
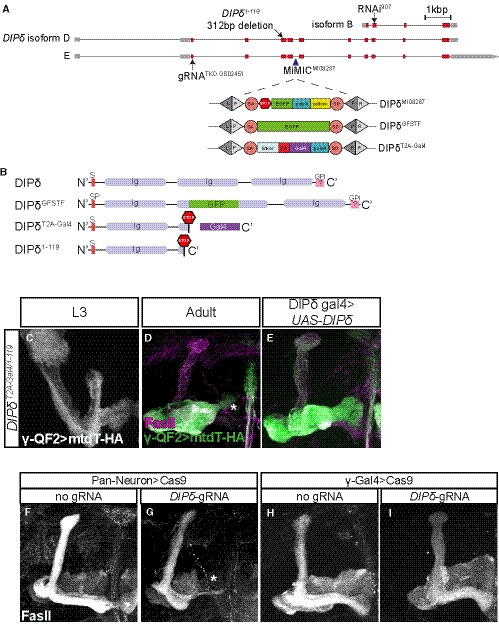
Figure EV3. Description of DIP‐δ alleles used in the study and additional DIP‐δ perturbation phenotypes, related to Fig 4.
-
AA schematic representation of the DIP‐δ locus showing introns (black line) and coding and non‐coding exons (red and gray, respectively). The location of DIP‐δ gRNA, DIP‐δ1‐119 mutation, DIP‐δ RNAi, and MiMICMI08287 is indicated. SA and SD are splice acceptor and donor sites, respectively. Recombination‐mediated cassette exchange was used to transform DIP‐δMI08287 into DIP‐δGFSTF and DIP‐δT2A‐Gal4.
-
BA schematic description of DIP‐δ protein variants. Signal peptide (SP), Immunoglobulin (Ig), and GPI anchor (GPI).
-
C–EConfocal z‐projections of DIP‐δ transheterozygotes (DIP‐δT2A‐Gal4/1‐119) at L3 (C; n = 20/20) and adult (D; n = 12/12, E; n = 22/23), in which γ neurons were marked by expressing membrane‐bound tandem tomato (mtdT‐HA) driven by the γ‐specific QF2 driver GMR71G10‐QF2 (γ‐QF2). In DIP‐δ transheterozygotes, as in homozygous mutants (see Fig 4), γ‐KCs do not extend into the distal end of the lobe. Expression of DIP‐δ in DIP‐δ+ cells (E) rescues the growth defect present in DIP‐δT2A‐Gal4/1‐119 brains. Green and white represent mtdT‐HA; magenta represents FasII staining.
-
F–IConfocal z‐projections of brains expressing UAS‐Cas9 alone (F, H) or together with DIP‐δ‐gRNA (G, I). DIP‐δ knockout by tsCRISPR in all postmitotic neurons (G; n = 22/24) resulted in a defect in γ4/5 innervation by γ‐axons, while DIP‐δ knockout by tsCRISPR in γ‐KCs (I; n = 28/28) did not affect γ‐axon regrowth. Expression of Cas9 alone (F, n = 10/10; H, n = 14/14) did not affect γ‐axon regrowth. White represents FasII staining.
Data information: Scale bar is 20 µm.