

Abstract
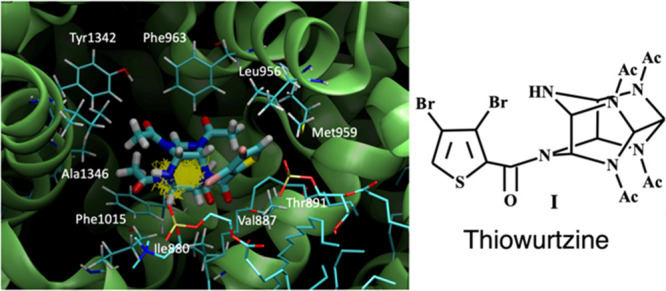
The number of candidate molecules for new non-narcotic analgesics is extremely limited. Here, we report the identification of thiowurtzine, a new potent analgesic molecule with promising application in chronic pain treatment. We describe the chemical synthesis of this unique compound derived from the hexaazaisowurtzitane (CL-20) explosive molecule. Then, we use animal experiments to assess its analgesic activity in vivo upon chemical, thermal, and mechanical exposures, compared to the effect of several reference drugs. Finally, we investigate the potential receptors of thiowurtzine in order to better understand its complex mechanism of action. We use docking, molecular modeling, and molecular dynamics simulations to identify and characterize the potential targets of the drug and confirm the results of the animal experiments. Our findings finally indicate that thiowurtzine may have a complex mechanism of action by essentially targeting the mu opioid receptor, the TRPA1 ion channel, and the Cav voltage-gated calcium channel.
Introduction
Synthesis of candidate molecules for the design of non-narcotic analgesics to relieve severe and moderate pain is a trending topic in pharmaceutics. Three types of treatments against pain of different etiologies are primarily used in out-patients and in-patients: (1) opioid and cannabinoid molecules with various analgesic activities, (2) nonsteroidal anti-inflammatory drugs (NSAIDs) acting as antalgic molecules, and (3) their various combinations in multimodal approaches for chronic pain, incorporating modalities such as physiotherapy, psychological therapy, patient education, and peripheral stimulation.1−5 An alternative to opiates for severe and moderate pain relief may reside in the use of non-narcotic analgesics acting on the central and peripheral nervous systems. However, the potential candidate molecules for non-narcotic analgesics are extremely limited in number. In this regard, one of the current successful approaches for the discovery of new molecules for biomedical research is the development of high-throughput methods of virtual screening for large databases of chemical compounds.6−8
Here, we report the development of a new patented analgesic molecule,9 hereafter referred to as thiowurtzine, based on the hexaazaisowurtzitane (CL-20) molecule, a substance commonly known as a component of rocket fuel and explosives. Although several explosives such as nitroglycerin or potassium nitrate have been used as drugs in medical history,10−13 the case of thiowurtzine is quite different, as it is directly derived from the CL-20 molecule but lacks all of the six nitro groups which confer its explosive properties to CL-20. As the six nitro groups are added only during the very last step of the chemical synthesis of CL-20,14 thiowurtzine can very well be considered more as a modified precursor rather than a revised explosive molecule.
Hexaazaisowurtzitane derivatives are caged polycyclic polynitrogen compounds and possess a framework structure determining their unique properties.15,16 Yet, numerous studies focused on the synthesis of new hexaazaisowurtzitane derivatives have until recently been primarily focused on their explosive and propelling properties for military purposes as promising components of solid rocket fuel and composite explosives.16,17
Only in recent years has hexaazaisowurtzitane been investigated as a new pharmacophore for the development of original pharmaceutical substances. In this more biological context, preliminary studies investigating the analgesic activity of hexaazaisowurtzitane derivatives have recently been described.18 The aim of the project was to conduct conclusive preclinical studies of drugs based on the hexaazaisowurtzitane molecule for the treatment of the pain syndrome caused by different etiologies.
Thiowurtzine is the first chemical compound in the class of hexaazaisowurtzitanes that presents a significant, experimentally validated bioactivity.15,19 After different patents of the molecule9,20 were published, we started to investigate its potential targets using computational techniques.
In this study, we report for the first time the synthesis of 4-(3,4-dibromothiophenecarbonyl)-2,6,8,12-tetraacetyl-2,4,6,8,10,12-hexaazatetracyclo[5,5,0,03,11,05,9]dodecane (thiowurtzine, I), whose properties were predicted using the PASS software package.21 Then, we use animal experiments to assess its analgesic activity in vivo upon chemical, thermal, and mechanical exposures, compared to the effect of several reference molecules. Finally, we provide the first investigation on the potential receptors of thiowurtzine in order to better understand its mechanism of action. We use docking, molecular modeling, and molecular dynamics (MD) simulations to identify and characterize the potential targets of the drug and confirm the results of the animal experiments.
Results and Discussion
Chemistry
A series of sequential reactions were used for the synthesis of thiowurtzine (I) as a final compound, as illustrated in Scheme 1.
Scheme 1. Synthesis of Thiowurtzine (I).
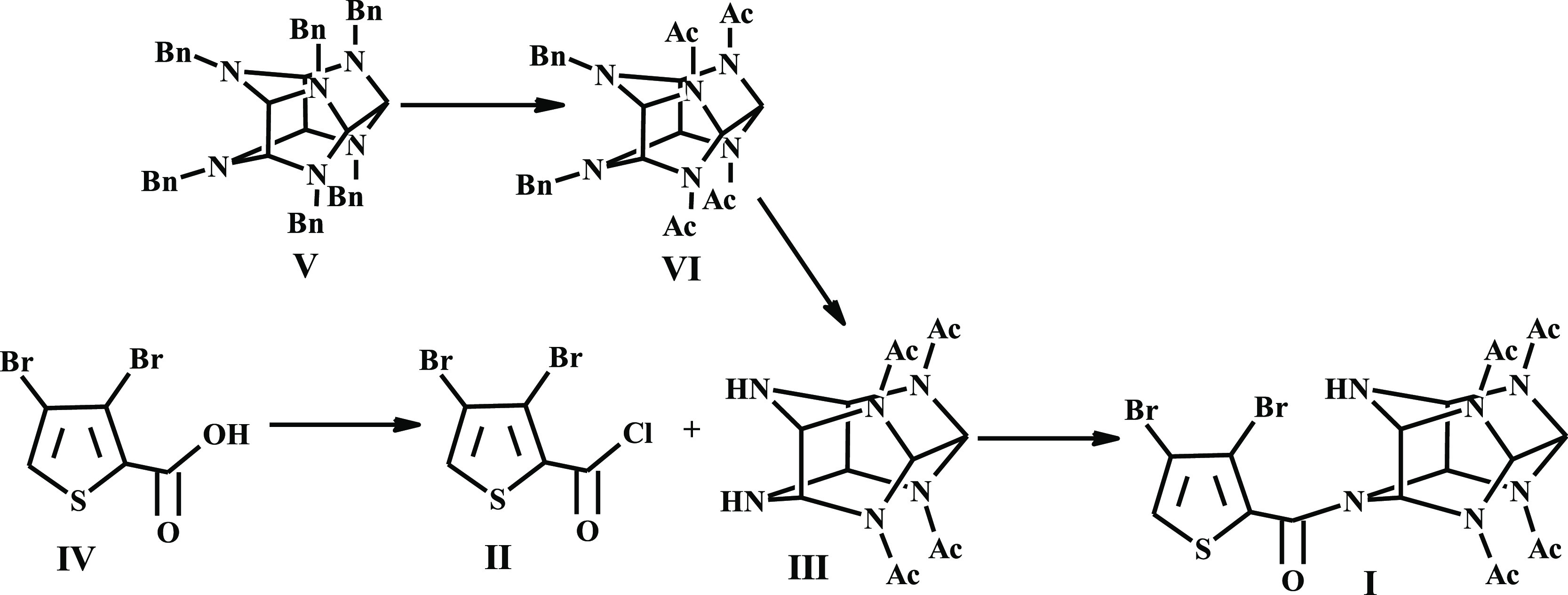
For the synthesis of compound IV, we optimized the procedure involving exhaustive bromination of thiophene, reduction with zinc dust to dibromothiophene, alkylation in the presence of titanium tetrachloride, and final oxidation of aldehyde to the desired dibromothiophene carboxylic acid IV.
The yield of IV on a thiophene (VII) basis was 37%. In case compound VIII was used, the yield of IV was 73%. These reactions are illustrated in Scheme 2. As pictured in Scheme 1, acylating agent II was obtained in a quantitative yield by treating 3,4-dibromothiophene carboxylic acid IV by boiling thionyl chloride for 1 h.
Scheme 2. Optimized Procedure for Dibromothiophene Carboxylic Acid (IV).

Synthetic methods for tetraacetyl hexaazaisowurtzitane (III) are commonly known and industrially employed.22−24 A cascade condensation of benzylamine with glyoxal furnished hexabenzyl hexaazaisowurtzitane (V). Two-stage hydrogenation of V over the Pd catalyst produced VI (Scheme 1).
The acylation reaction between starting III and chloroanhydride II was performed in boiling acetonitrile. The use of other organic solvents diminished the yield of product I. To lower the quantity of impurities, diamine III in acetonitrile was treated with acylating agent II, with compound I precipitating from the reaction mixture and not undergoing further reactions (Scheme 1).
No excess acylating agent II was required to complete the reaction. When II was used in excess from 0 to 65%, the yield of I ranged from 79 to 82%.
The structure of the obtained compound I was verified by a complex approach via physicochemical methods, including 1H and 13C NMR spectra (as illustrated in the Supporting Information).
Biological Evaluation
The predicted analgesic activity of the resulting compound I was confirmed on animals using the reference drugs tramadol, ketorolac, and diclofenac.
Two experimental series with chemical pain stimulation of the peritoneum showed a pronounced antinociceptive effect of thiowurtzine upon a single (100 mg/kg) and subchronic intragastric administration to mice in doses of 50 and 100 mg/kg (Table 1). After a single administration (100 mg/kg), thiowurtzine significantly relieved acute visceral pain by 56.4% and prolonged the pain response time (p < 0.05) in reference to the negative control and appeared to be equivalent to the reference drug, tramadol, which exhibits a 62.2% pain response inhibition (Table 1, series I). It should be noted that in the second series of experiments, after a 4-day compound administration in doses of 50 and 100 mg/kg, thiowurtzine reduced the number of writhings by 56.9% (p < 0.01) and 47.1% (p < 0.01) in comparison with the control, respectively (Table 1, series II). Thiowurtzine (50 mg/kg) exceeded the activity of reference drug diclofenac.
Table 1. Antinociceptive Effect of Thiowurtzine in the Acetic Acid Writhing Test (X ± m)a.
| group | number of writhings within 20 min | writhing latency, s | pain response inhibition, % |
|---|---|---|---|
| Series I: Inbred Male CBA Mice. Single Administration of Thiowurtzine and Tramadol | |||
| (1) negative control—vehicle (n = 9) | 24.1 ± 3.4 | 160.9 ± 86.4 | 0 |
| (3) tramadol, 20 mg/kg (n = 9) | 9.1 ± 2.0, **(1–3) | 444.8 ± 42.8, **(1–3) | 62.2 |
| (5) thiowurtzine, 100 mg/kg (n = 6) | 10.5 ± 3.0, *(1–5) | 382.5 ± 120.4, *(1–5) | 56.4 |
| Series II: Outbred Male Mice, Stock CD1. Drug Administration for 4 days | |||
| (1) negative control—vehicle (n = 10) | 27.4 ± 3.1 | 276 ± 15 | |
| (2) diclofenac, 10 mg/kg (n = 10) | 18.2 ± 1.2, **(1–2) | 270 ± 10 | 33.6 |
| (3) thiowurtzine, 50 mg/kg (n = 10) | 11.8 ± 2.9, **(1–3), *(2–3) | 270 ± 10 | 56.9 |
| (4) thiowurtzine, 100 mg/kg (n = 10) | 14.5 ± 3.0, **(1–4) | 273 ± 16 | 47.1 |
*p < 0.05, **p < 0.01 compared to the negative control and the reference group (Mann–Whitney U test).
The most pronounced analgesic effect of thiowurtzine was revealed in the central analgesia model, which is controlled by the cortical and subcortical structures of the brain. In the hot plate test, subchronic intragastric administration of thiowurtzine in a dose of 100 mg/kg caused a maximum increase in the pain response latency of 77.9%—by 1.8 times (p < 0.01)—relative to the control (Table 2, series I and II). Thiowurtzine (100 mg/kg) exceeded the activity of the reference drug diclofenac, where pain response inhibition proved to be 11.3%.
Table 2. Analgesic Activity of Thiowurtzine in the Hot Plate Test in Outbred Male Stock CD Rats (X ± m)a.
| group | pain response latency, s | pain response inhibition, % |
|---|---|---|
| Series I: Drug Administration for 4 days | ||
| (1) negative control—vehicle (n = 8) | 9.8 ± 1.0 | |
| (2) diclofenac, 5 mg/kg (n = 9) | 10.9 ± 1.5 | 11.3 |
| (3) thiowurtzine, 25 mg/kg (n = 9) | 10.4 ± 1.3 | 6.1 |
| (4) thiowurtzine, 50 mg/kg (n = 8) | 12.1 ± 1.6 | 23.5 |
| Series II: Drug Administration for 4 days | ||
| (1) negative control—vehicle (n = 10) | 11.3 ± 1.0 | |
| (2) thiowurtzine, 100 mg/kg (n = 10) | 20.1 ± 2.3, **(1–2) | 77.9 |
| (3) thiowurtzine, 200 mg/kg (n = 10) | 14.9 ± 2.1, *(2–3) | 31.9 |
| (4) thiowurtzine, 300 mg/kg (n = 10) | 13.2 ± 2.7 | 16.8 |
*p < 0.05, **p < 0.01 compared to the negative control and the reference group (Mann–Whitney U test).
Thiowurtzine showed a roughly constant increase in the nociceptive threshold throughout the study, which was noted to be significant in the 100 mg/kg group (Table 3). After a single administration (100 mg/kg), thiowurtzine significantly increased the supporting pressure in the left hind paw pad by 2.1 times (1 h, p < 0.01) and 1.6 times (4 h, p < 0.01), relative to the negative control.
Table 3. Analgesic Activity of Thiowurtzine in the Randall–Selitto Test in Outbred Male Stock CD1 Mice (X ± m)a.
| supporting
pressure in the left hind paw pad, g/condition |
supporting
pressure in the right hind paw pad with formalin edema, g/condition |
|||
|---|---|---|---|---|
| group | 1 h after drug administration | 4 h after drug administration | 1 h after drug administration, 1–5 min after injection of formalin | 1 h after drug administration, 40–50 min after injection of formalin |
| (1) negative control—vehicle (n = 10) | 294.9 ± 48.4 | 373.2 ± 70.9 | 141.1 ± 23.7 | 305.2 ± 72.6 |
| (2) tramadol, 10 mg/kg (n = 9) | 645.2 ± 40.8, **(1–2) | 606.7 ± 53.9, **(1–2) | 318.9 ± 84.1, *(1–2) | 518.0 ± 85.0, *(1–2) |
| (3) ketorolac, 6 mg/kg (n = 8) | 521.2 ± 107.9, **(1–3) | 449.7 ± 90.4 | 491.4 ± 88.2, *(1–3) | 396.6 ± 94.1 |
| (4) thiowurtzine, 100 mg/kg (n = 9) | 631.5 ± 43.9, **(1–4) | 612.9 ± 43.8, **(1–4) | 620.8 ± 60.0, **(1–4) | 611.3 ± 58.8, **(1–4) |
| (5) thiowurtzine, 200 mg/kg (n = 9) | 586.1 ± 68.6, **(1–5) | 455.8 ± 81.7 | 419.7 ± 94.2, *(1–5) | 366.3 ± 88.3 |
*p < 0.05, **p < 0.01 compared to the negative control and the reference group (Mann–Whitney U test).
Interestingly, the Randall–Selitto test performed on the right paw (100 mg/kg) with formalin edema also revealed a significant increase in the nociceptive threshold. In this case, increased supporting pressure was evidenced by 4.4 times (1 h, p < 0.01) and 2.0 times (4 h, p < 0.01) compared to that of the negative control.
The assessed effect of thiowurtzine (100 mg/kg) on thresholds of response to mechanical pressure stimulation appeared to be comparable with tramadol but exceeded the activity of the reference drug ketorolac.
The preclinical study demonstrated that chronic administration of thiowurtzine (abstinence syndrome) did not evoke drug abuse. There was no impact on respiration and the central nervous system, indicating that thiowurtzine is not a morphine-like action compound and does not cause ulcerogenic damage to the gastrointestinal mucosa of the test animals.
Target Identification
Previous studies investigating the action of naloxone and naloxone methiodide demonstrated that naloxone has a higher affinity than naloxone methiodide for the mu, kappa, and delta opioid receptors in the brain of the mouse.25 Therefore, naloxone targets the brain tissue, whereas naloxone methiodide, which has limited access to the brain, targets more specifically the peripheral opioid receptors.
Interestingly, injection of nonselective opioid receptor antagonist naloxone was shown to weaken the analgesia caused by a single thiowurtzine administration in a dose of 100 mg/kg,18 suggesting an action on the mu, kappa, and delta opioid receptors in the central opioid system of the mouse.
On the contrary, naloxone methiodide did not abolish analgesia, suggesting that thiowurtzine has no affinity for the peripheral opioid receptors.
These results are consistent with the central opioid system being involved in the mediation of the analgesic effect of thiowurtzine. For this reason, different opioid receptors (mu, kappa, and delta) and the opioid-like receptor 1 (ORL1) were investigated as potential targets using computational methods.
However, the antinociceptive effect of thiowurtzine was not completely abolished by naloxone, which suggests a complex mechanism of action of the compound under study. In this regard, the opioid analgesia pathway seems to be only one component of the mechanism of action of thiowurtzine; and consequently, there may be other targeted receptors.
Another target of choice for the action of thiowurtzine may be the transient receptor potential (TRP) ion channel family, whose discovery was an important step in exploring the nature of pain sensation at the molecular level. These channels are tetramers assembled from subunits with six membrane-spanning domains and are permeable to monovalent cations and calcium. This family of receptors is currently considered as the pharmacologically most important biotargets of the human nervous system for analgesia.
Recent results proved that thiowurtzine, when administered in a preventive single per os dose of 100 and 200 mg/kg, was found to effectively block nociceptive reactions normally caused by the activation of the TRPA1 and TRPV1 ion channels. That being said, the thiowurtzine analgesic activity turned out to be comparable and/or superior to that of ketorolac and diclofenac, depending on the model situation. Besides, thiowurtzine (200 mg/kg per os) was found to be equivalent to diclofenac sodium (10 mg/kg per os) and superior in anti-inflammatory expressiveness to ketorolac (6 mg/kg per os) in the formalin test. The test results suggest that the pronounced antinociceptive action of our innovative compound in the capsaicin and formalin tests is governed by the interaction with the TRPV1 and TRPA receptors. Moreover, all the data previously obtained are consistent with the analgesic action of thiowurtzine, triggering the activity modulation of the TRP ion channels. In this regard, the TRPA1 and TRPV1 ion channels were also investigated as potential targets using docking techniques.
It should also be noted that the preventive single intragastric gavage administration of thiowurtzine at a dose of 100 mg/kg led to a statistically significant decrease in the number of myoclonic seizures relative to the similar value of the carbamazepine group. This also resulted in a comparative increase in the life expectancy of animals while exceeding the main indicator of the effectiveness of an anticonvulsant action—an increase in the number of surviving animals (41.7% relative to 33.3% of the carbamazepine group).20
The mechanism of the anticonvulsant action can probably be attributed to both allosteric interactions with components of the γ-aminobutyric acid type A receptor complex and the effect on Ca2+-dependent K+ currents.
Finally, in order to identify new potential targets for the action of thiowurtzine, we screened the DrugBank database,26 looking for structurally similar drugs and their well-known receptors. Interestingly, we identified several drugs classified as calcium antagonists and acting as calcium channel blockers (namely, pranidipine, nicardipine, and especially lacidipine, which exhibited the highest score of 86% for structural similarity with thiowurtzine). Lacidipine, like other dihydropyridines, targets another family of calcium channels, namely, the voltage-dependent calcium channels, and has a selective activity on the Cav 1.2. channel. Dihydropyridines have a high affinity for these receptors ranging from 0.1 to 50 nM, and they specifically interact with their α1c subunit.27 This led us to investigate this class of voltage-gated calcium channels as potential targets for thiowurtzine. Indeed, calcium channel blockers have the ability to inhibit voltage-gated calcium channels, and thus, reduce the release of neurotransmitters at the presynaptic level, which may lead to pain reduction. As a consequence, voltage-gated calcium channels have been described as targets of choice for potential analgesics in the previous years,28−32 especially when used concomitantly with opioids in attenuation of clinical pain.33−35 In this particular case, calcium channel blockers have been shown to increase morphine analgesia.36,37
As a result, we used the Genetic Optimization for Ligand Docking (GOLD) software38 to perform docking experiments for the thiowurtzine molecule on the mu, kappa, delta, and ORL1 opioid receptors, the TRPA1 and TRPV1 ion channels, and voltage-dependent calcium channel Cav 1.2.
The mu, kappa, delta, and ORL1 opioid receptors were constructed using the corresponding UniProtKB human sequences and the Iterative Threading ASSEmbly Refinement (I-TASSER) software from the Zhang Lab.39 The models were then checked, charged, and minimized with the Molecular Operating Environment (MOE) software using the Amber 14:ETH force field.
The structure of the TRPA1 human receptor was directly extracted from the PDB file 6PQO, and the structure of the human TRPV1 ion channel was calculated as a homology model using the rat structure of the PDB file 3J5Q as a template and the Uniprot human sequence Q8NER1.
For human calcium channel Cav 1.2, we built a homology model using UniProtKB sequence Q13936 (isoform 1) of human voltage-gated calcium channel subunit α1c Cav 1.2 and the PDB structure 6JP5 of the rabbit Cav 1.1 calcium channel interacting with the dihydropyridine compound nifedipine. As the calcium channel was an unexpected target, we thoroughly checked the structure as follows: the RMSD calculated between the final model and the template was 0.738 Å. The root-mean-square deviation (RMSD) between the binding sites (residues with atoms within a sphere of 4.5 Å around the nifedipine ligand) of 6JP5 and the homology model was 0.32 Å, which indicates that the two binding sites are extremely similar. For supplemental validation, the Ramachandran diagram of the model was also checked with 93% of residues in the most favorable regions, 6% in the allowed regions, and only a few outlier residues. The homology model was further evaluated by docking nine ligands extracted from BindingDB (http://bindingdb.org). The docking scores were in very good agreement with the experimentally measured inhibitor constants (Ki) for each of the nine ligands, thus validating the robustness of our model.
Docking of Thiowurtzine
The docking of thiowurtzine into the models of the seven receptors (mu, kappa, delta, ORL1, TRPA1, TRPV1, and Cav 1.2) was performed using the GOLD software. We used control drugs (tramadol, ketorolac, and diclofenac) as references. As tramadol is commercially available as a racemic mix (1R-2R and 1S-2S), we separately tested each of the two enantiomers and reported their respective scores. All the results are summarized in Table 4.
Table 4. Docking Scores of Thiowurtzine and Control Drugs against the Mu, Kappa, Delta, and ORL1 Opioid Receptors, the TRPA1 and TRPV1 Ion Channels, and the Cav 1.2 Calcium Channela.
| drug | GOLD score with the mu receptor | GOLD score with the kappa receptor | GOLD score with the delta receptor | GOLD score with the ORL1 receptor | GOLD score with the TRPA1 receptor | GOLD score with the TRPV1 receptor | GOLD score with the Cav 1.2 receptor |
|---|---|---|---|---|---|---|---|
| tramadol 1S-2S | 19,546 | 27,003 | 21,797 | 20,520 | 24,965 | 21,236 | 24,499 |
| tramadol 1R-2R | 19,188 | 28,349 | 20,435 | 19,272 | 23,058 | 21,497 | 23,597 |
| ketorolac | 19,529 | 26,150 | 23,640 | 19,467 | 23,712 | 19,717 | 22,065 |
| diclofenac | 20,823 | 31,603 | 24,274 | 17,567 | 22,406 | 20,449 | 24,154 |
| thiowurtzine | 8,465 | 2,730 | 6,407 | 2,153 | 14,831 | 8,414 | 15,562 |
Interestingly, the docking scores of thiowurtzine are lower than those of the reference drugs for all the investigated receptors. However, these scores are high enough to unequivocally demonstrate an effective binding of thiowurtzine, especially with the mu and delta opioid receptors, the TRPA1 and TRPV1 ion channels, and the Cav calcium channel, which exhibits the highest docking score with thiowurtzine in our study.
For the mu and delta receptors, the scores are about 2.5 to 4 times lower than those of tramadol, ketorolac, or diclofenac. However, the animal experiments proved that the doses of thiowurtzine needed to be typically five times higher than those of tramadol (20 mg/kg vs 100 mg/kg) and diclofenac (10 mg/kg vs 50 mg/kg) to produce similar effects with the acetic acid writhing test (Table 1). The same ratio was also observed for the hot plate test (diclofenac 5 mg/kg and thiowurtzine 25 mg/kg, Table 2) to produce similar results. Apart from the typical bioavailability issues, the animal experiments are thus in very good agreement with our docking results. The best docking pose for thiowurtzine in complex with the model of the mu opioid receptor is displayed in Figure 1.
Figure 1.

Molecular representation of the best docking pose of the thiowurtzine molecule interacting with the calculated model of the human mu opioid receptor. Thiowurtzine is displayed as sticks, and the mu receptor is displayed as red ribbons. The water molecules are visible, as well as the molecular surface of the cavity surrounding the thiowurtzine molecule. The side chains of the residues interacting with thiowurtzine are labeled and displayed as sticks.
For the TRPA1 ion channel, the results with thiowurtzine are much better than for any of the opioid receptors, with a docking score almost twice as high as the one obtained by the mu receptor; and all the reference drugs exhibit similar scores. Despite being about 35% lower than those of the different reference drugs, the docking score of thiowurtzine, in this case, demonstrates a high affinity and a strong binding, as illustrated in Figure 2, which displays the best docking pose for thiowurtzine in complex with the 6PQO PDB structure of the human TRPA1 ion channel.
Figure 2.

Molecular representation of the best docking pose of the thiowurtzine molecule interacting with the human TRPA1 ion channel from the PDB file 6PQO. Thiowurtzine is displayed as sticks, and the TRPA1 receptor is displayed as red ribbons. The molecular surface of the cavity surrounding the thiowurtzine molecule is displayed with a gray surface. The side chains of the residues interacting with thiowurtzine are labeled and displayed as sticks.
For the TRPV1 receptor, the results are very similar to those obtained by the mu opioid receptor, with a very close docking score, and similar scores are exhibited by the reference drugs as well. In this regard, TRPV1 appears to be a target for thiowurtzine about as good as the mu opioid receptor but significantly less valuable than TRPA1.
For the Cav receptor, the docking score of thiowurtzine is better than for any of the other investigated receptors (Table 4), and it is close to those of the other reference drugs (albeit being about 30% lower). Consequently, thiowurtzine appears to be a significantly better ligand for the Cav receptor than for any of the investigated opioid receptors and even slightly better than for the TRPA1 ion channel.
All these results are consistent with a complex mechanism of action, which could target at the same time the opioid receptors, the TRP channels, and the voltage-gated calcium channels.
Molecular Dynamics
In order to validate whether our hypothesis of thiowurtzine binding to the mu opioid receptor and the Cav 1.2 calcium channel was stable over time, MD experiments were carried out for the mu receptor and the Cav 1.2 calcium channel in the presence of thiowurtzine. We used the best docking pose of thiowurtzine with the model of the mu receptor and the Cav 1.2 calcium channel as starting points. A lipid bilayer surrounding the receptors was generated with the MOE lipid generator for each protein. Water molecules were added, as well as Na+ and Cl– ions, in order to simulate a 0.1 M ionic force, and the pH was set at 7.4. Standard Amber 14:EHT parameters were used for MD. A quick minimization was performed prior to the start of the MD simulations. After a preliminary equilibration phase, 200 ns molecular simulations were performed. A frame was saved every 200 ps, leading to a total of 1000 frames. The last frame of each simulation is displayed in Figures 3 and 4 for the mu receptor and the Cav 1.2 calcium channel, respectively.
Figure 3.

Molecular representation of the last frame of the 200 ns molecular dynamic simulation of thiowurtzine in complex with the model of the mu opioid receptor. Thiowurtzine is displayed as thick sticks, the mu receptor as gray ribbons, and the lipids as cyan sticks. The trajectory of the thiowurtzine mass center throughout the entire simulation is displayed as yellow lines. The side chains of the residues interacting with thiowurtzine are displayed as sticks, and their name and number are indicated on the structure.
Figure 4.

Molecular representation of the last frame of the 200 ns molecular dynamic simulation of thiowurtzine in complex with the model of the Cav receptor. Thiowurtzine is displayed as thick sticks, the Cav calcium channel as lime ribbons, and the lipids as cyan sticks. The trajectory of the thiowurtzine mass center throughout the entire simulation is displayed as yellow lines. The side chains of the residues interacting with thiowurtzine are displayed as sticks, and their name and number are indicated on the structure.
The molecular complex formed by thiowurtzine with either the mu receptor or the Cav calcium channel model appeared to be extremely stable over time. The calculated RMSD for each of the receptors stayed quite stable along the simulations (as indicated in the Section S10). Moreover, for each receptor, thiowurtzine stayed within the interaction site during the 200 ns simulation, with only limited relative motions around its initial position at the beginning of the simulation. The RMSDs calculated for thiowurtzine in regard to each receptor are low and extremely stable over the simulation time (see the Section S10). The entire trajectory of the thiowurtzine mass center during the complete simulation is displayed as yellow sticks in Figures 3 and 4 and indicates great stability of the complexes over time. These results are in excellent agreement with the good docking scores previously obtained with the GOLD software for the thiowurtzine interaction with each of the receptors (Table 4).
Conclusions
In summary, we provided the chemical synthesis, biological evaluation, and in silico target identification of a potent new hexaazaisowurtzitane-based analgesic, referred to here as thiowurtzine. This nonaddictive analgesic does not exhibit any of the typical adverse effects of NSAIDs and may very well lead to new compounds of the same class with practical applications.
It is worth noting that the devised original method for the synthesis of 3,4-dibromothiophene carboxylic acid holds promise for further use, as it provides quite a high yield under mild synthesis conditions.
The experimental evidence (behavioral tests with thermal and chemical stimuli and mechanical compression) demonstrates an analgesic effect of thiowurtzine through its inhibitory action on the peripheral and central mechanisms of development and maintenance of pain syndrome. These findings highlight a promising potential of the new compound as an analgesic against pain syndromes of different etiologies.
The software-based evaluation of thiowurtzine, which was validated by the animal studies, gave some interesting perspectives about the potential molecular receptors of thiowurtzine. We highlighted the role of the opioid receptors as primary targets, which confirmed the results of the experiments using thiowurtzine in conjunction with naloxone. However, the antinociceptive effect of thiowurtzine was not completely abolished by naloxone, which suggests that the opioid analgesia pathway may not be the only component of the mechanism of action of thiowurtzine; consequently, there may be other targeted receptors as well.
Then, we investigated the TRP class of ion channels as targets for the thiowurtzine molecule. The docking experiments with the TRPA1 and TRPV1 receptors allowed us to highlight the strong binding capabilities of this family of ion channels, especially for TRPA1, which exhibited an excellent docking score and thus proved to be an extremely valuable target for thiowurtzine.
We also used molecular comparisons with other drugs, associated with molecular modeling and MD simulations, which allowed us to also consider voltage-gated calcium channels as potential targets. Interestingly, the possibility that calcium channel blockers might induce analgesia by reducing the release of neurotransmitters and thus increasing morphine analgesia has been investigated and confirmed in several studies.33−37 These results are consistent with thiowurtzine having a primary action on the central opioid system and suggest that this primary action may very well be potentiated by a secondary action on the voltage-gated calcium channels as well. These results were confirmed by the docking scores of thiowurtzine against the opioid and the calcium receptors. These findings might provide a solid basis for the explanation of the complicated mechanism of action of thiowurtzine.
Our docking results also provide a molecular explanation of the non-narcotic properties of thiowurtzine. As we proved that ion channels such as the TRP receptors and the Cav calcium channels are much better targets for thiowurtzine than any of the opioid receptors, this might partly explain why thiowurtzine does not have a morphine-like action and subsequently does not exhibit typical adverse effects of NSAIDs, as demonstrated by the animal studies.
New rational design studies involving further chemical modifications of hexaazaisowurtzitane derivatives are currently under way and might lead to the discovery of a whole new class of biologically active molecules of interest.
Experimental Section
General Methods
High-performance liquid chromatography (HPLC) was performed on an Agilent Technologies 1260 Infinity instrument with a 2.1 × 15 mm precolumn, Zorbax SB-18 sorbent, and 3 μm fractions; a 3.0 × 150 mm column, Zorbax SB-18 sorbent, and 3.5 μm fractions using gradient elution. Eluent A: 0.2% orthophosphoric acid solution and eluent B: acetonitrile. 1H and 13C NMR spectra were recorded on a Bruker AM-400 spectrometer operating at 400.13 MHz for 1H and at 100.61 MHz for 13C; DMSO-d6 was used as solvent. IR spectra were recorded on an Infralum FT-801 spectrophotometer in KBr pellets (1 mg substance in 200 mg potassium bromide) in the range of 4000 to 400 cm–1. The melting point was measured on a Stuart SMP 30 melting point apparatus.
Synthesis of 2,4,6,8,10,12-Hexabenzyl-2,4,6,8,10,12-hexaazatetra-cyclo[5,5,0,03,11,05,9]dodecane (V)
Into a stirred flask were put benzylamine (170 mL, 1.56 mol), distilled water (130 mL), acetonitrile (1430 mL), and 98% formic acid (5.4 mL). Then, 40% aqueous glyoxal (94.25 g, 0.65 mol) was added portionwise for 1 h at a temperature not above 20 °C. The reaction mixture was maintained at room temperature for 17 h. The resultant crystalline product was collected by filtration and washed in situ with cold acetonitrile. Yield: 121 g (76% on a benzylamine basis). mp: 145–150 °C.
For deresination, the resultant crude product was stirred in acetonitrile (240–250 mL) at 50 °C for 15–20 min, cooled to room temperature, filtered, and then washed with acetonitrile to furnish 115–118 g of the product (mp: 150–153 °C) after drying. Calcd for C48H48N6 (%): 81.32; H, 6.82; N, 11.85; found (%): C, 82.13; H, 6.79; N, 11.09. IR (ν/cm–1): 3082, 3061, 3023, 2923, 2857, 2823, 1493, 1452, 1351, 1323, 1302, 1244, 1140, 1072, 987, 898, 835, 733, 699, 625. 1H NMR (DMSO-d6, δ, ppm): 3.48 (s, 2H, CH), 4.01 (s, 4H, CH2), 4.04 (s, 4H, CH2), 4.15 (s, 4H, CH), 7.14–7.26 (m, 30H, CHap).
Synthesis of 4,10-Dibenzyl-2,6,8,12-tetraacetyl-2,4,6,8,10,12-hexaazatetracyclo[5,5,0,03,11,05,9]dodecane (VI)
Into a 300 mL autoclave equipped with an electromagnetic stirrer and a hydrogen gas delivery system were loaded hexabenzylhexaazaisowurtzitane (10 g), bromobenzene (0.18–0.20 mL), a catalyst, and DMF (40 mL). Lastly, acetic anhydride (15 mL) was added, and the autoclave was closed. The autoclave was purged with hydrogen three times. Hydrogen was then supplied under 5–6 kgf/cm2, stirring was turned on (550–600 rpm), and the autoclave was heated to 50–55 °C. The hydrogen absorption was controlled against the autoclave pressure drop. If the autoclave pressure went down below 2.5–2 kgf/cm2, hydrogen was again fed at 5–6 kgf/cm2.
After the process was complete (an autoclave pressure drop of 0.1–0.2 kgf/cm2 within 1 h), the heating was turned off, cold water was poured into the bath, and the reactor was cooled to 20–25 °C.
Hydrogen depressurization was then performed, and the product suspension with the catalyst was filtered. The mixture of dibenzyltetraacertylhexaazaisowurtzitane (DBTA) and catalyst was washed with ethanol (3 × 10 mL), squeezed, and air-dried to furnish DBTA not allowing for the catalyst weight. Yield: 5.8–6 g (80–83%). Calcd for C28H32N6O4 (%): C, 65.10; H, 6.24; N, 16.27; O, 12.39; found (%): C, 64.79; H, 6.18; N, 16.25. IR (ν/cm–1): 3083, 3044, 3022, 3005, 2876, 2829, 1687, 1650, 1455, 1412, 1358, 1319, 1300, 1239, 1147, 1068, 985, 893, 834, 730, 687, 627. 1H NMR (DMSO-d6, δ ppm): 1.85–1.95 (m, 12H, CH3), 4.09 (s, 4H, CH2), 5.71 (br s, 4H, CH), 6.51 (br s 4H, CH), 7.37–7.49 (m, 10H, CHap).
Synthesis of 2,6,8,12-Tetraacetyl-2,4,6,8,10,12-hexaazatetracyclo[5,5,0,03,11,05,9] dodecane (III)
Into an autoclave were loaded DBTA (25 g), 5% Pd/C (5 g), and 50% acetic acid (150 mL). The autoclave was closed and purged three times with nitrogen and two times with hydrogen. Hydrogen was then fed at 5 kgf/cm2 and stirring was turned on. The reaction was carried out at 70–75 °C for 3 h. The catalyst was then filtered, and the reaction mixture was evaporated in a rotary evaporator until it was viscous. To the evaporated mixture was added alcohol (150–200 mL), and the whole mixture was stirred for 15–20 min. The crystalline product was collected by filtration and air-dried to give tetraacetylhexaazaisowurtzitane (TA). Yield: 15 g (92%), purity 98% (as per HPLC). mp: 360 °C (decomposition). Calcd for C14H20N6O8 (%): C, 49.99; H, 5.99; N, 24.99; O, 19.03; found (%): C, 50.16; H, 6.04; N, 24.72. IR (ν/cm–1): 3369, 3329, 3047, 3019, 2931, 1659, 1402, 1359, 1293, 1255, 1140, 1111, 1019, 974, 909, 801, 710, 609, 509. 1H NMR (DMSO-d6, δ ppm): 1.94–2.07 (m, 12H, CH3CO), 4.59–4.99 (m, 2H, NH), 5.29 (br s, 2H, CH), 5.99–6.23 (m, 4H, CH).
Synthesis of 3,4-Dibromothiophene Carboxylic Acid (IV)
Into a round-bottom flask, thiophene (48 mL, 0.6 mol), chloroform (24 mL), and bromine (128 mL, 2.4 mol) were slowly added dropwise. The dosing time was 4 h. At the beginning of dosing, the solution was observed to be decolored and hydrogen bromide evolved vigorously. Hydrogen bromide was entrapped and absorbed with 20% NaOH solution. After dosing was completed, the reaction mixture was cooled on an ice bath and held for 12 h.
Upon completion of holding, to the reaction mixture was added chloroform (20 mL), the whole mixture was heated on a silicone bath at 85 °C, and 15% KOH solution in ethanol was added. The mixture was refluxed for 3 h with vigorous stirring, cooled down, and then poured out onto ice (400 g). After holding for 1 h, the product was filtered, washed with water, and air-dried.
To a 1 L flask fitted with a distiller was added zinc powder (180.6 g, 2.76 mol) in water (240 mL), and then, glacial acetic acid (180 mL, 5.2 mol) was added. The reaction mixture was heated to boil, and the crude product was added in a few portions. The distilled organic fraction was washed with water and then dried over MgSO4 to yield 3,4-dibromothiophene as a colorless liquid. Yield: 74.07 g (51%).
To a mixture of the resultant 3,4-dibromothiophene (0.024 mol) and dichloromethyl methyl ether (2.98 g, 2.35 mL, 0.026 mol) in dry dichloromethane (15 mL) was added TiCl4 (7.59 g, 4.39 mL, 0.04 mol) with stirring at −12 °C for 40–50 min. The reaction mixture was stirred for 1 h, and water (30 mL) was added drop by drop within 15 min. After 30 min, the layers were separated, and the water layer was extracted with dichloromethane (3 × 30 mL). The combined extract was dried over MgSO4 and evaporated. The product was recrystallized from heptane, dried, and used in the subsequent stage.
The resultant 3,4-dibromo-2-thiophene-aldehyde (47.1 g, 0.174 mol) was dissolved in acetone (550 mL). A potassium permanganate (37.8 g) solution in distilled water (720 mL) was prepared separately by heating.
To the aldehyde acetone solution was added potassium permanganate in portions of 15–20 mL at not above 20 °C. The whole solution was held for 3–5 min after adding each portion. The dosing was stopped after the solution turned a stable pink color. The resultant suspension was decolored by adding 10% sodium sulfite solution. The reaction mixture was filtered off of MnO2 and washed with water.
The filtrate was evaporated by half and acidified with HCl until the pH value was less than 2. The product was collected by filtration, washed with water and then recrystallized from dichloroethane to yield 3,4-dibromothiophene carboxylic acid. Yield: 42.29 g (85%). mp: 205–207 °C. Calcd for C5H2Br2O2S (%): C, 21.00; H, 0.70; Br, 55.89; O, 11.19; S, 11.21; found (%): C, 21.09; H, 0.69; S, 11.24. IR (ν/cm–1): 3102, 3098, 1663, 1478, 1389, 1321, 1123, 920, 857, 784, 685. 1H NMR (DMSO-d6, δ ppm): 7.44 (c, 1H, CH), 10.93 (br s, 1H, OH).
Synthesis of 3,4-Dibromothiophenecarbonyl chloride (II)
3,4-Dibromothiophene carboxylic acid (10 g, 0.035 mol) was added to thionyl chloride (50 mL) and heated to the boiling point of thionyl chloride. The mixture was maintained at the same temperature for 1 h and then cooled down and evaporated in a rotary evaporator. Yield: 9.2 g (86%). mp was not measured because the product was immediately used in further reaction.
Synthesis of 4-(3,4-Dibromothiophenecarbonyl)-2,6,8,12-tetraacetyl-2,4,6,8,10,12-hexaazatetracyclo[5,5,0,03,11,05,9]dodecane (I)
To a solution of chloroanhydride V (9.2 g, 0.03 mol) in dry acetonitrile (60 mL) was added 2,6,8,12-tetraacetyl-2,4,6,8,10,12-hexaazatetracyclo[5,5,0,03,11,05,9]dodecane (5.04 g, 0.015 mol). The resultant mixture was refluxed for 2 h and then cooled down; the precipitated product was collected by filtration and washed two times with acetonitrile.
The dried creamy-colored sediment was recrystallized from 70% (by vol) aqueous ethanol. The resultant product was washed with water three times and air-dried to furnish 4-(3,4-dibromothiophenecarbonyl)-2,6,8,12-tetraacetyl-2,4,6,8,10,12-hexaazatetracyclo[5,5,0,03,11,05,9]dodecane as a colorless crystalline product with an assay of 99% (as per HPLC). Yield: 7.19 g (79.4%). mp: 332–334 °C. Calcd for C19H20Br2N6O5S (%): C, 37.76; H, 3.34; Br, 26.45; N, 13.91; O, 13.24; S, 5.31; found (%): C, 37.68; H, 3.24; N, 13.82; S, 5.37. IR (ν/cm–1): 3287, 3119, 3005, 1657, 1504, 1425, 1356, 1323, 1289, 1250, 1162, 1045, 984, 893, 878, 742, 715, 623. 1H NMR (DMSO-d6, δ ppm): 1.85–2.15 (m, 12H, CH3CO), 4.75–5.12 (m, 1H, NH), 5.45–5.67 (m, 2H, CH), 5.92–6.85 (m, 4H, CH), 8.08–8.18 (m, 1H, CH). 13C NMR (DMSO-d6, δ ppm): 21.16, 21,47, 22.19, 22.43, 22.60, 22.90 (C, CH3, CH3CO); 65.07, 67.70, 69.3, 70.31, 71.3, 73.12 (C, CH); 113.13, 114.63 (C, arom.); 127.5 (C, CH, arom.); 132.14 (C, arom.); 162.63, 162.78 (C, CO); 166.7, 167.1, 167.5, 167.9 (C, CO, CH3CO).
Animals
The experiments were carried out on adult inbred male CBA mice (n = 24), outbred male stock CD1 mice (n = 45), outbred female stock CD1 mice (n = 40), and outbred male stock CD rats (n = 74) (1st category conventional animals). The animals were obtained from the Department of Experimental Biomodelling, E. D. Goldberg Research Institute of Pharmacology and Regenerative Medicine (animal health certificate). Animal maintenance and experimental design were approved by the Bioethics Committee of the E. D. Goldberg Research Institute of Pharmacology and Regenerative Medicine (JACUC protocol no. 96092015) and complied with the directive 2010/63/EU of the European Parliament and the Council of the European Union on the protection of animals used for scientific purposes, Order no. 199n of the Ministry of Health of the Russian Federation (August 1, 2016).
Study Design
When the maximum possible single injection volume of thiowurtzine was used, LD50 (or animal death) was not achieved. The compound was daily administered per os through a probe in a dose range of 50–200 mg/kg (mice) and 25–300 mg/kg (rats) for 1–4 days (Tables 1–3); the last administration was carried out 1 h prior to the pain sensitivity test. The reference drugs were NSAID diclofenac (Hemofarm), administered orally in a dose of 10 mg/kg in a volume of 0.2 mL/mouse and in a dose of 5 mg/kg in a volume of 0.5 mL/rat, NSAID ketorolac (Dr. Reddy’s Laboratories Ltd.), administered orally in a dose of 10 mg/kg, and Tramadol (Organic), administered in a dose of 20 mg/kg via a probe in the form of solution in purified water in a volume of 0.2 mL/mouse. The suggested doses of reference drugs matched the average therapeutic doses in humans.42 The animals of the negative control group received vehicle (water–Tween mixture) in an equivalent volume via the same route.
Animal Methods
The analgesic activity of thiowurtzine was assessed in behavioral tests with thermal and chemical exposures and in the Randall–Selitto test with mechanical compression of the normal paw pad and with formalin edema.
Hot Plate Test
The hot plate test is basic in analgesic activity assessment studies; it involves behavioral responses to pain, controlled by cortical and subcortical structures of the brain.43 One hour after compound administration, the test was performed using a Hot Plate Analgesia Meter (Columbus Instruments). The rats were placed on a plate heated at 54.0 ± 0.5 °C, after which pain reaction latency was recorded (licking front and hind paw pads). Analgesic activity was measured by the mean latency in the group and percentage of the pain response inhibition (%PRI), according to the formula (Tcontrol – Texperiment)/Tcontrol × 100%, where T is the pain response latency in the corresponding group.
Acetic Acid Writhing Test
The acetic acid writhing test is aimed at the assessment of acute visceral deep pain.42−47 The specific response to pain (writhing) was caused by intraperitoneal injection of 0.75% acetic acid solution in a volume of 0.1 mL/10 g body weight. The analgesic effect was evaluated by the ability of the compound (within 20 min after the injection) to reduce the number of writhings (in %) in comparison with the control group.
Randall–Selitto Test
The Randall–Selitto test (Randall and Selitto, 1957), intended to serve as a tool to assess the effect of analgesic agents on the response thresholds to mechanical pressure stimulation, is usually used by a number of investigators to evaluate inflammatory painful responses.42−47 An Ugo Basile electronic device based on the Randall–Selitto principle, which allows testing pain in a quantitative manner by pressuring the hind limbs of animals, has been used. Hyperalgesia was induced by subcutaneous injection of 2% formalin solution in a volume of 50 μL intraplantarily into the right hind paw pad after 1 h of compound administration. The Randall–Selitto test was performed twice on each individual; 1 and 4 h after compound administration on the left hind paw pad and in the first 1–5 and 40–50 min after inducing hyperalgesia on the right hind paw pad. Analgesic activity was evaluated by the ability of the compound to change the response threshold in comparison with the positive and negative control groups.
The rats were sacrificed by CO2 inhalation, and the mice were killed by craniocervical dislocation.
Statistical Processing
Statistical processing of the results was performed using ANOVA (Statistica 6.0). For all data, the mean (X) and standard error of the mean (m) were calculated (shown in tables); n is the number of variants in the group. Differences in the studied parameters were assessed with the nonparametric Mann–Whitney U test for testing hypotheses about the homogeneity of the means. The differences were significant at p < 0.05.
Homology Modeling
Several structures for the opioid receptors are available in the PDB. However, they usually are not complete, have average resolutions, lack residues, or are crystallized in complex with other proteins. This does not make them great choices for docking studies. In an attempt at consistency, the mu, kappa, delta, and ORL1 opioid receptors were constructed using the corresponding human sequences (respective UniProtKB identifier P35372, P41145, P41143, and P4114) and the online software I-TASSER from the Zhang Lab.39 This iterative modeling software can use several different partial templates when no single valid template is available. Five models for each receptor were computed using the software default options, and the five best ones were further imported into the MOE software to be checked, charged, and minimized using the Amber 14:ETH force field.
The structure of the TRPA1 human receptor was directly extracted from the PDB file 6PQO. The homology model for the human TRPV1 ion channel was built using the UniProtKB sequence Q8NER1 of the human protein and the rat PDB structure 3J5Q as a template. The model was calculated using the MOE software and the homology modeling module. The PDB file 5IS0 was then used to determine the position of the ligand in the complex.
The homology model for the Cav 1.2 calcium channel was built using the UniProtKB sequence Q13936 (isoform 1) of the human voltage-gated calcium channel subunit α1c Cav 1.2 and the PDB structure 6JP5 of the rabbit Cav1.1/ nifedipine complex (resolution 2.90 Å) as a template. The alignment statistics were calculated as follows: 66.4% identity, 75.3% similarity, and 4.7% gaps for a total length of 1359 residues. Prior to the homology modeling, the geometry of all the molecules, bonds, and charges of the PDB structure 6JP5 was checked using the structure preparation tool of the MOE software (MOE 2020, Chemical Computing Group, Köln, Germany) and minimized with the Amber 14:EHT forcefield implemented within the MOE software. The homology model was generated using the α1C subunit as the template and the MOE standard parameters in the protein/homology model module. Several loops (215–232, 446–509, 776–892, 941–962, and 1326–1374) were added as the corresponding residues did not exist in the template, but their structure and position were checked and none of them were involved in the interaction with nifedipine.
Docking
The docking procedures were performed with the GOLD38 software (version 2020.3) using the HERMES48 interface (CCDC, Cambridge Crystallographic Data Centre) and the implemented GOLD Wizard. We used the GOLD standard parameters for the docking, with the exploration of a spherical site (10 Å radius) centered on the previously cocrystallized ligands. The settings allowed for protein flexibility for the side chains of the residues but not for the backbone of the proteins. The genetic algorithm parameter was set to a number of 200 poses with the activation of the early stop function. We used the Astex scoring potential fitness function49 for calculating the docking scores and the Chemscore scoring function40,41 for the rescore of the results.
Molecular Dynamics
MD simulations were performed for the model of the mu receptor and the homology model of the Cav 1.2 calcium channel in the presence of thiowurtzine using the Amber 14:EHT force field implemented in the MOE software. A lipid bilayer surrounding the calcium channel composed of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine and 1,2-dioleoyl-sn-glycero-3-phospho-rac-1-glycerol with a respective ratio of 3:1 was generated with the MOE lipid generator, using the PACKMOL-Memgen approach.50−52 TIP3P water molecules53,54 were added, as well as Na+ and Cl– ions in order to simulate a 0.1 M ionic force, and the pH was set at 7.4. Standard Amber 14:EHT parameters were used for MD. A quick minimization was performed prior to the start of the MD simulations. The geometry of all the molecules, bonds, and charges was then checked using the structure preparation tool in MOE, in order to start the molecular simulations. After a preliminary equilibration phase, 200 ns molecular simulations were performed. A frame was saved every 200 ps, leading to a total of 1000 frames.
Acknowledgments
The authors would like to thank Johnny Truong for his work on the homology model of the Cav 1.2 calcium channel and the subsequent docking of thiowurtzine.
Glossary
Abbreviations
- GOLD software
Genetic Optimization for Ligand Docking software
- MD
molecular dynamics
- MOE software
Molecular Operating Environment software
- NSAIDs
Nonsteroidal anti-inflammatory drugs
- ORL1
opioidlike receptor 1
- vs
versus
- TRP
transient receptor potential
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01786.
1H and 13C NMR spectra for compounds I, IV, VIII, and IX, IR spectra for I, RMSD plots for the MD calculations, and structural PDB data for the different models of the receptors (mu, kappa, delta, ORL1, and Cav 1.2) (PDF)
Author Contributions
⊥ S.A. and S.M. contributed equally to this work as first coauthors. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The authors declare no competing financial interest.
Supplementary Material
References
- Varrassi G. Severe Chronic Pain – the Reality of Treatment in Europe. Curr. Med. Res. Opin. 2011, 27, 2063–2064. 10.1185/03007995.2011.619426. [DOI] [PubMed] [Google Scholar]
- Piper B. J.; Shah D. T.; Simoyan O. M.; McCall K. L.; Nichols S. D. Trends in Medical Use of Opioids in the U.S., 2006–2016. Am. J. Prev. Med. 2018, 54, 652–660. 10.1016/j.amepre.2018.01.034. [DOI] [PubMed] [Google Scholar]
- Cawich S. O.; Deonarine U.; Harding H. E.; Dan D.; Naraynsingh V.. Chapter 46—Cannabis and Postoperative Analgesia. In Handbook of Cannabis and Related Pathologies; Preedy V. R., Ed.; Academic Press: San Diego, 2017; pp 450–458. [Google Scholar]
- Karateev A. E.; Nasonov E. L.; Yakhno N. N.; Ivashkin V. T.; Chichasova N. V.; Alekseeva L. I.; Karpov Y. A.; Evseev M. A.; Kukushkin M. L.; Danilov A. B.; Vorobyeva O. V.; Amelin A. V.; Novikova D. S.; Drapkina O. M.; Kopenkin S. S.; Abuzarova G. R. Clinical guidelines “Rational use of nonsteroidal anti-inflammatory drugs (NSAIDs) in clinical practice”. Mod. Rheumatol. J. 2015, 9, 4–23. 10.14412/1996-7012-2015-1-4-23. [DOI] [Google Scholar]
- Argoff C. E.; Albrecht P.; Irving G.; Rice F. Multimodal Analgesia for Chronic Pain: Rationale and Future Directions. Pain Med. 2009, 10, S53–S66. 10.1111/j.1526-4637.2009.00669.x. [DOI] [PubMed] [Google Scholar]
- Pogodin P. V.; Lagunin A. A.; Filimonov D. A.; Nicklaus M. C.; Poroikov V. V. Improving (Q)SAR Predictions by Examining Bias in the Selection of Compounds for Experimental Testing. SAR QSAR Environ. Res. 2019, 30, 759–773. 10.1080/1062936X.2019.1665580. [DOI] [PubMed] [Google Scholar]
- Macarron R.; Banks M. N.; Bojanic D.; Burns D. J.; Cirovic D. A.; Garyantes T.; Green D. V. S.; Hertzberg R. P.; Janzen W. P.; Paslay J. W.; Schopfer U.; Sittampalam G. S. Impact of High-Throughput Screening in Biomedical Research. Nat. Rev. Drug Discovery 2011, 10, 188–195. 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- Singh N.; Chaput L.; Villoutreix B. O. Virtual Screening Web Servers: Designing Chemical Probes and Drug Candidates in the Cyberspace. Briefings Bioinf. 2020, 22, 1790. 10.1093/bib/bbaa034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylova S. G.; Amosova E. N.; Zueva E. P.; Razina T. G.; Rybalkina O. Y.; Lopatina K. A.; Sysolyatin S. V.; Kalashnikov A. I.; Malykhin V.; Dygai A. M.; Zhdanov V. V.; Vorozhtsov A. B.; Zhukov A. S.. 4-(3,4-Dibromothiophene Carbonyl)-2,6,8,12-Tetraacetyl-2,4,6,8,10,12-Hexaazatetracyclo[5,5,0,03,11,05,9]Dodecane as Analgesic Agent and Method for Production Thereof. RU 2565766 C1, October 20, 2015.
- Solovev V. N. Effect of nitroglycerin and menthol on vascular dilatation. Klin. Med. 1949, 27, 57–62. [PubMed] [Google Scholar]
- Lepow H.; Turner R. A. Peripheral Vasodilatation in Response to Controlled-Release Nitroglycerin. Southwest. Med. 1966, 47, 190–192. [PubMed] [Google Scholar]
- Durlach J.; Lemerre L.; Lepage F. Treatment of premenstrual syndrome & pelvic pain syndrome with sugar-coated enteric tablets of potassium nitrate. Rev. Fr. Gynecol. Obstet. 1958, 53, 17–24. [PubMed] [Google Scholar]
- Collins J. F.; Gingold J.; Stanley H.; Simring M. Reducing Dentinal Hypersensitivity with Strontium Chloride and Potassium Nitrate. Gen. Dent. 1984, 32, 40–43. [PubMed] [Google Scholar]
- Nair U. R.; Sivabalan R.; Gore G. M.; Geetha M.; Asthana S. N.; Singh H. Hexanitrohexaazaisowurtzitane (CL-20) and CL-20-Based Formulations (Review). Combust., Explos. Shock Waves 2005, 41, 121–132. 10.1007/s10573-005-0014-2. [DOI] [Google Scholar]
- Tolstikova T. G.; Morozova E. A.; Sysolyatin S. V.; Kalashnikov A. I.; Zhukova Y. I.; Surmachev V. N. Synthesis and Biological Activity of the Derivatives of 2,4,6,8,10,12-Hexaazatetracyclo[5.5.0.03,11.05,9]Dodecane. Chem. Sustainable Dev. 2010, 4, 431–436. [Google Scholar]
- Sysolyatin S. V.; Lobanova A. A.; Chernikova Y. T.; Sakovich G. V. Methods of Synthesis and Properties of Hexanitrohexaazaisowurtzitane. Russ. Chem. Rev. 2005, 74, 757. 10.1070/RC2005v074n08ABEH001179. [DOI] [Google Scholar]
- Menke K. Organic Chemistry of Explosives, J. P. Agrawal, R. D. Hodgson. Pyrotechnics 2007, 32, 182. 10.1002/prep.200790002. [DOI] [Google Scholar]
- Krylova S. G.; Povet’eva T. N.; Zueva E. P.; Suslov N. I.; Amosova E. N.; Razina T. G.; Lopatina K. A.; Rybalkina O. Y.; Nesterova Y. V.; Afanas’eva O. G.; Kiseleva E. A.; Sysolyatin S. V.; Kulagina D. A.; Zhdanov V. V. Analgesic Activity of Hexaazaisowurtzitane Derivatives. Bull. Exp. Biol. Med. 2019, 166, 461–465. 10.1007/s10517-019-04372-9. [DOI] [PubMed] [Google Scholar]
- Lin G.; Tsai H.-J.; Tsai Y.-H. Cage Amines as the Stopper Inhibitors of Cholinesterases. Bioorg. Med. Chem. Lett. 2003, 13, 2887–2890. 10.1016/S0960-894X(03)00599-7. [DOI] [PubMed] [Google Scholar]
- Krylova S. G.; Povetyeva T. N.; Sysolyatin S. V.; Suslov N. I.; Zue-va E. P.; Nesterova Y. V.; Afanasyeva O. G.; Lopatina K. A.; Kulpin P. V.; Zhdanov V. V.; Kulagina D. A.; Malykhin V. V.; Vorozhtsov A. B.. 4-(3,4-Dibromothiophene Carbonyl)-2,6,8,12-Tetraacetyl-2,4,6,8,10,12-Hexaazatetracyclo[5,5,0,03.11,05.9]Dodecane Used as an Anticonvulsant. Patent RU 2684107 C1 Russian Federation, IPC C07D, A61K, A61P, January 12, 2018.
- Sample Size Software|Power Analysis Software|PASS|NCSS.com. https://www.ncss.com/software/pass/ (accessed Feb 19, 2021).
- Bellamy A. J. Reductive Debenzylation of Hexabenzylhexaazaisowurtzitane. Tetrahedron 1995, 51, 4711–4722. 10.1016/0040-4020(95)00155-2. [DOI] [Google Scholar]
- Kalashnikov A. I.; Sysolyatin S. V.; Sakovich G. V.; Surmacheva I. A.; Surmachev V. N.; Lapina Y. T. Debenzylation of 2,6,8,12-Tetraacetyl-4,10-Dibenzyl-2,4,6,8,10,12-Hexaazatetracyclo[5.5.0.03,11.05,9]Dodecane. Russ. Chem. Bull. 2009, 58, 2164–2168. 10.1007/s11172-009-0295-9. [DOI] [Google Scholar]
- Ananikov V. P.; Khemchyan L. L.; Ivanova Y. V.; Bukhtiyarov V. I.; Sorokin A. M.; Prosvirin I. P.; Vatsadze S. Z.; Medved’ko A. V.; Nuriev V. N.; Dilman A. D.; Levin V. V.; Koptyug I. V.; Kovtunov K. V.; Zhivonitko V. V.; Likholobov V. A.; Romanenko A. V.; Simonov P. A.; Nenajdenko V. G.; Shmatova O. I.; Muzalevskiy V. M.; Nechaev M. S.; Asachenko A. F.; Morozov O. S.; Dzhevakov P. B.; Osipov S. N.; Vorobyeva D. V.; Topchiy M. A.; Zotova M. A.; Ponomarenko S. A.; Borshchev O. V.; Luponosov Y. N.; Rempel A. A.; Valeeva A. A.; Stakheev A. Y.; Turova O. V.; Mashkovsky I. S.; Sysolyatin S. V.; Malykhin V. V.; Bukhtiyarova G. A.; Terent’ev A. O.; Krylov I. B. Development of New Methods in Modern Selective Organic Synthesis: Preparation of Functionalized Molecules with Atomic Precision. Russ. Chem. Rev. 2014, 83, 885–985. 10.1070/RC2014v83n10ABEH004471. [DOI] [Google Scholar]
- Lewanowitsch T.; Irvine R. J. Naloxone and its quaternary derivative, naloxone methiodide, have differing affinities for μ, δ, and κ opioid receptors in mouse brain homogenates. Brain Res. 2003, 964, 302–305. 10.1016/S0006-8993(02)04117-3. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Feunang Y. D.; Guo A. C.; Lo E. J.; Marcu A.; Grant J. R.; Sajed T.; Johnson D.; Li C.; Sayeeda Z.; Assempour N.; Iynkkaran I.; Liu Y.; Maciejewski A.; Gale N.; Wilson A.; Chin L.; Cummings R.; Le D.; Pon A.; Knox C.; Wilson M. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi G. W.; Striessnig J.; Koschak A.; Dolphin A. C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. 10.1124/pr.114.009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou X.; Yu X.; Bai D.; Tan B.; Cao P.; Qian M.; Zheng X.; Chen L.; Shi Z.; Li Y.; Ye F.; Liang Y.; Ni J. Pharmacology and Mechanism of Action of HSK16149, a Selective Ligand of α2δ Subunit of Voltage-Gated Calcium Channel with Analgesic Activity in Animal Models of Chronic Pain. J. Pharmacol. Exp. Ther. 2020, 376, 330. 10.1124/jpet.120.000315. [DOI] [PubMed] [Google Scholar]
- Bourinet E.; Zamponi G. W. Voltage Gated Calcium Channels as Targets for Analgesics. Curr. Top. Med. Chem. 2005, 5, 539–546. 10.2174/1568026054367610. [DOI] [PubMed] [Google Scholar]
- Weiss N.; Waard M. D. Les canaux calciques dépendants du voltage au cœur de la douleur. Méd./Sci. 2006, 22, 396. 10.1051/medsci/2006224396. [DOI] [PubMed] [Google Scholar]
- Todorovic S. M.; Meyenburg A.; Jevtovic-Todorovic V. Mechanical and Thermal Antinociception in Rats Following Systemic Administration of Mibefradil, a T-Type Calcium Channel Blocker. Brain Res. 2002, 951, 336–340. 10.1016/s0006-8993(02)03350-4. [DOI] [PubMed] [Google Scholar]
- Todorovic S. M.; Pathirathna S.; Meyenburg A.; Jevtovic-Todorovic V. Mechanical and Thermal Anti-Nociception in Rats after Systemic Administration of Verapamil. Neurosci. Lett. 2004, 360, 57–60. 10.1016/j.neulet.2004.02.049. [DOI] [PubMed] [Google Scholar]
- Feliks K. B.; Wrońska D.. Voltage-Gated Calcium Channel Antagonists: Potential Analgesics for Jejunal Pains. Pain Relief—From Analgesics to Alternative Therapies; IntechOpen, 2017. [Google Scholar]
- Ray B.; Kumar R.; Mehra R. L-Type Calcium Channel Blockers, Morphine and Pain: Newer Insights. Indian J. Anaesth. 2010, 54, 127–131. 10.4103/0019-5049.63652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado W. A. Involvement of Calcium in Pain and Antinociception. Braz. J. Med. Biol. Res. 2001, 34, 449–461. 10.1590/S0100-879X2001000400003. [DOI] [PubMed] [Google Scholar]
- Del Pozo E.; Caro G.; Baeyens J.M. Analgesic Effects of Several Calcium Channel Blockers in Mice. Eur. J. Pharmacol. 1987, 137, 155–160. 10.1016/0014-2999(87)90216-0. [DOI] [PubMed] [Google Scholar]
- Ray S. B.; Mishra P.; Verma D.; Gupta A.; Wadhwa S. Nimodipine Is More Effective than Nifedipine in Attenuating Morphine Tolerance on Chronic Co-Administration in the Rat Tail-Flick Test. Indian J. Exp. Biol. 2008, 46, 219–228. [PubMed] [Google Scholar]
- GOLD—Protein Ligand Docking Software—The Cambridge Crystallographic Data Centre (CCDC). https://www.ccdc.cam.ac.uk/solutions/csd-discovery/Components/Gold/ (accessed Feb 9, 2021).
- I-TASSER server for protein structure and function prediction. https://zhanglab.ccmb.med.umich.edu/I-TASSER/ (accessed Feb 16, 2021).
- Eldridge M. D.; Murray C. W.; Auton T. R.; Paolini G. V.; Mee R. P. Empirical Scoring Functions: I. The Development of a Fast Empirical Scoring Function to Estimate the Binding Affinity of Ligands in Receptor Complexes. J. Comput.-Aided Mol. Des. 1997, 11, 425–445. 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- Baxter C. A.; Murray C. W.; Clark D. E.; Westhead D. R.; Eldridge M. D. Flexible Docking Using Tabu Search and an Empirical Estimate of Binding Affinity. Proteins 1998, 33, 367–382. . [DOI] [PubMed] [Google Scholar]
- Manual for Preclinical Studies of New Pharmacological Substances. Part I; Mironov A. N., Ed.: Moscow, 2013. (in Russian). [Google Scholar]
- Rice A. S. C.; Cimino-Brown D.; Eisenach J. C.; Kontinen V. K.; Lacroix-Fralish M. L.; Machin I.; Mogil J. S.; Stöhr T.; Animal Models and the Prediction of Efficacy in Clinical Trials of Analgesic Drugs: A Critical Appraisal and Call for Uniform Reporting Standards. Pain 2008, 139, 243–247. 10.1016/j.pain.2008.08.017. [DOI] [PubMed] [Google Scholar]
- Woolfe G.; Macdonald A. D. The Evaluation of the Analgesic Action of Pethidine Hydrochloride (Demerol). J. Pharmacol. Exp. Ther. 1944, 80, 300–307. [Google Scholar]
- Koster R.; Anderson M.; De Beer E. Acetic Acid-Induced Analgesic Screening. Fed. Proc. 1959, 18, 412. [Google Scholar]
- Randall L. O.; Selitto J. J. A Method for Measurement of Analgesic Activity on Inflamed Tissue. Arch. Int. Pharmacodyn. Ther. 1957, 111, 409–419. [PubMed] [Google Scholar]
- Barrot M. Tests and Models of Nociception and Pain in Rodents. Neuroscience 2012, 211, 39–50. 10.1016/j.neuroscience.2011.12.041. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Mooij W. T. M.; Verdonk M. L. General and Targeted Statistical Potentials for Protein-Ligand Interactions. Proteins 2005, 61, 272–287. 10.1002/prot.20588. [DOI] [PubMed] [Google Scholar]
- Martínez J. M.; Martínez L. Packing Optimization for Automated Generation of Complex System’s Initial Configurations for Molecular Dynamics and Docking. J. Comput. Chem. 2003, 24, 819–825. 10.1002/jcc.10216. [DOI] [PubMed] [Google Scholar]
- Martínez L.; Andrade R.; Birgin E. G.; Martínez J. M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. 10.1002/jcc.21224. [DOI] [PubMed] [Google Scholar]
- Schott-Verdugo S.; Gohlke H. PACKMOL-Memgen: A Simple-To-Use, Generalized Workflow for Membrane-Protein-Lipid-Bilayer System Building. J. Chem. Inf. Model. 2019, 59, 2522–2528. 10.1021/acs.jcim.9b00269. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Neria E.; Fischer S.; Karplus M. Simulation of Activation Free Energies in Molecular Systems. J. Chem. Phys. 1996, 105, 1902–1921. 10.1063/1.472061. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.