

Abstract
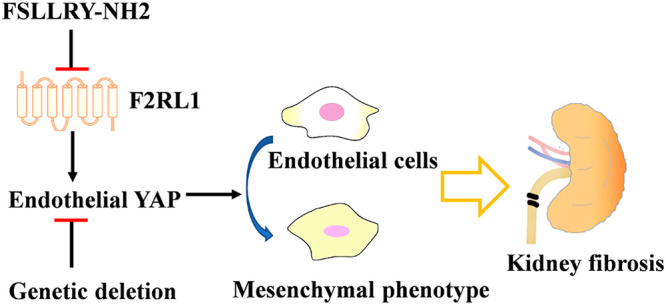
Kidney fibrosis is accompanied by vascular dysfunction. Discovering new ways to ameliorate dysfunctional angiogenesis may bypass kidney fibrosis. YAP (Yes-associated protein) plays a multifaceted role during angiogenesis. Here, we found that selectively targeting YAP signaling in the endothelium ameliorates unilateral ureteral obstruction (UUO)-induced kidney fibrosis. Genetic deletion of Yap1, encoding YAP protein, in VE-cadherin+ endothelial cells inhibited endothelial-to-mesenchymal transition (EndMT) and dysfunctional angiogenesis and improved obstructive nephropathy and kidney fibrosis. Treatment with the systemic YAP inhibitor verteporfin worsened kidney fibrosis symptoms because of its lack of cell specificity. In an attempt to identify endothelial-specific YAP modulators, we found that G-protein-coupled receptor coagulation factor II receptor-like 1 (F2RL1) was highly expressed in vessels after UUO-induced kidney fibrosis. The F2RL1 peptide antagonist FSLLRY-NH2 selectively blocked YAP activity in endothelial cells and ameliorated kidney fibrosis. Thus, selective antagonization of endothelial YAP activity might bypass kidney fibrosis and provide new avenues for the design of antifibrotic therapies.
Keywords: kidney fibrosis, Hippo/YAP signaling, EndMT, dysfunctional angiogenesis, G-protein-coupled receptor, F2RL1
Fibrosis is a process characterized by excessive accumulation of extracellular matrix, which leads to disruption of tissue structure and loss of normal function. This pathology is usually caused by aberrant healing response (scarring) to wounds as a result of repeated or chronic damage.1 Kidney fibrosis is a common feature of chronic kidney disease (CKD) and contributes markedly to end-stage renal failure. The only treatments available for renal failure consist of the replacement of renal function by dialysis or kidney transplantation.2 Currently, few therapies for kidney fibrosis are attempted, and their efficacy is limited. Hence, there is an urgent need to understand how to reduce or reverse kidney fibrosis and to identify potential therapeutic approaches.
The kidney is permeated by a highly complex vascular system with glomerular and peritubular capillary networks, which are essential for maintaining the normal functions of glomerular and tubular epithelial cells.3 In progressive CKD, renal hypoxia is intricately linked to persisting capillary loss, mainly due to dysregulated angiogenesis. Abnormal capillary formation and dysfunctional angiogenesis in the kidney may cause morphological changes in glomeruli as well as infiltration of inflammatory cells, which all constitute potential targets for therapeutic intervention.4 Angiogenic tissue-specific endothelial cells form specialized vascular niches, which participate to induction, specification, and control of organ regeneration as well as homeostasis and metabolism.5
Fibroblasts are key mediators of fibrosis in the kidney, and in addition to primary resident fibroblasts, activated fibroblasts are also produced from endothelial cells via endothelial-to-mesenchymal transition (EndMT). During this transition, endothelial cells lose their identity and acquire mesenchymal characteristics such as compromised cell polarity, loss of cell–cell junctions, and acquisition of fibroblast-like morphology.6 EndMT is reported in animal models of obstructive and diabetic nephropathies, as evidenced by the coexpression of endothelial (CD31) and mesenchymal (α-SMA, FSP1) markers, and contribute to the early development of kidney fibrosis.6−8 Its complex regulation is orchestrated by several signaling pathways. Inhibiting endothelial TGF-β signaling is sufficient to attenuate EndMT and fibrosis in CKD,9,10 and silencing of Snail and Twist transcription factors also limits the acquisition of the EndMT phenotype.6 Blockade of EndMT by a specific inhibitor of Smad3 could delay the early development of streptozotocin-induced diabetic nephropathy.11 Therefore, suppression of EndMT and dysfunctional angiogenesis could alleviate kidney fibrosis and potentially lead to new therapies.
The Hippo/YAP pathway controls organ size in animals and is essential for organ regeneration and development.12 However, abnormal activation of YAP signaling is also involved in organ fibrosis and tumorigenesis.13−15 YAP plays a multifaceted role for endothelial behaviors, for instance in the control of proliferation and metabolic activity during sprouting angiogenesis and maturation.16 Endothelial YAP activity could be regulated by several pathways, including integrin–Gα13–RhoA, and is involved in atherosclerosis and neovascular diseases.16−18 In this study, we showed that YAP expression is induced in kidney sections after unilateral ureteral obstruction (UUO). Genetic deletion of vascular endothelial YAP reduces EndMT and alleviates kidney fibrosis. We further discovered that a coagulation factor II receptor-like 1 (G-protein-coupled receptor F2RL1, also known as PAR2) antagonist selectively blocks the pro-fibrotic activity of YAP in endothelial cells and alleviates kidney fibrosis, which represents a potential therapeutic target for kidney fibrotic diseases.
The Expression of YAP is Increased in Sections from Kidneys Affected by UUO-Induced Fibrosis
To study the pathological changes accompanying kidney fibrosis, we used a mouse model of UUO mimicking obstructive nephropathy. Mouse kidney samples were harvested and analyzed. UUO kidneys showed tubular dilation, epithelial desquamation, and tubulointerstitial fibrosis evidenced by hematoxylin and eosin (H&E), Masson’s, and α-smooth muscle actin (α-SMA) staining (Figure 1A). Immunofluorescence analysis of the endothelial marker CD31 showed angiogenesis was strongly increased in UUO-induced kidney fibrosis (Figure 1B), and EndMT, visualized by the colocalization of CD31 and the fibroblast marker α-SMA,7 was striking. These results demonstrated that dysfunctional angiogenesis and EndMT occurred during the kidney fibrosis. YAP protein has been reported to regulate sprouting angiogenesis and vascular maturation.16 Immunohistochemical and immunofluorescence analyses showed that the expression of YAP was UUO-induced and partly localized in vascular endothelial cells (Figure 1C and D). As a functionally reductant protein of YAP, TAZ protein showed little downregulation in UUO-induced kidney fibrosis and almost was not localized with endothelial cells (Figure S1). These data show that UUO-induced kidney fibrosis promotes dysfunctional angiogenesis, EndMT, and YAP expression.
Figure 1.

Increased expression of YAP correlates with the emergence of EndMT in the mouse model of UUO. (A) Kidney sections were analyzed after 8 days of UUO. The pictures display representative photomicrographs of H&E, Masson’s, and α-SMA stained kidney sections. H&E images show the number of damaged tubules; Masson-stained blue areas and α-SMA staining typify fibrosis. (B) Costaining of UUO or control kidney sections for the vascular endothelial marker CD31 and the mesenchymal marker α-SMA. (C) Immunohistochemical analysis of YAP expression in kidney sections from the mice in panel A. (D) Costaining for CD31 and YAP in kidney sections from the mice in panel A. Large image represents a big vision. Scale bars = 50 μm. The results are from four repeated experiments and are shown as the mean ± SD. *p < 0.05; **p < 0.01.
Genetic Deletion of Vascular Endothelial YAP Inhibits EndMT and Ameliorates UUO-Induced Kidney Fibrosis
To evaluate the potential implication of endothelial YAP in regulating kidney fibrosis, we exploited tamoxifen-inducible endothelial-specific Yap1 (encoding YAP protein) knockout mice. Tamoxifen injection specifically activated CreERT2 activity in endothelial cells and induced endothelial-specific deletion of Yap1 in YAPEnd-cko mice. YAPloxP/loxP mice were used as control (Figure 2A). H&E and Masson’s staining showed that YAPEnd-cko mice had significantly reduced kidney tubular damages upon UUO induction. Consistently, α-SMA expression was also significantly reduced in the YAPEnd-cko mice kidney sections by immunohistochemistry, indicating that kidney fibrosis was alleviated (Figure 2B). Immunofluorescence imaging of kidney tissues to detect CD31 and mesenchymal markers (α-SMA and FSP1) showed that the proportion of endothelial cells undergoing EndMT was significantly reduced in YAPEnd-cko mice compared to the controls (Figure 2C). To further delineate the fibrogenic function of endothelial YAP, we performed RNA sequencing analysis (RNA-seq) on kidney endothelial cells isolated from the different UUO-induced transgenic mice (Figure 2D, Table S1). The expression of proinflammatory cytokines in endothelial cells was significantly upregulated after UUO induction in control mice and reversed in YAPEnd-cko mice. This result is consistent with a previous report showing that deletion of endothelial YAP suppresses inflammatory response.17 Together, these results indicate that genetic deletion of endothelial YAP inhibits EndMT and limits kidney fibrosis (Figure 2E).
Figure 2.

Conditional deletion of YAP in endothelial cells ameliorates UUO-induced kidney fibrosis. (A) Schematic diagram of endothelial cell-specific deletion of YAP in mouse induced by tamoxifen injection. (B) Photomicrographs of H&E, Masson’s, and α-SMA stained kidney sections from YAPloxP/loxP, YAPEnd-cko, and WT (C57BL/6J) mice. Staining was quantified as measurement of fibrosis. (C) Costaining of sections from the indicated kidneys for the vascular endothelial marker CD31 and mesenchymal markers α-SMA and FSP1 and quantification of the numbers of CD31+α-SMA+ and CD31+FSP1+double-positive cells per visual field. (D) Comparison of RNA sequencing data from kidney endothelial cells. (E) Endothelial cell-specific deletion of YAP ameliorates UUO-induced fibrosis by inhibiting endothelial-to-mesenchymal transition. Scale bars = 50 μm. The results are from at least four repeated experiments and are shown as the mean ± SD. *p < 0.05; **p < 0.01.
The Systemic YAP Inhibitor Verteporfin Fails to Ameliorate Obstructive Nephropathy and Aggravates Kidney Fibrosis Symptoms
Verteporfin, a YAP inhibitor, disrupts YAP-TEAD interaction (Figure 3A).19 We first verified that verteporfin inhibited the expression of mesenchymal biomarkers (α-SMA and vimentin) in human umbilical vein endothelial cells (HUVECs) treated with TGF-β (Figure 3B). Next, we investigated the effects of verteporfin on UUO-induced kidney fibrosis in vivo. To our surprise, H&E, Masson’s, and α-SMA staining showed that treatment with verteporfin failed to ameliorate obstructive nephropathy and slightly worsened kidney tubular damages (Figure 3C and D). Meanwhile, verteporfin-treated UUO kidneys had lower frequency of EndMT as in YAP deficient mice, identified by the coexpression of CD31 and α-SMA or FSP1 in comparison to control mice, but single α-SMA and FSP1 were expressed at a higher level (Figure 3E). The results of this study are inconsistent with previous studies,14 and it may be due to the use of different doses of verteporfin (our 10 mg/kg vs their 100 mg/kg). We think that the slight deterioration of kidney fibrosis treated with verteporfin is likely related to its lack of tissue selectivity. Our result is consistent with previous reports that noncell-specific targeting of YAP by small interfering RNA (siRNA) has a paradoxical effect compared to selective YAP inhibition,20 and deletion of YAP in podocytes worsens glomerulosclerosis in response to injury.21 Therefore, pharmacological targeting of endothelial-specific Hippo/YAP signaling is needed to generate more relevant approaches and treat kidney fibrotic pathologies (Figure 3F).
Figure 3.

Systemic YAP inhibitor verteporfin blocks EndMT but also worsens obstructive nephropathy and kidney fibrosis symptoms. (A) Chemical structure of the verteporfin. (B) Effects of verteporfin on the expression of the mesenchymal markers α-SMA and vimentin induced in TGF-β-treated HUVEC cells for 24 h incubation. (C) Schematic diagram of detecting the effects of verteporfin injected intravenously in a mouse model of UUO-induced kidney fibrosis. (D) Photomicrographs of H&E, Masson’s, and α-SMA stained kidney sections. Staining quantification was used as the measurement of fibrosis severity. Contralateral kidneys served as healthy controls. (E) Costaining of sections from the indicated kidneys for the vascular endothelial marker CD31 and the mesenchymal markers α-SMA and FSP1. Quantification of the number of CD31+α-SMA+ and CD31+FSP1+ double-positive cells per visual field. (F) The YAP inhibitor verteporfin worsens obstructive nephropathy and aggravates kidney fibrosis symptoms due to low tissue selectivity. Scale bars = 50 μm. The results are from at least four repeated experiments and are shown as the mean ± SD. *p < 0.05; **p < 0.01.
Coagulation Factor II Receptor-like 1 (F2RL1) Regulates Hippo/YAP Signaling in Vascular Endothelial Cells
G-protein-coupled receptor (GPCR) signaling regulates YAP activity in a receptor class-specific manner.22,23 This knowledge led us to hypothesize that GPCRs could be attractive therapeutic targets because of their cell-specific expression pattern. F2RL1 is associated with new blood vessel formation during retinal development and in ischemic retinopathy.24 Immunohistochemical and immunofluorescence analyses showed that F2RL1 expression was upregulated after UUO induction and restricted to the vascular endothelium (Figure 4A and B). To investigate whether F2RL1 could regulate Hippo/YAP signaling in endothelial cells, we used the RNA interference (RNAi) method and constructed lentiviral vectors driving the production of short hairpin RNAs (shRNAs) targeting F2RL1. We obtained stable F2RL1-knockdown HUVEC cell lines by gene resistance selection and successfully silenced F2RL1 mRNA, with the highest efficacy obtained in the cell line shF2RL1-2 (Figure 4C). F2RL1 silencing reduced YAP expression in HUVECs (Figure 4D). Knockdown of F2RL1 decreased HUVEC proliferation, tubulogenesis, and expression of mesenchymal biomarkers (α-SMA and vimentin) (Figure 4E and F). Furthermore, silencing of F2RL1 and YAP in HUVECs prevented the induction of proinflammatory cytokines upon LPS stimulation (Figure 4G). These data suggest that targeting of F2RL1 might selectively modulate YAP activity in endothelial cells (Figure 4H).
Figure 4.

G-protein-coupled receptor F2RL1 is selectively upregulated in vascular endothelial cells after UUO induction and targets YAP signaling in HUVECs. (A) Immunohistochemical analysis showing UUO-induced F2RL1 expression in kidney sections from mice. Scale bars = 50 μm. (B) Costaining of sections from the indicated kidneys for the vascular endothelial marker CD31 and F2RL1 indicates selective upregulation of F2RL1 in the vascular endothelium after UUO induction. Scale bars = 50 μm. (C) Knockdown efficiency by shRNA targeting of endogenous F2RL1 in HUVECs confirmed by quantitative PCR. (D) Knockdown of endogenous F2RL1 in HUVECs reduces YAP expression. (E) Effects of endogenous F2RL1 or YAP knockdown on the expression of the mesenchymal markers α-SMA and vimentin in HUVECs treated with TGF-β for 24 h. (F) Knockdown of endogenous F2RL1 reduces HUVEC proliferation and tubulogenesis. Scale bars = 100 μm. (G) Knockdown of endogenous F2RL1 in HUVECs reduces the expression of proinflammatory cytokines after stimulation with 200 ng/mL LPS for 6 h. (H) F2RL1 targets YAP signaling in HUVECs. The results are from at least three repeated experiments and are shown as the mean ± SD. *p < 0.05; **p < 0.01.
The F2RL1 Antagonist FSLLRY-NH2 Attenuates EndMT and Ameliorates Kidney Fibrosis
FSLLRY-NH2 is a selective peptide antagonist targeting F2RL1.25 Our results showed that it inhibited YAP activity by promoting its phosphorylation in HUVECs (Figure 5A and B). The in vivo effects of FSLLRY-NH2 on YAP activity and kidney fibrosis were evaluated in a UUO-induced kidney fibrosis mouse model (Figure 5C). The FSLLRY-NH2-treated group exhibited a reversal of established kidney fibrosis compared to the control group, as revealed by H&E and Masson’s staining. Immunohistochemistry showed that the α-SMA positive area in the kidneys of treated mice was reduced, as compared to the control group (Figure 5D). Costaining of CD31 and YAP showed that treatment with FSLLRY-NH2 mainly inhibited YAP expression in the vascular endothelium and preserved its expression in other regions (Figure 5E). Meanwhile, CD31 and α-SMA or FSP1 costaining of kidney tissues showed a lower frequency of EndMT in the FSLLRY-NH2-treated group compared to the control group (Figure 5F). Together, these data indicate that the F2RL1 antagonist FSLLRY-NH2 can selectively block YAP activity in vascular endothelial cells and afford significant protection in UUO-induced kidney fibrosis (Figure 5G).
Figure 5.

The F2RL1 antagonist FSLLRY-NH2 ameliorates kidney fibrosis symptoms by inhibiting EndMT and dysfunctional angiogenesis. (A) Schematic diagram of the F2RL1 antagonist FSLLRY-NH2. (B) FSLLRY-NH2 induces YAP phosphorylation in HUVECs treated with TGF-β for 24 h. (C) Schematic diagram of detecting the effects of FSLLRY-NH2 intravenously injected in mice with UUO-induced kidney fibrosis. (D) Photomicrographs of H&E, Masson’s, and α-SMA stained kidney sections. Staining quantification was performed to evaluate fibrosis severity. Contralateral kidneys were used as negative controls. (E) Costaining of sections from the indicated kidneys for CD31 and YAP shows the selective inhibition of YAP expression in vascular endothelial cells achieved by F2RL1 targeting. (F) Costaining of sections from the indicated kidneys for the vascular endothelial marker CD31 and the mesenchymal markers α-SMA and FSP1 and quantification of the numbers of CD31+α-SMA+ and CD31+FSP1+ double-positive cells per visual field. (G) The F2RL1 antagonist FSLLRY-NH2 selectively inhibits YAP activity in vascular endothelial cells and ameliorates kidney fibrosis. Scale bars = 50 μm. The results are from at least four repeated experiments and are shown as the mean ± SD. *p < 0.05; **p < 0.01.
In this study, we show that genetic deletion of YAP in VE-cadherin+ endothelial cells inhibits the emergence of EndMT and improves kidney fibrosis in a UUO mouse model. Although verteporfin could inhibit dysfunctional angiogenesis and EndMT, it aggravates obstructive nephropathy. We speculated that verteporfin lacks selectivity toward endothelial-specific YAP and impairs the pro-regenerative functions of YAP in other cell lineages.12,21 Indeed, our immunofluorescence analysis also showed that YAP expression was not completely restricted to the vascular endothelium but also covered other regions (Figure 1D). Widespread expression and pleiotropic roles of YAP suggested that specific targeting strategies are required to selectively block its pathological activities.20,26
GPCRs represent the largest family of membrane receptors and are readily targetable by therapeutic drugs. Meanwhile, GPCR expression varies across organs and between adjacent cell types, suggesting that GPCR-directed therapeutics might be utilized for cell-specific pharmacological targeting of YAP.23 Our results showed that GPCR F2RL1 expression was upregulated after UUO induction and dominantly confined to the vascular endothelium. The F2RL1 antagonist FSLLRY-NH2 markedly blocked YAP activity in vascular endothelial cells and preserved its pro-regenerative functions in other regions. Therefore, targeting endothelial cell-specific YAP signaling via GPCR modulation might help development of new approaches to ameliorate kidney fibrosis.
Materials and Methods
Mouse Models
Mice carrying transgenic loxP sites flanking the Yap1 locus were obtained from Jackson Laboratories (Yap1loxP/loxP mice, stock #032192). Mice expressing endothelial-specific VE-cadherin-CreERT2 were provided by R. Adams. Yap1loxP/loxP mice were crossed with VE-cadherin-CreERT2 mice to obtain Yap1loxP/loxP-VE-cadherin-CreERT2+ mice (referred to as YAPEnd-cKO mice) and Yap1loxP/loxP -VE-cadherin-CreERT2- mice (referred to as YAPloxP/loxP mice). Mice were genotyped by polymerase chain reaction (PCR) with tail DNA.
UUO was performed as previously described.27 Sham animals underwent the same procedure without ligation of the ureter. For therapies, verteporfin (Selleck, catalog S1786, 10 mg/kg) and FSLLRY-NH2 (peptide was synthesized in GL Biochem, 10 mg/kg) were injected intravenously into the mice every other day after UUO. At indicated time points, mice were killed, and whole kidney tissues were collected for analysis. Animal welfare supervision and experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China) and the Animal Experiment Administration Committee of State Key Laboratory of Biotherapy, Sichuan University.
Immunostaining and Histological Assays of Mice Kidneys
The experiments were performed as previously described.6
Silencing of Endogenous Expression of YAP and F2RL1
Construction of shRNA recombinant lentiviruses targeting human YAP and F2RL1 and silencing experiments were adapted from a previous report.28 The shRNA sequences used in this study are reported in the Supporting Information (Table S2).
Statistical Analysis
Statistical analysis was conducted using the GraphPad Prism 6 software. All data are expressed as mean ± SD. Statistical analysis of differences between two groups was performed using a two-tailed t test. p-Values <0.05 and <0.01 were considered significant and very significant, respectively, and shown as *p < 0.05; **p < 0.01.
More methods, including antibody information, are available in the Supporting Information.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grants 81700542, 22077106, and 81601213); Sichuan Province Science and Technology Innovation Talent Project (Grant 2020JDRC0113); Science and Technology Strategic Cooperation Project of Luzhou Municipal People’s Government-Southwest Medical University (Grant 2019LZXNYDJ31); Luzhou Municipal-Southwest Medical University Joint Special Grant for the Introduction of High-Level Talents (Lan Hui-Yao Team); Luzhou-Southwest Medical Joint Platform Project (Grant 2018LZXNYD-PT03); Joint Project of Southwest Medical University and Affiliated Traditional Medicine Hospital of Southwest Medical University (Grants 2018XYLH-022 and 2017XYLH-028), and the Natural Science Project of Southwest Medical University (Grant 2020ZRQNA036). We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00010.
Supporting Methods: cell lines and antibodies, isolation of mouse kidney endothelial cells, and RNA quantification and sequencing; Table S1: RNA sequencing data from kidney endothelial cells; Table S2: ShRNA targeting sequence; Table S3: all primers used for RT-PCR; Figure S1: detection of the expression of renal TAZ in the mouse model of UUO (PDF)
Table S1: RNA sequencing analysis on kidney endothelial cells isolated from the UUO-induced YAPEnd-cko or control mice (XLS)
Author Contributions
□ Y.R., Y.Z., and Lu W. contributed equally to this work. J.Q., Li W., and B.-S.D. designed the study. J.Q., Li W., B.-S.D., Y.R., and Y.Z. wrote the manuscript. J.Q., Y.R., Y.Z., Lu W., M.Y., X.L., Y.O., T.B., and S.W. performed experiments. J.Q., Y.R., Y.Z., F.H., and Q.W. analyzed the data. J.L. revised the manuscript and provided some experimental conditions. J.Q., Li W., and B.-S.D. supported the research financially.
The authors declare no competing financial interest.
Supplementary Material
References
- Jun J. I.; Lau L. F. (2018) Resolution of organ fibrosis. J. Clin. Invest. 128, 97–107. 10.1172/JCI93563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande M. T.; Sanchez-Laorden B.; Lopez-Blau C.; De Frutos C. A.; Boutet A.; Arevalo M.; Rowe R. G.; Weiss S. J.; Lopez-Novoa J. M.; Nieto M. A. (2015) Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21, 989–997. 10.1038/nm.3901. [DOI] [PubMed] [Google Scholar]
- Tanabe K.; Wada J.; Sato Y. (2020) Targeting angiogenesis and lymphangiogenesis in kidney disease. Nat. Rev. Nephrol. 16, 289–303. 10.1038/s41581-020-0260-2. [DOI] [PubMed] [Google Scholar]
- Liu L.; You Z.; Yu H.; Zhou L.; Zhao H.; Yan X.; Li D.; Wang B.; Zhu L.; Xu Y.; Xia T.; Shi Y.; Huang C.; Hou W.; Du Y. (2017) Mechanotransduction-modulated fibrotic microniches reveal the contribution of angiogenesis in liver fibrosis. Nat. Mater. 16, 1252–1261. 10.1038/nmat5024. [DOI] [PubMed] [Google Scholar]
- Rafii S.; Butler J. M.; Ding B. S. (2016) Angiocrine functions of organ-specific endothelial cells. Nature 529, 316–325. 10.1038/nature17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovisa S.; Fletcher-Sananikone E.; Sugimoto H.; Hensel J.; Lahiri S.; Hertig A.; Taduri G.; Lawson E.; Dewar R.; Revuelta I.; Kato N.; Wu C. J.; Bassett R. L.; Putluri N.; Zeisberg M.; Zeisberg E. M.; LeBleu V. S.; Kalluri R. (2020) Endothelial-to-mesenchymal transition compromises vascular integrity to induce Myc-mediated metabolic reprogramming in kidney fibrosis. Sci. Signaling 13, eaaz2597. 10.1126/scisignal.aaz2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg E. M.; Potenta S. E.; Sugimoto H.; Zeisberg M.; Kalluri R. (2008) Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 19, 2282–2287. 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Qu X.; Bertram J. F. (2009) Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 175, 1380–1388. 10.2353/ajpath.2009.090096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier S.; Vasko R.; Matsumoto K.; Zullo J. A.; Chen R.; Maizel J.; Chander P. N.; Goligorsky M. S. (2015) Curtailing endothelial TGF-beta signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J. Am. Soc. Nephrol. 26, 817–829. 10.1681/ASN.2013101137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley B. C.; Nevado J.; Mellad J.; Yang D.; St Hilaire C.; Negro A.; Fang F.; Chen G.; San H.; Walts A. D.; Schwartzbeck R. L.; Taylor B.; Lanzer J. D.; Wragg A.; Elagha A.; Beltran L. E.; Berry C.; Feil R.; Virmani R.; Ladich E.; Kovacic J. C.; Boehm M. (2014) TGF-beta signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci. Transl. Med. 6, 227ra234. 10.1126/scitranslmed.3006927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Qu X.; Yao J.; Caruana G.; Ricardo S. D.; Yamamoto Y.; Yamamoto H.; Bertram J. F. (2010) Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 59, 2612–2624. 10.2337/db09-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya I. M.; Halder G. (2019) Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 20, 211–226. 10.1038/s41580-018-0086-y. [DOI] [PubMed] [Google Scholar]
- Johnson R.; Halder G. (2014) The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment, Nature reviews. Nat. Rev. Drug Discovery 13, 63–79. 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M.; Yu M.; Xia R.; Song K.; Wang J.; Luo J.; Chen G.; Cheng J. (2017) Yap/Taz Deletion in Gli(+) Cell-Derived Myofibroblasts Attenuates Fibrosis. J. Am. Soc. Nephrol. 28, 3278–3290. 10.1681/ASN.2015121354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M. B.; Sun H.; Robichaux J.; Pfeifer M.; McDermott U.; Travers J.; Diao L.; Xi Y.; Tong P.; Shen L.; Hofstad M.; Kawakami M.; Le X.; Liu X.; Fan Y.; Poteete A.; Hu L.; Negrao M. V.; Tran H.; Dmitrovsky E.; Peng D.; Gibbons D. L.; Wang J.; Heymach J. V. (2020) A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci. Transl. Med. 12, eaaz4589. 10.1126/scitranslmed.aaz4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Kim Y. H.; Kim J.; Park D. Y.; Bae H.; Lee D. H.; Kim K. H.; Hong S. P.; Jang S. P.; Kubota Y.; Kwon Y. G.; Lim D. S.; Koh G. Y. (2017) YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Invest. 127, 3441–3461. 10.1172/JCI93825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Luo J. Y.; Li B.; Tian X. Y.; Chen L. J.; Huang Y.; Liu J.; Deng D.; Lau C. W.; Wan S.; Ai D.; Mak K. K.; Tong K. K.; Kwan K. M.; Wang N.; Chiu J. J.; Zhu Y.; Huang Y. (2016) Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540, 579–582. 10.1038/nature20602. [DOI] [PubMed] [Google Scholar]
- Wang K. C.; Yeh Y. T.; Nguyen P.; Limqueco E.; Lopez J.; Thorossian S.; Guan K. L.; Li Y. J.; Chien S. (2016) Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. U. S. A. 113, 11525–11530. 10.1073/pnas.1613121113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chittenden Y.; Huang B.; Shim J. S.; Chen Q.; Lee S. J.; Anders R. A.; Liu J. O.; Pan D. (2012) Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26, 1300–1305. 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haak A. J.; Kostallari E.; Sicard D.; Ligresti G.; Choi K. M.; Caporarello N.; Jones D. L.; Tan Q.; Meridew J.; Diaz Espinosa A. M.; Aravamudhan A.; Maiers J. L.; Britt R. D.; Roden A. C.; Pabelick C. M.; Prakash Y. S.; Nouraie S. M.; Li X.; Zhang Y.; Kass D. J.; Lagares D.; Tager A. M.; Varelas X.; Shah V. H.; Tschumperlin D. J. (2019) Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci. Transl. Med. 11, eaau6296. 10.1126/scitranslmed.aau6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J. J.; Goldstein L.; Chen Y. J.; Lee J.; Webster J. D.; Roose-Girma M.; Paudyal S. C.; Modrusan Z.; Dey A.; Shaw A. S. (2020) Single-Cell Transcriptome Profiling of the Kidney Glomerulus Identifies Key Cell Types and Reactions to Injury. J. Am. Soc. Nephrol. 31, 2341–2354. 10.1681/ASN.2020020220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F. X.; Zhao B.; Panupinthu N.; Jewell J. L.; Lian I.; Wang L. H.; Zhao J.; Yuan H.; Tumaneng K.; Li H.; Fu X. D.; Mills G. B.; Guan K. L. (2012) Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791. 10.1016/j.cell.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel P. A.; Snead A.; Murray F.; Zhang L.; Yokouchi H.; Katakia T.; Kwon O.; Dimucci D.; Wilderman A. (2012) GPCR expression in tissues and cells: are the optimal receptors being used as drug targets?. British journal of pharmacology 165, 1613–1616. 10.1111/j.1476-5381.2011.01434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyal J. S.; Nim S.; Zhu T.; Sitaras N.; Rivera J. C.; Shao Z.; Sapieha P.; Hamel D.; Sanchez M.; Zaniolo K.; St-Louis M.; Ouellette J.; Montoya-Zavala M.; Zabeida A.; Picard E.; Hardy P.; Bhosle V.; Varma D. R.; Gobeil F. Jr.; Beausejour C.; Boileau C.; Klein W.; Hollenberg M.; Ribeiro-da-Silva A.; Andelfinger G.; Chemtob S. (2014) Subcellular localization of coagulation factor II receptor-like 1 in neurons governs angiogenesis. Nat. Med. 20, 1165–1173. 10.1038/nm.3669. [DOI] [PubMed] [Google Scholar]
- Ocak U.; Eser Ocak P.; Huang L.; Zhang J. H. (2019) FSLLRY-NH2 Improves Neurological Outcome After Cardiac Arrest in Rats. Turk Neurosurg 30, 244–251. 10.5137/1019-5149.JTN.27714-19.1. [DOI] [PubMed] [Google Scholar]
- van Soldt B. J.; Qian J.; Li J.; Tang N.; Lu J.; Cardoso W. V. (2019) Yap and its subcellular localization have distinct compartment-specific roles in the developing lung. Development 146, dev175810. 10.1242/dev.175810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M.; Soubasakos M. A.; Kalluri R. (2005) Animal models of renal fibrosis. Methods Mol. Med. 117, 261–272. 10.1385/1-59259-940-0:261. [DOI] [PubMed] [Google Scholar]
- Qing J.; Wu M.; Luo R.; Chen J.; Cao L.; Zeng D.; Shang L.; Nong J.; Wu Q.; Ding B. S.; Chen X.; Rao Z.; Liu L.; Lou Z. (2020) Identification of Interferon Receptor IFNAR2 As a Novel HCV Entry Factor by Using Chemical Probes. ACS Chem. Biol. 15, 1232–1241. 10.1021/acschembio.9b00912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.