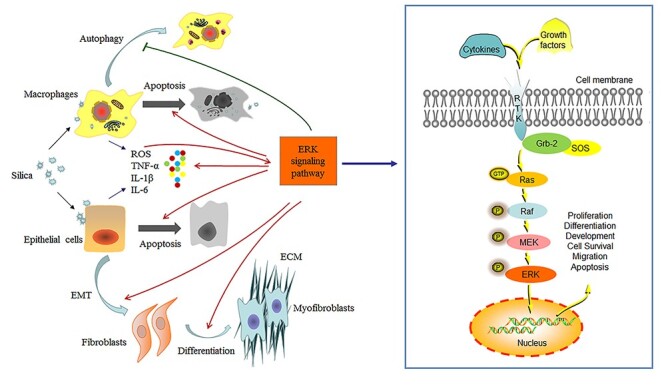
ERK signaling pathway and silicosis. ERK signaling pathway is involved in diverse cellular events. Growth factor and cytokines bind to RTK on the cell membrane, causing the combination of growth factor receptor bound protein 2 (Grb-2) and SOS to activate Ras. Active GTP-bound Ras triggers a cascade of Raf–MEK–ERK phosphorylation. ERK translocates into the nucleus to regulate the expression of genes. ERK signaling pathway participates in the formation and development of silicosis, including silica-elicited ROS activating the pathway and inducing the release of cytokines such as TNF-α, IL-6, and IL-8; differentiation of fibroblasts to myofibroblasts and accumulation of ECM; EMT process; and cell autophagy and apoptosis.
In the respiratory system, ERK signaling pathway is associated with lung development and the process of lung injury and repair [19, 23]. Multiple extracellular stimuli including environmental pollutants, cytokines, and oxidants, can activate ERK signaling pathway, leading to pulmonary inflammatory response and fibrosis [24–26]. Accumulating studies have indicated that ERK signaling pathway participates in the formation and development of silicosis [27–29], including (1) the crystalline silica-induced oxidative stress activating the pathway; (2) the release of large amounts of inflammatory cytokines; (3) the epithelial–mesenchymal transformation (EMT) and the proliferation and activation of fibroblasts induced by silica particles; and (4) autophagy and apoptosis process of cells (Fig. 1). In this review, we will focus on the current in vivo and in vitro studies to reveal the role of ERK signaling pathway in the formation and development of silicosis (Table 1).
Table 1.
The role of ERK signaling pathway in silicosis
| Effect on silicosis | Experimental model | Key findings | Reference |
|---|---|---|---|
| Activated by oxidative stress | Male BALB/c mice with silica instillation | HO-1, an inducible antioxidant, negatively regulates phosphorylation of ERK in silicosis mice. | [38] |
| RAW 264.7 (mouse macrophages) were exposed to silica | ROS blocking by the radical scavenger leads to the suppression of ERK activation induced by silica. | [38] | |
| 16HBE cells (human bronchial epithelial cells) were exposed to silica | Silica-derived ROS induces HO-1 via ERK1/2 activation. | [38] | |
| Rat2 fibroblasts were exposed to silica | Silica-induced ROS serves as a signal transduction element in activating ERK signaling pathway. | [36] | |
| Promote inflammatory response | HBE cells (human bronchial epithelial cells) were exposed to silica | Silica exposure activates ERK and increases the expression of inflammatory cytokines (IL-6 and IL-8) | [37] |
| T2 (primary rat type-II epithelial cells) were exposed to silica | Silica induces MIP-2 release via activating ERK signaling pathway | [46] | |
| A549 (human lung epithelial cells) were exposed to silica | Silica induces the release of IL-8 and COX-2 via activating ERK signaling pathway. | [45–47] | |
| Promote EMT process | Male C57BL/6 mice were treated with silica intratracheally | In a silica-induced lung fibrosis mouse model, ApoA1 overexpression inhibits the phosphorylation of ERK and reduces the process of EMT. | [59] |
| A549 (human lung epithelial cells) were exposed to silica | The expression of PAI-1, which contributes to the EMT process, is induced by silica through a ERK/AP-1-dependent mechanism. | [56]. | |
| 16HBE cells (human bronchial epithelial cells) were exposed to silica | NLRP3 inflammasomes modulate silica-induced EMT through the ERK/NF-κB pathway. | [50] | |
| Promote proliferation and transformation of cells | Male C57BL/6 mice were intratracheally exposed to silica | The activation of AP-1 stimulated by silica is mediated by ERK signaling pathway. | [71] |
| RAW 264.7 (mouse macrophages) were exposed to silica | Silica-induced TNF-α and TGF-β1 expression and nuclear factor Egr-1 activation are dependent on ERK signaling pathway. | [29, 62] | |
| A549 (human lung epithelial cells) were exposed to silica | Silica-induced activation of Egr-1 is mainly mediated by ERK signaling pathway | [77] | |
| HELF (human embryonic lung fibroblasts) were exposed to silica | Silica exposure causes cell cycle alternation through activating the ERK/AP-1/cyclin D1-CDK4 pathway. | [66, 72] | |
| Mouse lung fibroblasts from male C57BL/6 mice treated with 50 mg/kg silica. | LncRNAPCAT29 inhibits the proliferation of pulmonary fibroblasts and expression of fibronectin and collagen type I via blocking ERK signaling pathway. | [64] | |
| Against proliferation | HELF (human embryonic lung fibroblasts) were exposed to silica | The expression of cyclin D1 and CDK4 decrease after silica treatment through activating ERK signaling pathway. | [70, 73] |
| Promote apoptosis | BEAS-2B (human bronchial epithelial cells) were exposed to silica | Silica induced apoptosis of cells via activating ERK signaling pathway. | [78] |
| Against apoptosis | RAW 264.7 (mouse macrophages) were exposed to silica | ERK phosphorylation can protect against apoptosis of cells induced by silica. | [79] |
| Against autography | Male C57BL/6 mice were administrated with suspension of silica particles | Silica activates the ERK signaling pathway, leading to the inhibition of autophagy activity. | [63] |
AP-1: Activator protein-1; ApoA1: Apolipoprotein A1; CDK4: Cyclin-dependent kinase 4; COX-2: Cyclooxygenase-2; Egr-1: Early growth response protein-1; EMT: Epithelial–mesenchymal transformation; ERK: Extracellular signal-regulated kinase; HO-1: Hemeoxygenase-1; IL-6: Interleukin-6; IL-8: Interleukin-8; LncRNAPCAT29: LncRNA, prostate cancer-associated transcript 29; MIP-2: Macrophage inflammatory protein-2; PAI-1: Plasminogen Activator Inhibitor-1; ROS: Reactive oxygen species; TGF-β1: Transforming growth factor-β1; TNF-α: Tumor necrosis factor-α.
ERK Signaling Pathway in the Oxidative Stress Induced by Silica Particles
Oxidative stress, which is induced by imbalance between ROS production and ROS scavenging, is supposed to be initial part of the process of silicosis. ROS is a mediator that can help cells respond to external stimuli under physiological conditions, but silica-derived overproduction of ROS will result in pulmonary damage through causing the cell death and the release of inflammatory cytokines [30–33]. ERK signaling pathway has been reported to be activated by increasing ROS production and is considered as an important part in silicosis [34, 35].
Cho et al. [36] found that silica (diameter: 1–5 μm; 1 mg/well) caused elevation of ROS in Rat2 fibroblasts and activated the phosphorylation of ERK1/2. Catalase reduced the phosphorylation levels of ERK1/2 protein by suppressing ROS generation. Similarly, Ghio et al. [37] observed the effects of silica (median diameter: 1.6–1.7 μm; 100 μg/ml) on human bronchial epithelial (HBE) cells, and found that increased production of H2O2 (a kind of ROS) in HBE cells triggered ERK signaling pathway, which in turn caused the secretion of inflammatory cytokines. Nakashima et al. [38] detected that the phosphorylation of ERK was induced and the expression of Hemeoxygenase-1 (HO-1), an inducible antioxidant protein, was increased in the lung tissue of BALB/c mice with silica-induced (median diameter: 1.6–1.7 μm; 2.5 mg) lung injury, as well as in the mouse macrophage cell line RAW264.7 and HBE cells 16HBE treated with silica (median diameter: 1.6–1.7 μm; 0.1–0.5 mg/ml). Moreover, hydroxyl radical scavenger (tetramethylthiourea) could suppress the phosphorylation of ERK and induction of HO-1. However, the phosphorylation ERK was decreased in the lungs of silicosis mice pretreated with hemin (an inducer of HO-1), but increased after pretreated with zinc protoporphyrin (an inhibitor of HO-1). This study clarified that silica-induced ROS production triggered ERK signaling pathway to active the antioxidant system, while antioxidant inhibited the ERK pathway through negative feedback regulation. These above studies suggest that continuous production of oxidants will activate the ERK signaling pathway, and lead to the subsequent adverse effect.
ERK Signaling Pathway in the Inflammatory Response Induced by Silica Particles
Pulmonary inflammation is one of the most important features of silicosis. Silica stimulate tremendous induction of inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), which contribute to the subsequent processes of lung injury via diverse signal transduction pathways including ERK signaling pathway [39–42]. Besides, inhibition of ERK signaling pathway suppress the release of inflammatory cytokines to alleviate inflammatory response [43, 44].
Øvrevik et al. [45, 46] found that crystalline silica (median diameter: 1.6–1.7 μm; 30 μg/cm2) exposure stimulated primary rat type-II epithelial cells to release macrophage inflammatory protein-2 (MIP-2) and induced phosphorylation of ERK1/2. Although blocking ERK signaling pathway by PD98059 (the MEK1/2 inhibitor) inhibited the release of the MIP-2. Similarly, secretion of Interleukin-8 (IL-8) in human lung epithelial A549 cells treated with silica (median diameter: 1.6–1.7 μm; 40 μg/cm2) was dependent on activation of ERK signaling pathway. In addition, Tomaru and Matsuoka [47] showed that both mRNA and protein level of cyclooxygenase-2 (COX-2), an essential mediator of airway inflammation, were increased in A549 cells exposed to crystalline silica (median diameter: 1.6–1.7 μm; 60 μg/cm2), accompanied by an elevated level of ERK1/2 phosphorylation. While U0126, an inhibitor of MEK1/2, suppressed the expression of COX-2 via blocking the ERK signaling pathway. The other study by Li et al. [29] showed silica (diameter: 0.1–10 μm; 100 μg/ml) stimulated the secretion of TNF-α through activating ERK signaling pathway in RAW264.7 cells, and PD98059 significantly reduced the expression of TNF-α. All of these results suggest that ERK signaling pathway is involved in the inflammatory response caused by silica.
ERK Signaling Pathway in the Fibrosis Induced by Silica Particles
Myofibroblasts, which have a high capacity to produce ECM, are usually transformed from activated fibroblasts stimulated by external stimuli, but studies have proposed that EMT process contributes to the accumulation of myofibroblasts as well [48]. EMT helps to increase the number of fibroblasts and myofibroblasts, and the excessive proliferation and activation of fibroblasts and myofibroblasts lead to the production and accumulation of ECM, resulting in the formation of lung fibrosis [49–51].
EMT is the transformation of polarized epithelial cells into migrating mesenchymal cells, characterized by loss of epithelial markers E-cadherin and gain of mesenchymal markers like N-cadherin and α-smooth muscle actin. After epithelial cells damage, the cell–cell connection is destroyed and the integrity of epithelium is impaired. Meanwhile, the epithelial cells transform into fibroblasts and myofibroblasts to produce ECM [48, 50, 52]. Accumulating studies have confirmed that silica-induced EMT of airway and alveolar epithelia cells is a vital process of silicosis formation, and ERK signaling pathway is necessary for EMT [53–55]. Li et al. [50] found that silica (median diameter: 2.5 μm; 50 μg/cm2) exposure induced EMT in 16HBE cells, accompanied by continuous activation of NLRP3 inflammasome and elevated level of ERK1/2 phosphorylation. While inhibition of NLRP3 partially reversed the phosphorylation of ERK1/2 and decreased phosphorylation of its downstream transcription factor NF-κb, which in turn alleviate silica-induced EMT. It has been proved in A549 cells that blockage of ERK signaling pathway suppressed the silica-induced expression of plasminogen activator inhibitor-1 to inhibit EMT process, thus alleviating the fibrotic responses [56–58]. The study conducted by Baek et al. [59] also showed that A549 cells had a high level of ERK1/2 phosphorylation treated with transforming growth factor-β1 (TGF-β1), which could promoted EMT. Besides, they demonstrated that apolipoprotein A1 (ApoA1), an anti-fibrotic protein, could inhibit the process of EMT by blocking the ERK signaling pathway in silica-induced lung fibrosis mouse model (median diameter: 1–5 μm; 20 mg). All these researches suggest that ERK signaling pathway is involved in the silica-induced EMT process. However, it is hardly to draw the conclusion because the lack of in vivo studies. Whether ERK signaling pathway participates in EMT process in silicosis needs further investigation.
Fibroblasts are the key effector cells for tissue regeneration and repair under physiological conditions, but will result in chronic fibrosis of organs in a pathological state [49, 60]. It has been reported that ERK signaling pathway is involved in the proliferation and activation of fibroblasts induced by silica particles [61, 62]. Inhibition of activated fibroblasts via blocking the ERK signaling pathway can largely reduce the expression of fibronectin and collagen type I, which in turn alleviated the severity of silica-induced lung lesions and retarded the formation of pulmonary fibrosis [63]. Li et al. [29] found that the secretion of TGF-β1, which can promote activation and transformation of fibroblasts, increased in RAW264.7 cells exposed to silica (diameter: 0.1–10 μm; 100 μg/ml) via ERK signaling pathway. Liu et al. [64] demonstrated that lncRNA, prostate cancer-associated transcript 29 exerted important functions in pulmonary fibrosis induced by silica (50 mg/kg) via activation of ERK signaling pathway and may inhibit the expression of fibronectin and collagen type I by targeting the ERK signaling pathway in mouse lung fibroblasts.
Silica exposure can cause disorder of cell proliferation by altering cell cycle, in which the transformation from G1 to S phase is the checkpoint [65, 66]. Activator protein-1 (AP-1) controls gene expression and affect cell proliferation, involving in the initiation and development of silicosis [67–69]. Activated ERK signaling pathway induced by silica contributes to the alteration of cell cycle through activating AP-1, thereby promoting cell proliferation [66, 70]. Vallyathan et al. [71] found that in the lungs of silica-treated (mean diameter: 3.7 μm; 5 mg) mice, the activation of AP-1 was mediated by ERK signaling pathway. Shen et al. [72] revealed that silica (diameter: <5 μm; 50–400 μg/ml) triggered ERK signaling pathway in human embryonic lung fibroblasts (HELF), accompanied by activation of HELF transformation. Compared with HELF, the expression of cyclin D1 and cyclin-dependent kinase 4 (CDK4), enzymes that regulate the cell transition from G1 to S phase, were increased in silica-induced transformed HELF cells (S-HELF) that with silica (diameter: <5 μm; 160 μg/cm2) treatment for 72 h. While inhibition of ERK1/2 phosphorylation and AP-1 both reduced the expression of cyclin D1 and CDK4 in S-HELF. This study provided evidence for involvement of ERK/AP-1 signaling pathway in silica-induced high levels of cyclin D1 and CDK4. Similarly, Jia et al. [65, 66] found that cyclin D1 and CDK4 were highly expressed in HELF cells after silica (diameter: <5 μm; 200 μg/ml) exposure, which could explain the significantly increasing proportion of S phase cells and the decreasing proportion of G1 phase cells. Furthermore, they observed that overexpression of dominant-negative mutants of ERK inhibited AP-1 activation and attenuated the G1 phase cell reduction. These studies demonstrated that silica exposure could cause cell cycle alteration through the ERK signaling pathway, leading to fibroblasts abnormal proliferation. However, studies indicated that the phosphorylation levels of ERK were increased, while the expression levels of cyclin D1 and CDK4 were decreased in HELF cells after treated with silica (diameter: <5 μm; 400 μg/ml) for 2 h. And blocking ERK signaling pathway prevented the change [73]. Similarly, Wang et al. [70] found that phosphorylation of ERK increased significantly but the expression level of CDK4 was reduced in HELF exposed to silica (diameter: <5 μm; 200 μg/cm2). The reason for the inconsistencies of the above studies may be that short-time silica exposure may cause cells to stay in G1 phase for DNA damage repair by inhibiting cell cycle, but long-time exposure may lead to abnormal cell proliferation. More researches need to be performed to clarify the precise mechanism.
In addition to fibroblasts, the proliferation and activation of other cells are also associated with silicosis. Early growth response protein-1 (Egr-1), a transcription factor, plays a central role in silica-induced fibrogenic response by regulating cell proliferation and synthesis of ECM [74–76]. Zeng et al. [62] observed that exposure to silica (100 μg/ml) upregulated the level of ERK 1/2 phosphorylation in RAW264.7 rapidly, accompanied by overexpression of Egr-1 mRNA and protein. And nuclear expression and transcription of Egr-1 could be largely suppressed via using U0126. In this study, similar results were detected in the lungs of Wistar rats with silica (50 mg) treatment. Chu et al. [77] also found similar results in A549 cells after silica (100 μg/ml) administration. These researches suggest that Egr-1 plays a key role in silica-induced cell proliferation depended on ERK signaling pathway.
ERK Signaling Pathway in Apoptosis and Autophagy Induced by Silica
Apoptosis is a programmed cell death and it is responsible for the lung cells injury and repair. Researches have indicated that there is a causal relationship between activation of ERK signaling pathway and cell apoptosis, and inhibition of the pathway can abrogate cell apoptosis induced by silica, asbestos or oxidants [34, 78]. Antognelli et al. [78] indicated that crystalline silica (median diameter: 1.6–1.7 μm; 100 μg/cm2) induced apoptosis of human bronchial BEAS-2B cell via activating ERK signaling pathway and U0126 could abolish the effect of silica-induced apoptosis through negatively modulating Glyoxalase I, a cellular defense enzyme. However, Gambelli et al. [79] suggested ERK phosphorylation could protect against apoptosis of the mouse macrophage RAW 264.7 cells induced by silica (average diameter: 1.7 μm; 20 μg/cm2); while in the presence of PD98059 or dominant negative ERK mutants, the silica-induced apoptosis in RAW 264.7 macrophage significantly increased through inhibition of NF-κb activation. ERK signaling pathway has opposite effects on cell apoptosis in response to different time of silica exposure via diverse downstream factors.
Autophagy, a predominant cellular mechanism that regulates cell growth, survival and senescence, often occurs in the state of various cellular stresses such as starvation, hypoxia, and DNA damage [80]. Autophagy delays the occurrence of silica-induced lung inflammation and fibrosis through reducing the production of ROS and inflammatory cytokines, as well as the apoptosis of macrophages and alveolar epithelial cells [81, 82]. Deficiency of autophagy in macrophages can lead to inflammation after silica exposure [83]. Han et al. [63] found that the expression of the anti-autophagy protein B-cell lymphoma 2 (Bcl-2) was upregulated in silicosis mouse model (diameter: 0.5–10 μm; 50 mg/kg), while activated autophagy inhibited silica-induced fibrogenesis. In the follow-up study, they proved that TGF-β1 increased the expression of Bcl-2 by activating the ERK signaling pathway, leading to the inhibition of autophagy activity, whereas U0126 decreased the expression levels of Bcl-2. These results implied that ERK signaling pathway affected the silica-induced lung fibrosis by regulating the autophagy process of cells. Nevertheless, some studies suggested that silica can induce macrophage autophagy and the autophagy process activated by the ERK signaling pathway led to cells death; while decreasing level of autophagy attenuated the inflammatory responses [84, 85]. The controversy of these results may be linked to the different levels of autophagy. In physiological state, autophagy can hinder the progress of silicosis by leading to apoptosis resistance and reducing lung inflammation. While overactive and persistent autophagy will promote apoptosis of alveolar macrophages and proliferation and migration of pulmonary fibroblasts, leading to lung injury and fibrogenesis of silicosis [86, 87]. The role and mechanism of autophagy in silicosis are worth further exploration.
Conclusion
In summary, published studies have demonstrated that ERK signaling pathway is related to silicosis. The production of ROS elicited by silica can activate ERK signaling pathway, and the activation of the pathway induces the release of inflammatory factors, modulates autophagy and apoptosis of cells, promotes EMT process and proliferation and activation of fibroblasts to result in synthesis of ECM, leading to the formation of lung fibrosis. Meanwhile, the interaction among different mediators and the feedback regulation of ERK signaling pathway constitute a relatively complex regulatory system. To date, studies about the role of ERK signaling pathway in the progression of silicosis are mainly focused on experimental models, especially in vitro studies. In the future, relevant epidemiological studies and researches about cellular and molecular changes are still needed to be conducted to provide clues for the prevention and treatment of silicosis.
Ethics Approval and Consent to Participate
Not Applicable.
Consent for Publication
Not applicable.
Availability of Data and Supporting Materials Section
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Author Contributions
Y.X. and J.M. researched the literature and drafted this review. M.Y. and L.F. helped to organize and revised the manuscript. W.C. contributed to the design and critically revised this article. All authors read and approved the final manuscript.
Y.X. and J.M. contributed equally to thiswork.
Funding
This work was supported by the National Natural Science Foundation of China [grant number 81803205 and 81872593]; and China Postdoctoral Science Foundation (2019 T120665).
Conflicts of Interest
There are no conflicts of interest to declare.
Acknowledgments
Not applicable.
Abbreviations
- AP-1
activator protein-1
- ApoA1
apolipoproteinA1
- Bcl-2
B-cell lymphoma 2
- CDK4
cyclin-dependent kinase 4
- COX-2
cyclooxygenase-2
- ECM
extracellular matrix
- Egr-1
early growth response protein-1
- EMT
epithelial–mesenchymal transformation
- ERK
extracellular signal-regulated kinase
- Grb-2
growth factor receptor bound protein-2
- HBE
human bronchial epithelial
- HELF
human embryonic lung fibroblasts
- HO-1
hemeoxygenase-1
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- IL-8
interleukin-8
- MAPK
mitogen-activated protein kinase
- MIP-2
macrophage inflammatory protein-2
- ROS
reactive oxygen species
- S-HELF
silica-induced transformed HELF cells
- SOS
son of sevenless
- TGF-β1
transforming growth factor-β1
- TNF-α
tumor necrosis factor-α
References
- 1. Rose C, Heinzerling A, Patel K. et al. Severe silicosis in engineered stone fabrication workers-California, Colorado, Texas, and Washington, 2017-2019. MMWR Morb Mortal Wkly Rep 2019;68:813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. t Mannetje A, Steenland K, Attfield M. et al. Exposure-response analysis and risk assessment for silica and silicosis mortality in a pooled analysis of six cohorts. Occup Environ Med 2002;59:723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. The Lancet Respiratory M . The world is failing on silicosis. Lancet Respir Med 2019;7:283. [DOI] [PubMed] [Google Scholar]
- 4. GBD 2017 Risk Factor Collaborators. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet (London, England) 2018;392:1923–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet (London, England) 2018;392:1789–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. GBD 2017 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 359 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet (London, England) 2018;392:1859–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoy R, Chambers DC. Silicosis: an ancient disease in need of a dose of modern medicine. Respirology (Carlton, Vic) 2020;25:464–465. [DOI] [PubMed] [Google Scholar]
- 8. Hoy RF, Baird T, Hammerschlag G. et al. Artificial stone-associated silicosis: a rapidly emerging occupational lung disease. Occup Environ Med 2018;75:3–5. [DOI] [PubMed] [Google Scholar]
- 9. Perez-Alonso A, Cordoba-Dona JA, Millares-Lorenzo JL. et al. Outbreak of silicosis in Spanish quartz conglomerate workers. Int J Occup Environ Health 2014;20:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Y, Steenland K, Rong Y. et al. Exposure-response analysis and risk assessment for lung cancer in relationship to silica exposure: a 44-year cohort study of 34,018 workers. Am J Epidemiol 2013;178:1424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jindal SK. Silicosis in India: past and present. Curr Opin Pulm Med 2013;19:163–8. [DOI] [PubMed] [Google Scholar]
- 12. Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med 1998;157:1666–80. [DOI] [PubMed] [Google Scholar]
- 13. Leung CC, Yu IT, Chen W. Silicosis. Lancet (London, England) 2012;379:2008–18. [DOI] [PubMed] [Google Scholar]
- 14. Pollard KM. Silica, silicosis, and autoimmunity. Front Immunol 2016;7:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hoy RF, Chambers DC. Silica-related diseases in the modern world. Allergy 2020;75:2805–2817. [DOI] [PubMed] [Google Scholar]
- 16. Carneiro PJ, Clevelario AL, Padilha GA. et al. Bosutinib therapy ameliorates lung inflammation and fibrosis in experimental silicosis. Front Physiol 2017;8:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001;410:37–40. [DOI] [PubMed] [Google Scholar]
- 18. Cristea S, Sage J. Is the canonical RAF/MEK/ERK Signaling pathway a therapeutic target in SCLC? J Thorac Oncol 2016;11:1233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirata E, Kiyokawa E. ERK activity imaging during migration of living cells in vitro and in vivo. Int J Mol Sci 2019;20:679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu F, Yang X, Geng M. et al. Targeting ERK, an Achilles' heel of the MAPK pathway, in cancer therapy. Acta Pharmaceutica Sinica B 2018;8:552–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 2015;16:281–98. [DOI] [PubMed] [Google Scholar]
- 22. Sturm OE, Orton R, Grindlay J. et al. The mammalian MAPK/ERK pathway exhibits properties of a negative feedback amplifier. Sci Signal 2010;3:ra90. [DOI] [PubMed] [Google Scholar]
- 23. Boucherat O, Landry-Truchon K, Aoidi R. et al. Lung development requires an active ERK/MAPK pathway in the lung mesenchyme. Dev Dyn 2017;246:72–82. [DOI] [PubMed] [Google Scholar]
- 24. Puddicombe SM, Davies DE. The role of MAP kinases in intracellular signal transduction in bronchial epithelium. Clinical and experimental allergy: journal of the British Society for Allergy and. Clin Immunol 2000;30:7–11. [DOI] [PubMed] [Google Scholar]
- 25. Wang J, Huang J, Wang L. et al. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-kappaB signaling pathway. J Thorac Dis 2017;9:4398–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maik-Rachline G, Seger R. The ERK cascade inhibitors: towards overcoming resistance. Drug resistance updates: reviews and commentaries in antimicrobial and anticancer. Chemotherapy 2016;25:1–12. [DOI] [PubMed] [Google Scholar]
- 27. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science (New York, NY) 2002;298:1911–2. [DOI] [PubMed] [Google Scholar]
- 28. Ding M, Shi X, Dong Z. et al. Freshly fractured crystalline silica induces activator protein-1 activation through ERKs and p38 MAPK. J Biol Chem 1999;274:30611–6. [DOI] [PubMed] [Google Scholar]
- 29. Li X, Hu Y, Jin Z. et al. Silica-induced TNF-alpha and TGF-beta1 expression in RAW264.7 cells are dependent on Src-ERK/AP-1 pathways. Toxicol Mech Methods 2009;19:51–8. [DOI] [PubMed] [Google Scholar]
- 30. Nardi J, Nascimento S, Goethel G. et al. Inflammatory and oxidative stress parameters as potential early biomarkers for silicosis. Clin Chim Acta; Int J Clin Chem 2018;484:305–13. [DOI] [PubMed] [Google Scholar]
- 31. Zhang Z, Rong Y, Cui X. et al. Oxidative stress and mitochondrion-related cell apoptosis in human bronchial epithelial 16HBE cells induced by silica dust. Chin J Ind Hygiene Occup Dis 2015;33:801–5. [PubMed] [Google Scholar]
- 32. Nuvoli B, Camera E, Mastrofrancesco A. et al. Modulation of reactive oxygen species via ERK and STAT3 dependent signalling are involved in the response of mesothelioma cells to exemestane. Free Radic Biol Med 2018;115:266–77. [DOI] [PubMed] [Google Scholar]
- 33. Feno S, Butera G, Vecellio Reane D. et al. Crosstalk between calcium and ROS in pathophysiological conditions. Oxid Med Cell Longev 2019;2019:9324018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiménez LA, Zanella C, Fung H. et al. Role of extracellular signal-regulated protein kinases in apoptosis by asbestos and H2O2. Am J Physiol 1997;273:L1029–35. [DOI] [PubMed] [Google Scholar]
- 35. Fubini B, Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med 2003;34:1507–16. [DOI] [PubMed] [Google Scholar]
- 36. Cho YJ, Seo MS, Kim JK. et al. Silica-induced generation of reactive oxygen species in Rat2 fibroblast: role in activation of mitogen-activated protein kinase. Biochem Biophys Res Commun 1999;262:708–12. [DOI] [PubMed] [Google Scholar]
- 37. Ghio AJ, Tong H, Soukup JM. et al. Sequestration of mitochondrial iron by silica particle initiates a biological effect. Am J Physiol Lung Cell Mol Physiol 2013;305:L712–24. [DOI] [PubMed] [Google Scholar]
- 38. Nakashima K, Sato T, Shigemori S. et al. Regulatory role of heme oxygenase-1 in silica-induced lung injury. Respir Res 2018;19:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ding M, Chen F, Shi X. et al. Diseases caused by silica: mechanisms of injury and disease development. Int Immunopharmacol 2002;2:173–82. [DOI] [PubMed] [Google Scholar]
- 40. Mittal M, Siddiqui MR, Tran K. et al. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 2014;20:1126–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou T, Rong Y, Liu Y. et al. Association between proinflammatory responses of respirable silica dust and adverse health effects among dust-exposed workers. J Occup Environ Med 2012;54:459–65. [DOI] [PubMed] [Google Scholar]
- 42. Li W, Xie L, Ma J. et al. Genetic loss of Gas6/Mer pathway attenuates silica-induced lung inflammation and fibrosis in mice. Toxicol Lett 2019;313:178–87. [DOI] [PubMed] [Google Scholar]
- 43. Hsieh YH, Deng JS, Chang YS. et al. Ginsenoside Rh2 ameliorates lipopolysaccharide-induced acute lung injury by regulating the TLR4/PI3K/Akt/mTOR, Raf-1/MEK/ERK, and Keap1/Nrf2/HO-1 signaling pathways in mice. Nutrients 2018;10:1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou QL, Wang TY, Li M. et al. Alleviating airway inflammation by inhibiting ERK-NF-kappaB signaling pathway by blocking Kv1.3 channels. Int Immunopharmacol 2018;63:110–8. [DOI] [PubMed] [Google Scholar]
- 45. Øvrevik J, Refsnes M, Namork E. et al. Mechanisms of silica-induced IL-8 release from A549 cells: initial kinase-activation does not require EGFR activation or particle uptake. Toxicology 2006;227:105–16. [DOI] [PubMed] [Google Scholar]
- 46. Øvrevik J, Låg M, Schwarze P. et al. p38 and Src-ERK1/2 pathways regulate crystalline silica-induced chemokine release in pulmonary epithelial cells. Toxicol Sci 2004;81:480–90. [DOI] [PubMed] [Google Scholar]
- 47. Tomaru M, Matsuoka M. The role of mitogen-activated protein kinases in crystalline silica-induced cyclooxygenase-2 expression in A549 human lung epithelial cells. Toxicol Mech Methods 2011;21:513–9. [DOI] [PubMed] [Google Scholar]
- 48. Li J, Yao W, Hou JY. et al. The role of fibrocyte in the pathogenesis of silicosis. Biomed Environ Sci: BES 2018;31:311–6. [DOI] [PubMed] [Google Scholar]
- 49. Ng B, Dong J, Viswanathan S. et al. Fibroblast-specific IL11 signaling drives chronic inflammation in murine fibrotic lung disease. FASEB J 2020;34:11802–11815. [DOI] [PubMed] [Google Scholar]
- 50. Li X, Yan X, Wang Y. et al. NLRP3 inflammasome inhibition attenuates silica-induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells. Exp Cell Res 2018;362:489–97. [DOI] [PubMed] [Google Scholar]
- 51. Karsdal MA, Nielsen SH, Leeming DJ. et al. The good and the bad collagens of fibrosis - their role in signaling and organ function. Adv Drug Deliv Rev 2017;121:43–56. [DOI] [PubMed] [Google Scholar]
- 52. Rout-Pitt N, Farrow N, Parsons D. et al. Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res 2018;19:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ng B, Dong J, D'Agostino G. et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci Transl Med 2019;11:1237. [DOI] [PubMed] [Google Scholar]
- 54. Yan W, Xiaoli L, Guoliang A. et al. SB203580 inhibits epithelial-mesenchymal transition and pulmonary fibrosis in a rat silicosis model. Toxicol Lett 2016;259:28–34. [DOI] [PubMed] [Google Scholar]
- 55. Yang M, Wang N, Li W. et al. Therapeutic effects of scavenger receptor MARCO ligand on silica-induced pulmonary fibrosis in rats. Toxicol Lett 2019;311:1–10. [DOI] [PubMed] [Google Scholar]
- 56. Hu YB, Lin Z, Feng DY. et al. Silica induces plasminogen activator inhibitor-1 expression through a MAPKs/AP-1-dependent mechanism in human lung epithelial cells. Toxicol Mech Methods 2008;18:561–7. [DOI] [PubMed] [Google Scholar]
- 57. Flevaris P, Vaughan D. The role of plasminogen activator inhibitor type-1 in fibrosis. Semin Thromb Hemost 2017;43:169–77. [DOI] [PubMed] [Google Scholar]
- 58. Omori K, Hattori N, Senoo T. et al. Inhibition of plasminogen activator inhibitor-1 attenuates transforming growth factor-beta-dependent epithelial mesenchymal transition and differentiation of fibroblasts to myofibroblasts. PLoS One 2016;11:e0148969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Baek AR, Lee JM, Seo HJ. et al. Apolipoprotein A1 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition of alveolar epithelial cells. Tuberc Respir Dis 2016;79:143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bhattacharyya S, Fang F, Tourtellotte W. et al. Egr-1: new conductor for the tissue repair orchestra directs harmony (regeneration) or cacophony (fibrosis). J Pathol 2013;229: 286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yuan J, Li P, Pan H. et al. miR-542-5p attenuates fibroblast activation by targeting integrin alpha6 in silica-induced pulmonary fibrosis. Int J Mol Sci 2018;19:3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zeng QF, Chu L, Wang TS. et al. In vivo and in vitro silica induces nuclear factor Egr-1 activation mediated by ERK 1/2 in RAW264.7 cell line. Toxicol Mech Methods 2005;15:93–9. [DOI] [PubMed] [Google Scholar]
- 63. Han R, Ji X, Rong R. et al. MiR-449a regulates autophagy to inhibit silica-induced pulmonary fibrosis through targeting Bcl2. J Mol Med (Berl) 2016;94:1267–79. [DOI] [PubMed] [Google Scholar]
- 64. Liu X, Gao S, Xu H. lncRNAPCAT29 inhibits pulmonary fibrosis via the TGF-β1-regulated RASAL1/ERK1/2 signal pathway. Mol Med Rep 2018;17:7781–8. [DOI] [PubMed] [Google Scholar]
- 65. Jia X, Liu B, Ye M. et al. Silica induces cell cycle changes through PI-3K/AP-1 pathway in human embryo lung fibroblast cells. Cell Biochem Funct 2010;28:613–9. [DOI] [PubMed] [Google Scholar]
- 66. Jia X, Liu B, Shi X. et al. Roles of the ERK, JNK/AP-1/cyclin D1-CDK4 pathway in silica-induced cell cycle changes in human embryo lung fibroblast cells. Cell Biol Int 2011;35:697–704. [DOI] [PubMed] [Google Scholar]
- 67. Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol 2002;4:E131–6. [DOI] [PubMed] [Google Scholar]
- 68. Koo JH, Plouffe SW, Meng Z. et al. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev 2020;34:72–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang X, Jia X, Mei L. et al. Global DNA methylation and PTEN hypermethylation alterations in lung tissues from human silicosis. J Thorac Dis 2016;8:2185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang H, Xiao S, Tang Y. et al. Activation of MAPK and cyclin D1/CDK4 in malignant transformation of human embryonic lung fibroblasts induced by silica and Benzopyrene. Asian Pac J Cancer Prev 2020;21:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vallyathan V, Ding M, Shi X. et al. Molecular activation of activator Protein-1 in silica and asbestos-induced carcinogenesis. Inhal Toxicol 2000;12:353–7. [DOI] [PubMed] [Google Scholar]
- 72. Shen F, Fan X, Liu B. et al. Overexpression of cyclin D1-CDK4 in silica-induced transformed cells is due to activation of ERKs, JNKs/AP-1 pathway. Toxicol Lett 2006;160:185–95. [DOI] [PubMed] [Google Scholar]
- 73. Shen F, Fan X, Liu B. et al. Downregulation of cyclin D1-CDK4 protein in human embryonic lung fibroblasts (HELF) induced by silica is mediated through the ERK and JNK pathway. Cell Biol Int 2008;32:1284–92. [DOI] [PubMed] [Google Scholar]
- 74. Nemeth A, Mozes MM, Calvier L. et al. The PPARgamma agonist pioglitazone prevents TGF-beta induced renal fibrosis by repressing EGR-1 and STAT3. BMC Nephrol 2019;20:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bhattacharyya S, Wu M, Fang F. et al. Early growth response transcription factors: key mediators of fibrosis and novel targets for anti-fibrotic therapy. Matrix Biol: J Int Soc Matrix Biol 2011;30:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xiang F, Bai M, Jin Y. et al. Egr-1 mediates Si0(2)-driven transcription of membrane type I matrix metalloproteinase in macrophages. J Huazhong Univ Sci Technol Med Sci = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban 2007;27:13–6. [DOI] [PubMed] [Google Scholar]
- 77. Chu L, Wang T, Hu Y. et al. Activation of Egr-1 in human lung epithelial cells exposed to silica through MAPKs signaling pathways. PLoS One 2013;8:e68943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Antognelli C, Gambelunghe A, Muzi G. et al. Peroxynitrite-mediated glyoxalase I epigenetic inhibition drives apoptosis in airway epithelial cells exposed to crystalline silica via a novel mechanism involving argpyrimidine-modified Hsp70, JNK, and NF-κB. Free Radic Biol Med 2015;84:128–41. [DOI] [PubMed] [Google Scholar]
- 79. Gambelli F, Di P, Niu X. et al. Phosphorylation of tumor necrosis factor receptor 1 (p55) protects macrophages from silica-induced apoptosis. J Biol Chem 2004;279:2020–9. [DOI] [PubMed] [Google Scholar]
- 80. Ravanan P, Srikumar IF, Talwar P. Autophagy: the spotlight for cellular stress responses. Life Sci 2017;188:53–67. [DOI] [PubMed] [Google Scholar]
- 81. Du S, Li C, Lu Y. et al. Dioscin alleviates crystalline silica-induced pulmonary inflammation and fibrosis through promoting alveolar macrophage autophagy. Theranostics 2019;9:1878–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhao H, Wang Y, Qiu T. et al. Autophagy, an important therapeutic target for pulmonary fibrosis diseases. Clin Chim Acta; Int J Clin Chem 2020;502:139–47. [DOI] [PubMed] [Google Scholar]
- 83. Jessop F, Hamilton RF, Rhoderick JF. et al. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol Appl Pharmacol 2016;309:101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J 2010;277:2–21. [DOI] [PubMed] [Google Scholar]
- 85. Li D, Hu J, Wang T. et al. Silymarin attenuates cigarette smoke extract-induced inflammation via simultaneous inhibition of autophagy and ERK/p38 MAPK pathway in human bronchial epithelial cells. Sci Rep 2016;6:37751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liu H, Cheng Y, Yang J. et al. BBC3 in macrophages promoted pulmonary fibrosis development through inducing autophagy during silicosis. Cell Death Dis 2017;8:e2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Racanelli AC, Kikkers SA, Choi AMK. et al. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018;14:221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]