

ABSTRACT
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system, which has been found associated with dysfunctional mitochondria. In order to advance our understanding of the complex molecular mechanisms underlying this disease, we analyzed mitophagy, a process fundamental for the elimination of damaged mitochondria through the autophagic process, in peripheral blood mononuclear cells (PBMCs) of MS patients. Through a genetic analysis carried out on 203 MS patients and 1000 healthy controls, we identified a natural variant of CALCOCO2/NDP52, a well-known autophagic receptor, associated with and protective in MS. Structural modeling of the CALCOCO2 variant and functional studies highlighted an amino acid substitution (G140E) located near the LC3-interacting region (LIR) motif of CALCOCO2, crucial in controlling mitophagy. In addition, we found that among PBMCs, CALCOCO2 is mainly expressed in B cells and, by mediating mitophagy, it reduces pro-inflammatory cytokine production following stimulation of these cells. Here we summarize these recent findings, discuss the putative protective roles of CALCOCO2 in B cells and its novel association with an autoimmune disease such as MS.
Abbreviations: LIR: LC3-interacting region; MS: multiple sclerosis; PBMC: peripheral blood mononuclear cells; RR-MS: relapsing-remitting MS; TLR: toll like receptor.
KEYWORDS: B cells, CALCOCO2, inflammation, mitochondria, mitophagy, multiple sclerosis
Multiple sclerosis (MS) is a very debilitating chronic autoimmune disease. Depending on the region of the world, the incidence is of 50 to 300 per 100,000 individuals. MS generally affects young adults with a prevalence of 3 female patients for every male patient. Currently, the processes that trigger the disease are not known, but genetic predispositions, environmental agents (such as bacteria or virus infections) and social components (such as tobacco consumption) participate in the pathogenesis of MS.
There are three types of MS. A cyclic or remitting form (relapsing-remitting MS, RR-MS), which affects 80–85% of patients without progression of the handicap between relapses. This MS can progress to secondarily progressive MS (or MS-SP). There is also a primary progressive form (or PP-MS) in which the phases of the disease are not interspersed with phases of remission, affecting approximately 15% of patients.
In all cases, the disease is characterized by the onset of varying disabilities all due to the progressive infiltration of cells of the adaptive immune system, T and B lymphocytes, into the central nervous system. These activated cells are responsible for the destruction of the myelin sheath of neurons and the development of neuronal and axonal damage.
One characteristic of MS is the presence of oxidative stress and mitochondrial dysfunctions associated with the pathology. However, the role played by mitophagy – a process that selectively eliminates altered mitochondria through the autophagic process to regulate the functional quality of mitochondria and to reduce oxidative stress – remains completely obscure in MS pathogenesis.
In our recent article [1], we identified and characterized a variant of the autophagic receptor CALCOCO2/NDP52 that links mitophagy and MS pathogenesis. Indeed, by sequencing the DNA coming from peripheral blood mononuclear cells (PBMCs) of 203 RR-MS patients with specific probes for the genes encoding two main mitophagic receptors, CALCOCO2 and OPTN, we revealed a significant association of CALCOCO2 c.491 G > A mutation (rs550510, p.G140E) with a decreased susceptibility to MS, suggesting that the CALCOCO2 variant CALCOCO2G140E may confer protection to MS. Functional analyses were performed for the two CALCOCO2 forms, wild-type (WT) CALCOCO2 and protective CALCOCO2G140E, using overexpression of tagged protein variants in a human cell line under different conditions.
First, because enhanced immune response may benefit MS patients by eliminating microbes and because CALCOCO2 negatively regulates TLR (toll like receptor)-triggered NFKB activation, we thought to test whether the CALCOCO2G140E variant was protective in MS by favoring activation of NFKB, following TLR stimulation. However, our data indicate that the substitution of G140E of CALCOCO2 does not affect its function in regulating TLR signaling.
Second, because mitochondrial dysfunctions are related to MS pathogenesis and CALCOCO2 serves as a bridge between mitochondria and phagophores during degradation of damaged mitochondria by autophagy, we then decided to focus our attention on the role played by CALCOCO2G140E in mitophagy.
Our study showed that the CALCOCO2G140E variant is more efficient in removing damaged mitochondria compared with WT CALCOCO2. The single amino acid substitution in the CALCOCO2 sequence is crucial for extending its noncanonical LIR motif (cLIR) and promoting its binding with LC3C, thus favoring mitophagy.
Our structural and biochemical analysis indicated that the CALCOCO2G140E variant exhibits a better affinity with the LC3C protein. Further studies will be necessary to quantitatively estimate the propensity to form hydrogen bonds and/or to the formation of salt bridges induced by this point mutation.
We demonstrated that, despite the efficiency of the protective CALCOCO2G140E variant, the available data on the rs550510 variant (reported on 1000Genomes and GnomAD databases of genetic variants annotation) indicate that the protective allele is more common in some populations (i.e., East Asian) than others (i.e., European). This correlates with the global distribution of MS; however, from a genetic point of view, the cause of the lower distribution of CALCOCO2G140E in the European population will be an interesting point to examine in future studies.
We discovered that among PBMCs, CALCOCO2 is mainly expressed in B cells. In MS pathology, B lymphocytes are involved in the secretion of pro-inflammatory cytokines, in the presentation of autoantigens to T lymphocytes and in the secretion of autoantibodies. Depleting B-cells, using specific antibodies in RR-MS patients, leads to the suppression of an inflammatory response, thus reducing the formation of new lesions. Interestingly, our findings pointed out that, following B cell stimulation, CALCOCO2 ensures effective mitophagy thus reducing the release of a pro-inflammatory cytokine, TNF/TNF-α.
We do not exclude the possibility that, despite our findings that highlight a pro-mitophagy function of CALCOCO2G140E in MS, this variant may also emerge as a novel link among 1) xenophagy, a form of selective autophagy of pathogens (such as bacteria and virus), 2) B cells and 3) viral infections known to be associated with MS (i.e., Epstein-Barr virus, one of the main environmental risk factors for MS). Hence, future studies will be required to better characterize and understand the molecular mechanisms underlying the novel function of CALCOCO2 in B cells in the context of MS.
In conclusion, our results allow us to propose a model in which the variant G140E of CALCOCO2 may prevent the release of pro-inflammatory cytokines from B cells (and maybe mitochondrial factors such as mitochondrial DNA) that trigger inflammation in MS patients and most likely in other autoimmune diseases (See Figure 1).
Figure 1.
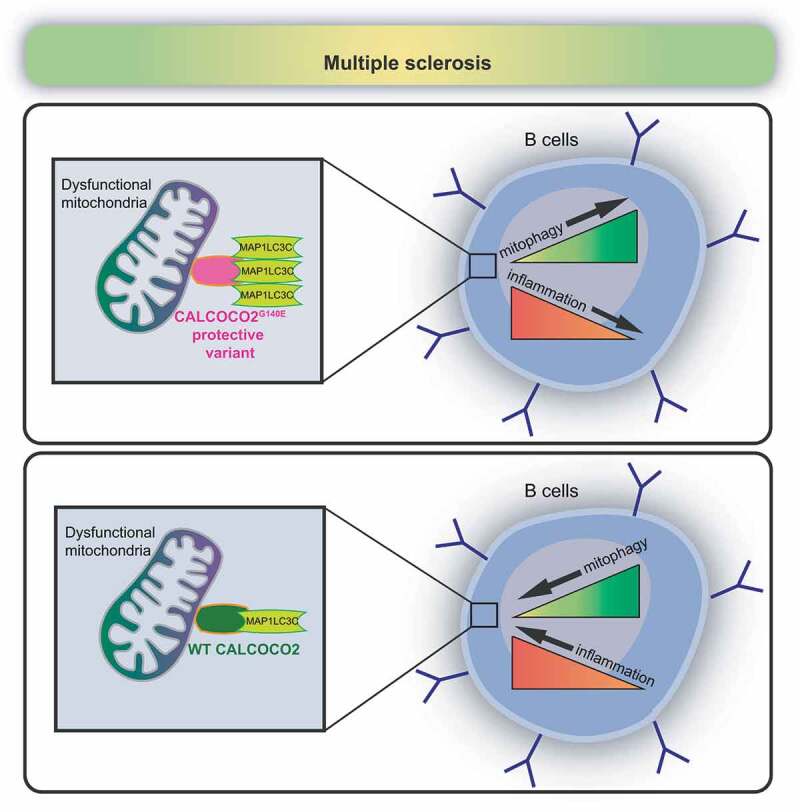
Hypothetical model of the protective role of CALCOCO2G140E variant in B cells of RR-MS patients. In multiple sclerosis, the protective variant of CALCOCO2 (CALCOCO2G140E) can inhibit pro-inflammatory cytokine production (TNF) in B cells by increasing its binding with MAP1LC3C and thus favoring the selective degradation of damaged mitochondria (mitophagy) (upper panel). WT CALCOCO2, instead, has a restricted interaction with MAP1LC3C, and this can reduce the elimination of dysfunctional mitochondria and enhance inflammation in MS patients (lower panel)
Altogether, this work highlights a potentially druggable mechanism that links CALCOCO2-mediated mitophagy to fine tuning pro-inflammatory signaling cascades in B lymphocytes. Small molecule development may focus on compounds enhancing/stabilizing LIR-dependent interactions between CALCOCO2 and human LC3 in B cells, thereby opening an important path for the generation of new therapeutic tools.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- [1].Di Rita A, Angelini DF, Maiorino T, et al. Characterization of a natural variant of human NDP52 and its functional consequences on mitophagy. Cell Death Differ. 2021. March 15. DOI: 10.1038/s41418-021-00766-3. [DOI] [PMC free article] [PubMed] [Google Scholar]