
Figure 7.
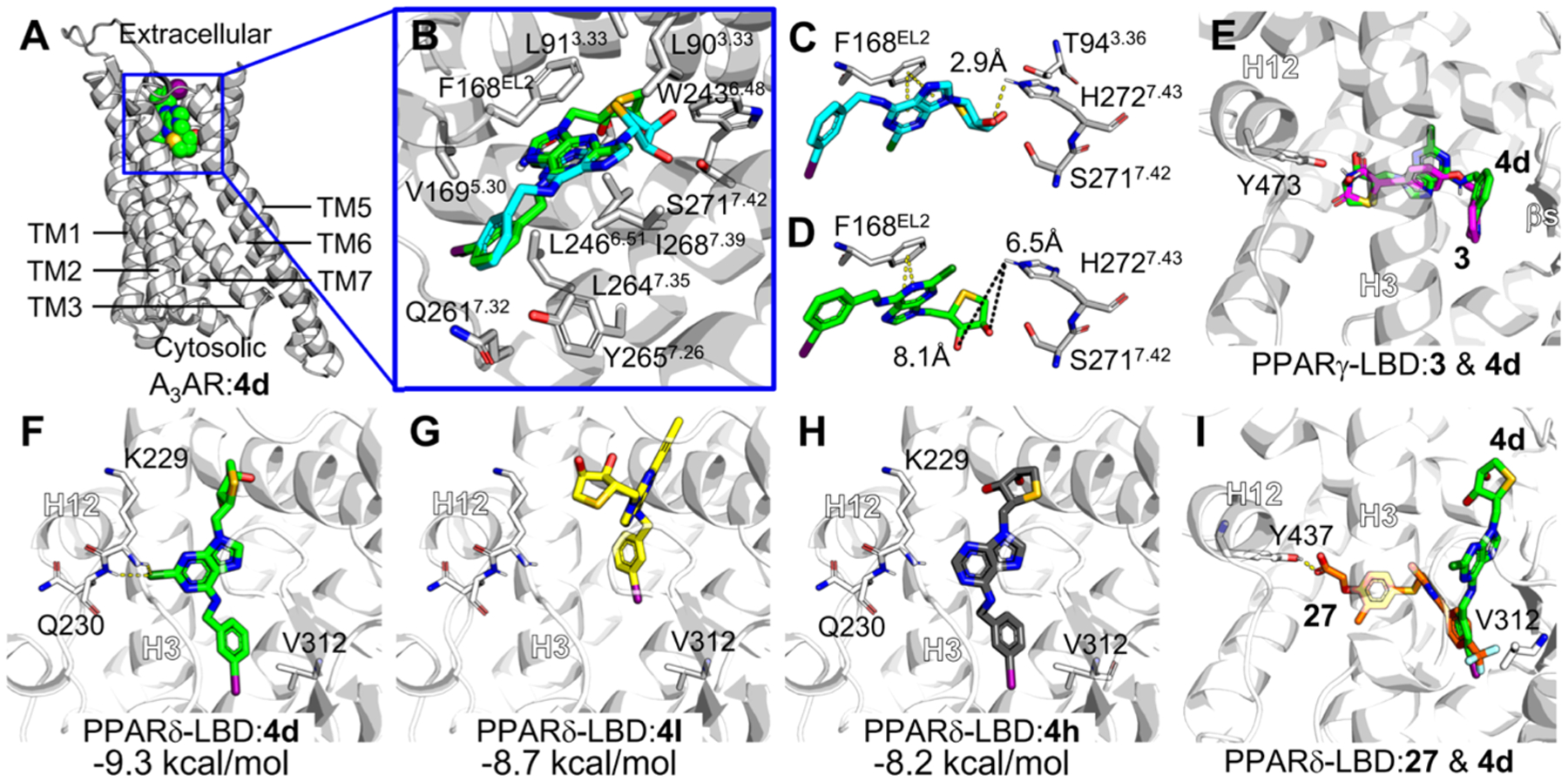
Molecular docking modes of compound 4d against A3AR, PPARγ, and PPARδ. (A) Optimum binding mode of compound 4d was generated in the A3AR homology model constructed based on the A2AAR structure (PDB ID: 5G53). TM, transmembrane. (B) Selected docking mode of compound 4d (green) for the comparison with A3AR antagonist 1 (cyan), forming π–π stacking with Phe168 with the adenine ring. Major amino acid residues interacting with compound 4d in the optimum docking mode against the A3AR homology model were labeled. The amino acid residues closely contacting with ligand (<van der Waals radii) or forming hydrogen bond with ligand are labeled. TM2 is shown transparently for clarity. The residue numbers were assigned following Ballesteros–Weinstein notation.27 (C,D) Comparison of binding models of 1 (cyan, C) and 4d (green, D) in the homology-modeled A3AR. Representative residues important for A3AR binding are shown in sticks. Distances between the hydroxyl group of thiosugar of ligands and hydrogen of His272 were measured, given in Ångstrom. (E) Putative binding modes suggested by docking simulation of 3 (magenta) and 4d against PPARγ-LBD (PDB ID: 5YCP). Binding models of 4d and 3 were overlaid. (F–I) Putative binding modes suggested by docking simulation of 4d (F), 4l (G), 4h (H) against PPARδ-LBD (PDB ID: 5U46). (I) Comparison of binding models of 27 (orange) and 4d in PPARδ-LBP. H, helix. Possible hydrogen bonds are shown in yellow dashes. Binding free energies calculated by AutoDock Vina are presented in kcal/ mol. All molecular graphics works were conducted using PyMOL software.