

Abstract
Gastrointestinal (GI) cancer encompasses a range of malignancies that originate in the digestive system, which together represent the most common form of cancer diagnosed worldwide. However, despite numerous advances in both diagnostics and treatment, the incidence and mortality rate of GI cancer are on the rise. Myeloid‐derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells that increase in number under certain pathological conditions, such as infection and inflammation, and this expansion is of particular relevance to cancer. MDSCs are heavily involved in the regulation of the immune system and act to dampen its response to tumors, favoring the escape of tumor cells from immunosurveillance and increasing both metastasis and recurrence. Several recent studies have supported the use of MDSCs as a prognostic and predictive biomarker in patients with cancer, and potentially as a novel treatment target. In the present review, the mechanisms underlying the immunosuppressive functions of MDSCs are described, and recent researches concerning the involvement of MDSCs in the progression, prognosis, and therapies of GI cancer are reviewed. The aim of this work was to present the development of novel treatments targeting MDSCs in GI cancer in the hope of improving outcomes for patients with this condition.
Keywords: gastrointestinal cancer, tumor immunology, myeloid‐derived suppressor cells, progression, prognosis, therapy
Gastrointestinal (GI) cancer encompasses a range of malignancies that originate in the digestive system, which together represent the most common form of cancer diagnosed worldwide.
Myeloid‐derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells that increase in number under certain pathological conditions such as infection and inflammation, and this expansion is of particular relevance to cancer.
In the present review, the mechanisms underlying the immunosuppressive functions of MDSCs are described, and recent research concerning the involvement of MDSCs in the progression, prognosis as well as therapies of GI cancer is reviewed.
Abbreviations
- GI
gastrointestinal
- MDSCs
myeloid‐derived suppressor cell
- WHO
World Health Organization
- TAMs
tumor associated macrophages
- IMCs
immature myeloid cells
- PMNs
pre‐metastatic niches
- PBMC
peripheral blood mononuclear cells
- CI
confidence interval
- EC
esophageal cancer
- ESCC
esophageal squamous‐cell carcinoma
- GC
gastric cancer
- CRC
colorectal cancer
- HCC
hepatocellular carcinoma
- PC
pancreatic cancer
- HNSCC
head and neck squamous cell carcinoma
- DCs
dendritic cells
- M‐MDSC
monocytic MDSCs
- G‐MDSC
granulocytic MDSCs
- ROS
reactive oxygen species
- GM‐CSF
granulocyte macrophage colony‐stimulating factor
- TDFs
tumor‐derived factors
- SLFN4+
Schlafen4+
- ARG1
arginase I
- NOS2
nitric oxide synthase 2
- ADP
adenosine diphosphate
- iNOS
inducible nitric oxide synthase
- IL‐6
interleukin 6
- IL‐8
interleukin 8
- PI3K‐AKT
phosphatidylinositol 3‑kinase (PI3K)/protein kinase B (AKT)
- NOX‐2
NADPH oxidase 2
- TGFβ
transforming growth factor β
- PD‐L1
programmed death‐ligand 1
- IDO
indoleamine 2, 3‐dioxygenase
- TCR
T‐cell receptor
- IFN‐γ
interferon γ
- CDK‐4
cyclin‐Dependent Kinase 4
- CCL2
C‐C motif chemokine ligand 2
- STAT1
signal transducer and activator of transcription 1
- ADCC
antibody‐dependent cellular cytotoxicity
- NF‐κB
nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- G‐CSF
granulocyte colony‐stimulating factor
- IL‐3
interleukin 3
- IL‐10
interleukin 10
- PDGF
platelet‐derived growth factor
- STAT3
signal transducer and activator of transcription 3
- Th
T helper
- T‐bet
T‐box transcription factor TBX21
- GATA3
GATA Binding Protein 3
- NKT
natural killer T
- IL‐13
interleukin 13
- IL‐4R
interleukin 4R
- STAT6
signal transducer and activator of transcription 6
- Treg
regulatory T cell
- NKG2D
natural killer group 2 member D
- IL‐12
interleukin 12
- TLR
Toll‐like receptors
- HIF‐1α
hypoxia‐inducible factor
- NKP30
natural killer protein 30
- FAS‐L
Fas ligand
- M‐CSF
macrophage colony‐stimulating factor
- VEGF
vascular endothelial growth factor
- ENTPD2
ectonucleoside triphosphate diphosphohydrolase 2
- ATP
adenosine triphosphate
- 5’‐AMP
adenosine 5'‐monophosphate
- CTLs
cytotoxic T lymphocytes
- MMPs
matrix metalloproteinases
- MAC‐1
macrophage‐1 antigen
- ICAM‐1
intercellular adhesion molecule‐1
- CTCs
circulating tumor clusters
- NETs
neutrophil extracellular traps
- NRF2
nuclear erythroid 2‐related factor 2
- ARE
antioxidant responsive element
- CAFs
cancer associated fibroblasts
- TDSFs
tumor‐derived suppressor factors
- FAP
fibroblast activation protein
- α‐SMA
alpha smooth muscle actin
- CXCL1
C‐X‐C motif chemokine ligand 1
- CXCR2
C‐X‐C motif chemokine receptor 2
- MCP‐1
monocyte chemoattractant protein 1
- CCL5
C‐C motif chemokine ligand 5
- CCL12
C‐C motif chemokine ligand 12
- CCL9
C‐C motif chemokine ligand 9
- CCL15
C‐C motif chemokine ligand 15
- CXCL17
C‐X‐C chemokine ligand 17
- VEGFA
vascular endothelial growth factor A
- MMP9
matrix metalloproteinases 9
- Bv8
bombina variegate peptide 8
- S100A8/A9
S100 calcium‐binding protein A8/A9
- TME
tumor microenvironment
- IL‐1β
interleukin‐1β
- Nlrp3
NOD‐like receptor family pyrin domain containing‐3 protein
- IL‐17
interleukin‐17
- TRAIL‐Rs
TNF‐related apoptosis–induced ligand receptors
- SCF
stem‐cell factor
- KIT
stem cell factor receptor
- GIST
gastrointestinal stromal tumors
- CAR‐T cells
chimeric antigen receptor T cells
- ATRA
all‐trans retinoic acid
- GSS
glutathione synthase
- 4‐NQO
4‐nitroquinoline 1‐oxide
- CCR7
C‐C chemokine receptor 7
- CpG ODN
CpG motif‐containing oligodeoxynucleotides
- pDCs
plasmacytoid DCs
- IFN‐α
interferon‐α
- CSF1R
colony‐stimulating factor 1 receptor
- CXCR2
C‐X‐C motif chemokine receptor 2
- STING
stimulator of interferon genes
- HSC
hepatic stellate cell
- SDF‐1
stromal cell‐derived factor 1
- PDE‐5
phosphodiesterase‐5
- CIKs
cytokine‐induced killers
- L‐NOHA
N omega‐Hydroxy‐L‐arginine
- L‐NMMA
NG‐monomethyl L‐arginine
- ENO1
α‐enolase
- LPS
lipopolysaccharides
- Tc17 cells
CD8+T cell with the production of interleukin 17
- IL‐23
interleukin‐23
- VEGFR2
vascular endothelial growth factor receptor 2
- MUC1
mucinous glycoprotein 1
1. INTRODUCTION
Gastrointestinal (GI) cancer accounts for a third of all new diagnoses and a similar proportion of cancer‐associated mortality worldwide, and its incidence and mortality rate are on the rise [1, 2]. A number of malignancies fall under the wide umbrella of GI cancer, including esophageal cancer (EC), gastric cancer (GC), colorectal cancer (CRC), hepatocellular carcinoma (HCC), and pancreatic cancer (PC) [3, 4]. Current research shows that the estimated number of increased GI cancer cases (not include pancreatic cancer) in both sexes and ages were 4,296,333 (23.8%) and the number of deaths was estimated at 2,953,693 (30.9%) in 2018, which promotes GI cancer to be the largest public health concern in general [1]. According to the World Health Organization (WHO), the number of deaths from cancer will increase by nearly 80% by 2030 worldwide, with most occurring in low‐ and middle‐income countries [5]. A broad range of therapeutic strategies has been developed to treat these diseases, which include surgery, chemotherapy, radiotherapy, and immunotherapy. While these approaches have improved outcomes for patients with GI cancer, the overall survival rate of these patients remains relatively poor [6, 7]. This is at least in part due to the tendency of GI cancer to resist chemotherapy and escape immune surveillance via the activation and expansion of the populations of immunosuppressive cells, such as myeloid‐derived suppressor cells (MDSCs), regulatory T‐cell (Treg) as well as tumor‐associated macrophages (TAMs), leading to immunosuppression and an increased propensity for metastasis [8, 9, 10].
MDSCs are a heterogeneous population of immature monocytes and granulocytes that have been observed in a number of murine tumor models, where they are identified by their expression of the CD11b and Gr‐1 markers [11]. In humans, similar populations of immature myeloid cells (IMCs) were initially described in head and neck squamous cell carcinoma (HNSCC) [12], and their presence has since been well‐documented in cancers of the esophagus, stomach, pancreas, colon, and skin [13, 14, 15]. MDSCs accumulate in tumor sites (both primary and metastatic), where they inhibit T‐cell‐mediated tumor clearance, and have also been found to suppress natural killer (NK) cell populations [11, 16, 17]. Furthermore, MDSCs appear to confer some level of protection to circulating tumor cells and to be involved in the formation of pre‐metastatic niches (PMNs), which encourage the invasion of cancer cells [18]. Their involvement in this process has been known for decades; in the 1970s, the “seed and soil” hypothesis was conceived by Paget [19], describing the link between tumor metastasis and MDSCs. More recently, in 2005, PMNs were defined for the first time as the specific cell microenvironment that favors the colonization of metastatic cancer cells in distant organs [20].
In GI cancer patients, MDSCs levels are closely associated with both therapeutic efficacy and overall clinical outcome [21]. For example, Limagne et al. [22] reported that accumulation of MDSCs in metastatic CRC patients was correlated with the efficacy of a 5‐fluorouracil (5‐FU), oxaliplatin, and bevacizumab treatment regimen. Markowitz et al. [23] studied the correlation between MDSC frequency in peripheral blood mononuclear cells (PBMCs) and disease progression among 16 chemotherapy‐treated PC patients and found that a higher chance of progression was observed in the patients with increased frequency of MDSCs in peripheral blood (P = 0.0013). A meta‐analysis showed that the combined hazard ratio for overall survival of GI cancer patients in the association between MDSC level and prognosis was 1.26 (95% confidence interval [CI] = 1.10‐1.44, P = 0.0003) [21]. Karakasheva et al. [24] reported that CD38+ monocytic MDSC (M‐MDSC) expansion could be associated with a subset of advanced CRC patients. Additionally, a recent study showed that peripheral circulating MDSCs were significantly correlated with the prognosis of ESCC patients [25]. Since substantial progress has been made in recent years towards understanding the function of these cells, as well as their genomic and biochemical characteristics [26, 27, 28], further investigation into their role in GI cancer is still required. In this review, we summarize the known functions and effects of MDSCs in GI cancer, as well as detail the involvement of MDSCs in immunosuppression, metastasis, and prognosis. The aim of this work was to present the development of novel treatments targeting MDSCs in GI cancer in the hope of improving outcomes for patients with this condition.
2. THE ORIGIN AND PHENOTYPE OF MDSCS IN CANCERS
MDSCs are a heterogeneous population of cells that include both myeloid progenitors and IMCs. Under healthy conditions, IMCs are formed in the bone marrow and rapidly differentiate into mature macrophages, dendritic cells (DCs), or granulocytes [11]. However, IMCs are often prevented from fully differentiating into mature cells under certain pathological conditions, including cancer [11, 12], infection [29], sepsis [30], trauma [31], and some autoimmune disorders [32], leading to an expansion of this population.
In tumor bearing mice, two main MDSC subtypes have been identified, termed granulocytic MDSCs (G‐MDSC) and monocytic MDSCs (M‐MDSC), respectively [33]. G‐MDSC is defined as CD11b+Ly6G+Ly6Clow/− cells, while M‐MDSC is defined as CD11b+Ly6Glow/−Ly6Chigh cells. Both of these cell subsets are expanded in murine tumor models and have primarily been documented in the bone marrow, peripheral blood, spleen, liver and lung tissues, and a range of tumor tissues [34].
In humans, MDSC populations are even more complex. In 1995, MDSCs bearing the cell surface marker CD34 was first reported in HNSCC patients [12]. Further researches defined the criteria for the phenotypic characterization of these cells by flow cytometry [35, 36]. Among the human PBMCs, M‐MDSC is defined as CD14+ while G‐MDSC is CD15+, and both these subsets are HLA‐DR−CD33+. A further subset of HLA‐DR−CD33+ lineage‐negative (Lin−) early MDSCs (e‐MDSCs) has since been proposed, which lack the expression of mature cell lineage markers such as CD3, CD14, CD15, CD19 and CD56. This subset contains mixed groups of MDSCs and is comprised of more immature progenitors [37]. In healthy individuals, IMCs with the phenotype described above comprise ∼0.5% of PBMCs [11]. Importantly, the evidence indicates that these subpopulations may have different functions in cancer [38], infections [39], and autoimmune diseases [32], with both G‐MDSC and M‐MDSC populations being expanded in those cases. Youn et al. [40] reported that the expansion of G‐MDSC population was greater than that of M‐MDSC subset in the spleen of 10 different tumor models of lung, breast, colon, melanoma, and sarcoma in three different strains of mice. They also found that the two subpopulations used different mechanisms to suppress T‐cell function: G‐MDSC mainly generated reactive oxygen species (ROS), whereas M‐MDSC mainly produced nitric oxide (NO). As MDSCs are a group of myeloid cells comprised of precursors of macrophages, granulocytes, and DCs at earlier stages of differentiation, these two subsets of MDSCs remained in different differentiation directions. Previously, it has been reported that 20% of G‐MDSCs differentiated to F4/80+ macrophages compared with more than 60% of cells differentiated from M‐MDSCs in an in vitro culture system with granulocyte macrophage colony‐stimulating factor (GM‐CSF). Additionally, 10% of G‐MDSC differentiated into CD11c+ DCs, whereas more than 50% of cells differentiated from M‐MDSC were CD11c+ cells [40]. Therefore, M‐MDSCs have been found to be restricted to mature macrophages and DCs in vitro more than G‐MDSCs, and various tumor‐derived factors (TDFs) produced by different tumor cells define the expansion of MDSC subsets.
From the perspective of GI cancer, multiple reports have shown involvement of MDSCs in tumor progression. For instance, Ding et al. [41] have demonstrated that bone marrow‐derived Schlafen4+(SLFN4+) MDSCs could migrate to the stomach during Helicobacter infection. Later, they found that high MiR‐130b expression in SLFN4+ MDSCs was responsible for neoplastic stimulation before GC. Mechanistically, miR‐130b‐mediated inhibition of cylindromatosis (Cyld) leads to release/activation of NF‐κB, which in turn promotes MDSC immunosuppression via up‐regulating the expression of arginase I (ARG1) and nitric oxide synthase 2 (NOS2). Suppression of miR‐130b impaired MDSCs function and restored CD8+ cytotoxic T‐cell infiltration of the stomach. Moreover, elevated levels of miR‐130b were found in the serum of both Helicobacter‐infected mice and gastric cancer patients that correlated with the respective metaplastic changes in the stomach, showing a strong correlation with pre‐neoplastic changes prior to GC [42]. CD38 is a transmembrane glycoprotein and belongs to the ADP‐ribosyl cyclase family, possessing both ectoenzyme and receptor functions. It was first reported by Karakasheva et al. [24] that CD38high MDSCs are more immature than MDSCs lacking CD38 expression, possess a greater capacity to suppress activated T cells, and promote tumor growth to a greater degree, with an increased inducible nitric oxide synthase (iNOS) production in EC. Later, Karakasheva et al. [43] also proved that CD38+ M‐MDSC expansion characterizes a subset of advanced CRC patients, and targeting M‐MDSCs with an anti‐CD38 monoclonal antibody would bring a benefit to the metastatic CRC patients. In GC patients, the nature of most MDSCs has been recognized as granulocyte‐like cells or monocyte‐like cells, which have been reported in many studies [44, 45]. Recently, a novel subset of CD45+CD33lowCD11bdim MDSCs in GC patients was identified by Zhuang's group [46]. Compared to healthy individuals, CD45+CD33lowCD11bdim MDSCs morphologically resembled neutrophils with the expression of CD66b, showing a more immunosuppressive function. Serum IL‐6 and IL‐8 derived from GC patients could activate granulocytic MDSCs to express ARG1 through the PI3K/AKT signalling pathway. Evidence showed that the percentage of CD45+CD33lowCD11bdim MDSCs in the peripheral blood and the levels of IL‐6, IL‐8, and ARG1 in the serum were positively correlated with GC progression and negatively correlated with overall patient survival.
Nevertheless, it remains unclear whether these special subsets of MDSCs would lead to a resistance to anti‐cancer therapy, such as chemoresistance, as current treatments for GI cancer patients retain little effect. From a personal opinion, apart from the reported agents or clinical trials, other cytokines, chemokine ligands/receptors, genes and molecules, especially those first reported in GI cancer that affect the number and function of MDSCs, could be potential therapeutic targets and promising drugs for GI cancer.
3. THE UNDERLYING MECHANISMS OF MDSC‐MEDIATED IMMUNOSUPPRESSION IN GI CANCER
Suppression of the immune system is a primary function of MDSCs in cancer and occurs both through direct targeting and release of soluble mediators that regulate immune response [11]. The main factors involved in MDSC‐mediated immune suppression includes ARG1, iNOS, NADPH oxidase 2 (NOX‐2), ROS, transforming growth factor β (TGFβ), programmed death‐ligand 1 (PD‐L1), and indoleamine 2,3‐dioxygenase (IDO), which specifically target T cells and other immune cells [34] (Figure 1).
FIGURE 1.
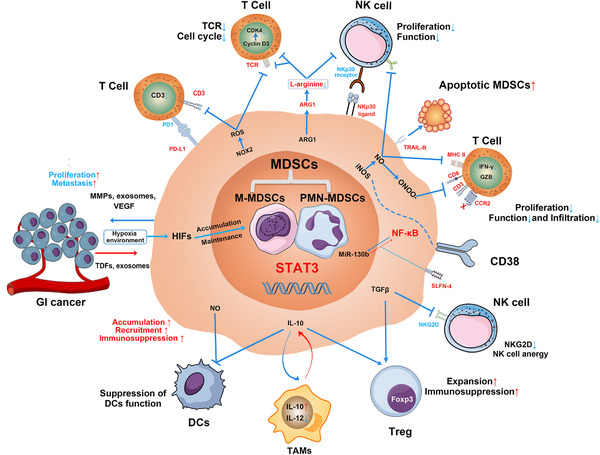
Molecular mechanisms involved in MDSC‐mediated immunosuppression in GI cancers. In GI cancer, MDSCs accumulate and expand in the tumor microenvironment that regulated by the expression of STAT3, with TDFs, exosomes, and hypoxia‐inducible factor 1α, etc. MDSCs suppress proliferation and function of T cells and NK cells through the enzymes of ARG1 and iNOS, ROS, and expression of PD‐L1. Peroxynitrite (ONOO–) causes the nitration of the CCL2 chemokine, which diminishes CD8+ T‐cell infiltration. At the same time, NO production can suppress DCs antigen presentation to CD4+ T cells. Additionally, the effect of ADCC function and anergy of NK cells are induced by the production of NO and the inhibition of NKG2D by TGFβ, respectively. While the expression of NKP30 ligand on MDSCs induces NK cell apoptosis, activation of TRAIL receptor could lead MDSC apoptosis conversely. MDSC‐derived IL‐10 suppresses DCs’ function, promotes M2 macrophage differentiation, and increases the number and immunosuppression of Treg. MDSC‐derived TGFβ can promote Treg expansion and immunosuppression as well. In return, MDSCs secret MMPs, exosomes, and VEGF to promote GI cancer cell proliferation and metastasis. The specific markers of CD38+ and SLFN4+ MDSCs were first reported in GI cancer. MDSCs, myeloid‐derived suppressor cells; M‐MDSC, monocytic MDSCs; G‐MDSC, granulocytic MDSCs; GI cancer, gastrointestinal cancer; TDFs, tumor derived factors; MMPs, matrix metalloproteinases; VEGFs, vascular endothelial growth factors; HIFs, hypoxia‐inducible factors; PD1, programmed cell death protein 1; PD‐L1, programmed death‐ligand 1; TCR, T‐cell receptor; CDK4, cyclin‐dependent kinase 4; ARG1, arginase I; NK cells, natural killer cells; NKP30, natural killer protein 30; iNOS, inducible nitric oxide synthase; NO, nitric oxide; ONOO‐, peroxynitrite; MHC II, major histocompatibility complex class 2; CCR2, C‐C motif chemokine receptor 2; NF‐κB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; MiR‐130b, microRNA 130b; SLFN4, Schlafen4; TGFβ, transforming growth factor β; NKG2D, natural killer group 2 member D; Foxp3, forkhead box P3; Treg, regulatory T‐cell; IL‐10, interleukin 10; IL‐12, interleukin 12; TAMs, tumor associated macrophages; DCs, dendritic cells; STAT3, signal transducer and activator of transcription 3
3.1. L‐arginine mechanisms
MDSC‐induced immunosuppression is associated with increased L‐arginine metabolism in the tumor microenvironment [38]. L‐arginine serves as a substrate for both arginases, particularly ARG1 [47, 48], and iNOS (generates NO) [49]. These enzymes are strongly expressed in MDSCs, where they function as T‐cell inhibitors. In GC patients, depletion of arginine reduces T‐cell receptor (TCR) expression and suppresses both T lymphocyte proliferation and interferon‐γ (IFN‐γ) production [50, 51, 52]. The mechanism of impaired proliferation of T cells was demonstrated previously that ARG1 converts arginine from the microenvironment into urea and L‐ornithine, leading to cell cycle arrest at G0‐G1 phase in T cells via the up‐regulation of cyclin D3 and cyclin‐dependent kinase 4 (CDK‐4), limiting T‐cell activation and proliferation [53]. In GC patients, ARG1+ MDSCs have been found to accumulate in the peripheral blood of patients and foster immunosuppression and cancer progression [54, 55]. Furthermore, MDSCs have been found to suppress CD8+ T‐cell activity through ARG1‐mediated IL‐6/IL‐8 axis in GC [46]. In a colon tumor model (CT26 cells), the presence of ARG1 impairs the proliferation of tumor‐infiltrating CD8+ T cells and NK cells, as well as the production of inflammatory cytokines and expression of interferon‐inducible genes [56, 57]. The link between immunosuppression and iNOS expression by MDSCs is well established in different cancer types. In GI cancer, NO not only mediates MDSCs’ inhibitory effects on the proliferation and function of immune cells [58] but also regulates the nitration of the CCL2 chemokine by diminishing CD8+ T‐cell infiltration [59]. Markowitz et al. [60] demonstrated that MDSC‐derived NO mediates the inhibition of antigen presentation from DCs to CD4+ T cells through STAT1 nitration in pancreatic tumor model. They have also demonstrated that MDSC‐derived NO impairs the Fc receptor‐mediated proliferation and function of NK cells through multiple mechanisms including the reduction of the antibody‐dependen T‐cellular cytotoxicity (ADCC) of NK cells and inhibition of IFN‐γ production in pancreatic tumor model [61]. In murine esophageal tumor model, CD38high MDSCs were found producing high iNOS level, likely due to NF‐κB activation in these cells [24]. In colitis‐associated colorectal cancers, granulocyte colony‐stimulating factor (G‐CSF) has been reported to increase the level of iNOS+ MDSCs, which in turn supported tumor immune escape. In contrast, blocking G‐CSF could reduce the expression of iNOS in MDSCs [62].
3.2. ROS generation
ROS is also produced by MDSCs and has emerged as one of the main immunosuppressive mechanisms in both tumor‐bearing mice and cancer patients [63, 64, 65, 66]. Increased generation of ROS by MDSCs is induced by TDFs, including TGFβ, IL‐3, IL‐6, IL‐10, platelet‐derived growth factor (PDGF) and GM‐CSF [67, 68]. High ROS levels in the colon carcinoma microenvironment down‐regulated the response of antigen‐specific T cells by altering their expression of CD3ζ chains; in PC patients, a high level of ROS was correlated with the reducing cytokine secretion [69, 70]. Nagaraj et al. [71] showed that the nitration of TCR on CD8 T cells was induced by ROS and peroxynitrite generated from MDSCs during direct T‐cell‐cell contact in a MC38 xenograft model. MDSCs also suppressed T‐cell proliferation to favor tumor cell growth via ROS in CRC [72]. Again, in colon tumor models (CT26 and MC38 cells), the up‐regulated activity of NOX‐2, primarily p47phox and gp91phox subunits, produces ROS in MDSCs, which is mediated by STAT3 [73].
3.3. Cytokine secretion
TGFβ and IL‐10 are two other soluble cytokines that are secreted by MDSCs and have been found to be involved in the suppression of T cells and NK cells and in macrophage polarization, respectively. Previously, TGFβ was reported to target CD4+ T helper (Th) lymphocytes undergoing differentiation towards Th1 and Th2 phenotypes through inhibiting T‐box transcription factor TBX21 (T‐bet) and GATA‐binding protein 3 (GATA3) expression [74, 75, 76]. In an early stage of GI cancer, CD1d‐restricted natural killer T cells have been reported to induce TGFβ production by MDSCs through an IL‐13‐IL4R‐STAT6 signaling pathway; abrogation of this population of cells partially enhanced the antitumor immunity in a CT26 colon tumor model [77]. In the MCA26 colon tumor model, MDSCs were reported to suppress Ag‐specific CD4+ T cells by promoting the development of inducible CD4+CD25+Foxp3+ Treg via the production of TGFβ as well as IL‐10 [17]. In return, Treg cells support MDSC functions to promote an immunosuppressive environment that is suitable for tumor progression as well [78, 79]. In a hepatocellular tumor model, membrane‐bound TGFβ1 in MDSCs can directly inhibit natural killer group 2 member D (NKG2D) expression and IFN‐γ production of NK cells, inducing anergy of NK cells [16]. For IL‐10, a study showed that IL‐10 secretion by MDSCs induced macrophage polarization towards the M2 type with reduced IL‐12 secretion [80]. Similarly, in a hepatocellular tumor model, the secretion of IL‐10 from MDSCs suppressed the function of DCs as well, which inhibited Toll‐like receptor (TLR)‐induced IL‐12 production and reduced DC‐mediated activation of T cells [81].
3.4. Other factors
Other factors involved in the immunosuppressive function of MDSCs, such as hypoxia‐inducible factor 1‐alpha (HIF‐1α), PD‐L1, natural killer protein 30 (NKP30) ligand, IDO, and NOX‐2, have all been well reported. For example, Noman et al. [82] demonstrated that MDSCs at the tumor site showed a differential expression of PD‐L1 as compared with MDSCs from the spleen, which was caused by the HIF‐1α expression that selectively up‐regulated PD‐L1 on splenic MDSCs in tumor‐bearing mice. Further blockade of PD‐L1 under hypoxia was accompanied by the down‐regulation of IL‐6 and IL‐10 in MDSCs and enhanced T‐cell activation. Meanwhile, PD‐L1 and Fas ligand (FAS‐L) were also found to be expressed on the surface of MDSCs, promoting T‐cell apoptosis [83, 84, 85]. As shown in in vitro culture system and clinical investigation, PD‐L1+ MDSCs could be induced by macrophage colony‐stimulating factor (M‐CSF) and vascular endothelial growth factor (VEGF) released by a liver cancer cell line; peripheral blood collected from HCC and CRC patients had significantly higher frequency of PD‐L1+ MDSCs compared to those from healthy donors and patients after treatment [86, 87]. Again, in a hepatocellular tumor model, HIF‐1α induced ectonucleoside triphosphate diphosphohydrolase 2 (ENTPD2) expression in the liver cancer cells; ENTPD2 converted extracellular adenosine triphosphate (ATP) to adenosine 5'‐monophosphate (5’‐AMP), which prevented the differentiation of MDSCs and maintained their immunosuppression [88]. In HCC patients, MDSCs inhibited autologous NK cell cytotoxicity and IFN‐γ release through contact with NKP30 ligand and receptor, instead of ARG1 and iNOS [89]. Both IDO and NOX‐2 in MDSCs played an important role in the inhibition of cytotoxic T lymphocytes (CTLs), DCs, and NK cells, in addition to inducing the expansion of Treg, to make an immunosuppressive tumor microenvironment [11, 90‐94].
4. THE INVOLVEMENT OF MDSCS IN THE MALIGNANT PROGRESSION OF GI CANCER
MDSC‐mediated tumor metastasis is a complex, multi‐stage process characterized by the secretion of chemokines, cytokines, and enzymes, all of which contribute to tumor cell proliferation, invasion, survival, and chemoattraction [33]. MDSCs favor tumor cell invasion and metastasis through remodeling of the basal membrane via the secretion of matrix metalloproteinases (MMPs), which opens a route for tumor cell migration and invasion [95]. This process may also involve macrophage‐1 antigen (MAC‐1)/ligand intercellular adhesion molecule‐1 (ICAM‐1) interactions [96, 97] and TGFβ1 [98]. In addition, MDSCs protect circulating tumor cells and promote their extravasation [99], serving a critical role in the formation of circulating tumor clusters (CTCs). These CTCs are strongly associated with immune escape. Furthermore, MDSCs also support tumor cell extravasation and invasion through the release of neutrophil extracellular traps (NETs) [100] and promote tumor cell survival via the ROS‐nuclear erythroid 2‐related factor 2 (NRF2)/antioxidant responsive element (ARE) axis and Notch signaling pathway [101, 102]. With CTCs being favored in this way by MDSCs, patients with GI cancer are more likely to experience rapid metastasis and poor prognosis [103, 104].
In addition, MDSCs have been found to promote the generation of PMNs in different cancer patients, including GI cancer. Prior to the arrival of tumor cells to a secondary site, MDSCs infiltrate the healthy tissue and induce their transformation to future metastatic sites. The accumulation, expansion, and recruitment of MDSCs in distant organs may be induced by a range of cytokines and chemokines, which are derived from the distant organs, tumor cells, and cancer‐associated fibroblasts (CAFs). In the tumor microenvironment, several tumor‐derived suppressor factors (TDSFs) have been found to influence the accumulation and expansion of myeloid precursors in PMNs, including G‐CSF, GM‐CSF, IL‐6, and Flt3‐ligand [105, 106]. Meanwhile, chemokines such as CCL2, which is produced by both tumor cells and target organs, drive MDSCs into normal tissues to form PMNs [107]. Expression of CAFs, such as fibroblast activation protein (FAP) and alpha smooth muscle actin (α‐SMA), further promote the recruitment and sustainment of MDSCs in PMNs via the STAT3‐CCL2 signaling pathway [108].
TAMs have also been found to impact the formation of liver PMNs in a murine colon tumor model by releasing CXCL1, which attracts CXCR2‐expressing MDSCs [109]. Other chemokines, such as monocyte chemoattractant protein 1 (MCP‐1) [110], CCL5 [111], CCL12 [112], CCL9 [113], CCL15 [114], and CXCL17 [115], have also been reported to promote the transport of MDSCs to PMNs, although the source of these cytokines in PMNs remains unclear. Furthermore, MDSCs initiate and sustain the development of PMNs by autocrine signaling: secreting a variety of regulatory molecules including PDGF [115], vascular endothelial growth factor A (VEGFA) [116], matrix metalloproteinase 9 (MMP9) [117], bombina variegate peptide 8 (Bv8) [118], and S100 calcium‐binding protein A8/A9 (S100A8/A9) [119]. For example, S100A9‐deficient mice showed reduced accumulation of MDSCs in PMNs during colonic metastasis [120]. Notably, MDSCs have also been found to differentiate into fibrocytes and osteoclasts in tumor mice, which may further enhance tumor growth and metastasis in cancer patients [121, 122]. In the clinic, recent investigations have documented a correlation between MDSC frequency in GI cancer patients and progression, which is summarized in Table 1.
TABLE 1.
Studies evaluating MDSC frequency as a prognostic biomarker in patients with GI cancer
| Cancer type (stage) | Study type | Objective | Specimen type | No. of patients/samples | MDSC definition | Detection method | Results and conclusion | Reference a |
|---|---|---|---|---|---|---|---|---|
| CRC (stage IV) | Experimental study | To further characterize BM‐MDSC‐mediated immune suppression. | PBMC | 50 patients vs. 0 controls | HLA‐DRlow/–Lin–CD33+ | Flow cytometry |
Comparison between patients with high and low MDSCs levels: The significance of overall survival: P = 0.0338. |
Solito et al., 2011. [124] |
| GC (stage IIIa) | Single center, prospective study | Evaluation of the role of immune cells (MDSCs, T‐cell and B cell) in the progression of GC. | Tumor tissue | 100 patients vs. 0 controls | CD33+/p‐STAT1+ | Immunohistochemistry |
Comparison between patients with high and low MDSCs levels: The significance of overall survival: P <0.0001; The significance of disease‐free survival: P <0.0001. |
Dong et al., 2013. [167] |
| CRC | Single center, prospective study | To investigate the frequency and clinical significance of the MDSCs in CRC patients. | PBMC, tumor tissue | 49 patients vs. 40 controls | HLA‐DR–CD33+ | Flow cytometry |
Comparison between patients with high and low MDSCs levels: For PBMC, the significance of metastasis significance: P = 0.0229; For tumor tissue, the significance of lymph node metastasis, P = 0.0120. |
Sun et al., 2012. [168] |
| CRC | Experimental study | Reveal the clinical relevance of circulating MDSCs in CRC patients with cancer progression. | PBMC | 64 patients vs. 32 controls | Linlow/–HLA‐DR–CD11b+CD33+ | Flow cytometry |
Comparison between Stage I/II and Stage III/IV: The significance of MDSCs frequency: P = 0.0109 Comparison between patients with extensive metastasis and limited metastasis: The significance of MDSCs frequency: P = 0.1342; The significance of MDSCs absolute count in blood P = 0.0411. |
Zhang et al., 2013. [169] |
| HCC | Prospective study | Clarified the relationship between a feature of MDSCs and host factors in HCC patients. | PBMC | 123 patients vs. 13 controls | HLA‐DR–Linlow/–CD14+ | Flow cytometry |
Comparison between Stage I/II and Stage III/IV: The significance of MDSCs frequency: P <0.01; The significance of overall survival: P = 0.008; The significance of advanced TNM stage: P = 0.0035. |
Arihara et al., 2013. [170] |
| CRC | Experimental study | Examination of the correlation between MDSCs and CRC patients progression and during routine treatment. | PBMC | 42 patients vs. 32 controls | CD33+CD11b+ HLA‐DR− | Flow cytometry |
Comparison between patients with high and low MDSCs levels: The significance of lymph node metastasis: P = 0.0067; The significance of advanced TNM stage: P = 0.0242. |
Ouyang et al., 2015. [72] |
| RC | Single center, prospective study | Examination of immunosuppressive molecules alterations during CRT and their associations with clinical features and prognosis in RC patients. | Tumor tissue | 62 patients vs. 0 controls | HLA‐DRlow/–Lin–CD33+CD11b+ | Immunohistochemistry |
Comparison between patients with high and low MDSCs levels: The significance of poor response after neoCRT, P = 0.0062. |
Teng et al., 2015. [171] |
| GC | Single center, prospective study | Explore the role of MDSCs in the occurrence and development of GC patients. | PBMC | 77 patients vs. 20 controls | HLA‐DR–CD14+ | Flow cytometry |
Comparison between Stage I and Stage II/III/IV: The significance of MDSCs frequency: P = 0.046; Comparison between patients with or without lymph node metastasis: The significance of MDSCs frequency: P = 0.075. |
Ni et al., 2015. [172] |
| PA | Prospective study | Investigation of the relationship between elevated cytokines and levels of MDSCs with progressive disease in PA patients. | PBMC | 16 patients vs. 9 controls | HLA‐DR–CD33+ | Flow cytometry |
Comparison between patients with stable disease and progressive disease: The significance of MDSCs frequency: P = 0.013. |
Markowitz et al., 2015. [23] |
| EC | Prospective study | Detection of the level of CD14+ HLA‐DR−/low cells in EC patients and analyzed its clinical significance. | PBMC | 78 patients vs. 35 controls | HLA‐DR–CD14+ | Flow cytometry |
Comparison between Stage I/II and Stage III/IV: The significance of MDSCs frequency: P = 0.0486; Comparison between patients with or without lymph node metastasis: The significance of MDSCs frequency: P = 0.0093. |
Huang et al., 2015. [173] |
| Metastatic CRC | Single center, prospective study | Compared the level of MDSCs in healthy volunteers and metastatic CRC patients treated with 5‐FU, oxaliplatin, and bevacizumab, and tested the association of immune parameters with patients' outcome. | PBMC | 25 patients vs. 20 controls | Lin–CD33highHLA‐DR– for total MDSCs | Flow cytometry |
Comparison between patients with high and low G‐MDSC levels: The significance of progression‐free survival: P = 0.04. |
Limagne et al., 2016. [22] |
| CRC | Single center, prospective study | Evaluation of MDSCs frequencies and CD38 expression patterns in PBMCs from CRC patients. | PBMC | 41 patients vs. 8 controls |
M‐MDSC: CD14+HLA‐DRlow/– PMN‐MDSC: CD33+CD11b+CD14–CD15+SSChi |
Flow cytometry |
Comparison between healthy donor and CRC patients: The significance of M‐MDSC levels: P = 0.005; Comparison between untreated and treated CRC patients: The significance of M‐MDSC and CD38+M‐MDSC level: P = 0.004 and P = 0.012, respectively. |
Karakasheva et al., 2018. [43] |
| CRC | Prospective study | Investigation of the relationship of prognosis and inflammatory parameters of CRC patient, regarding circulating MDSCs. | PBMC | 35 patients vs. 0 controls | CD14−CD11b+CD33+ | Flow cytometry |
Comparison between patients with MDSCs (≥ 3.2%) and MDSCs (<3.2%): The significance of recurrence‐free survival among stage 2 and 3: P = 0.038. |
Shimura et al., 2018. [174] |
| Advanced CRC | Prospective study | Evaluation of MDSCs as a potential biomarker and immune regulator in CRC progression. | PBMC | 10 patients vs. 0 controls |
M‐MDSC: CD45+CD11b+CD33+HLA‐DRlow/− CD14+CD15–; G‐MDSC: CD45+CD11b+CD33+HLA‐DRlow/− CD14–CD15+ |
Flow cytometry |
Comparison between patients with high and low PMN‐MDSC levels: The significance of distant metastasis: P = 0.014. |
Fedorova et al., 2018. [175] |
| GC (stage II and III) | Single center, prospective study | Investigation of the relationship between peripheral MDSCs in the perioperative period and prognosis in GC patients. | PBMC | 75 patients vs. 0 controls | CD14+ HLA‐DR− CD11b+ CD33+ | Flow cytometry |
Comparison between pre‐ and post‐operative patients: The significance of MDSCs: P = 0.001; The significance of recurrence‐free survival among stage 2 and 3: P = 0.0092 and P = 0.063, respectively. |
Urakawa et al., 2019. [176] |
| ESCC | Single center, retrospective study | Examined the correlations of the NLR and MDSCs with the prognoses in patients with ESCC. | PBMC | 94 patients vs. 20 controls | CD11b+CD33+HLA‐DR– | Flow cytometry | Percentage of MDSCs was significantly elevated in ESCC patients who developed disease failure after treatment: P = 0.008. | Chen et al., 2019. [25] |
Information listed in the table are arranged in an ascending chronological order.
Abbreviations: MDSCs, myeloid‐derived suppressor cells; M‐MDSC, monocytic MDSCs; G‐MDSC, granulocytic MDSCs; GI cancer, gastrointestinal cancer; CRC, colorectal cancer; GC, gastric cancer; HCC, hepatocellular carcinoma; RC, rectal cancer; PA, pancreatic adenocarcinoma; EC, esophageal cancer; ESCC, esophageal squamous cell carcinoma; PBMC, peripheral blood mononuclear cells; TNM, tumor nodes metastases; nCRT, neoadjuvant chemoradiotherapy; BM, bone marrow; STAT1, signal transducer and activator of transcription 1; NLR, neutrophil‐to‐lymphocyte ratio.
5. MDSCS AS A POTENTIAL PROGNOSTIC MARKER IN GI CANCER
Several clinical studies have suggested a strong relationship between prognosis and an increased frequency of MDSCs in the peripheral blood or tumor tissue of patients with GI cancer. For example, in HCC patients, a high MDSC frequency is correlated with the early recurrence after resection [123]. For CRC patients, a number of studies have demonstrated that the frequency of circulating MDSCs is correlated with reduced overall survival both before and after chemotherapy and radiotherapy, and this correlation was of clinical relevance [124]. A study including both healthy donors and ESCC patients showed that MDSC levels (CD11b+CD14+HLA–DR–) in the PBMCs of patients with stage IIIb or IV tumors were increased compared with those of patients with stage II or IIIa disease [125]. Also, the authors reported that 14 of the 23 patients (60.9%) in the low MDSC frequency group were responders, while only 1 of 12 patients (8.3%) in the high MDSC frequency group responded to the chemotherapy [125]. A further clinical report showed that increased peripheral blood MDSCs level was correlated with a 22% increased risk of death, based on data from 60 patients with EC, 25 patients with GC, and 54 healthy controls [13]. Another study also showed the correlation among MDSC frequency, cancer stage, and survival rates of GC patients [52]. Current studies concerning the relationship between the frequency of MDSCs and cancer progression, as well as prognosis, are summarized in Table 1. These findings suggest that the frequency of MDSCs could be used as a prognostic marker in patients with GI cancer and may also be a useful tool when selecting individual treatment regimens.
6. MDSC‐TARGETED STRATEGIES FOR TREATING GI CANCER
As described above, many studies have demonstrated that the existence of MDSCs in the tumor microenvironment remained an obstacle for the efficacy of cancer treatment. Fortunately, to date, many therapeutic approaches against GI cancer by targeting MDSCs, including small molecules, vitamins, conjugates, nucleotide, and immunotherapy, have been developed. These therapies work by affecting the accumulation, recruitment, differentiation, and immunosuppression of MDSCs (Table 2).
TABLE 2.
MDSC‐targeted strategies for treating GI cancer
| Therapeutic compound | Target or drug type | Targeted process | Tumor model | Implication | Reference a |
|---|---|---|---|---|---|
| NCX‐4016 | Small molecule | NO aspirin affects ARG1 activity and iNOS activity of MDSCs in tumor‐bearing mice. | CRC (mice) | Inhibition of the immunosuppression of MDSCs. | De Santo et al., 2005. [161] |
| Sildenafil | Small molecule | Inhibition of iNOS and ARG1 activity by downregulation of IL‐4Ra. | CRC, MA and melanoma (mice) | Inhibition of the immunosuppression of MDSCs. | Serafini et al., 2006. [154] |
| Celecoxib | Small molecule | Celecoxib decreased the levels of MDSCs. | CRC (mice) | Depletion of MDSCs. | Talmadge et al., 2007. [177] |
| Gemcitabine | Small molecule | Eliminating MDSCs via a cytotoxic pathway. | CRC and BC (mice) | Depletion of MDSCs. | Ko et al., 2007. [178] |
| L‐NAME | Small molecule | L‐NAME affects ARG1 activity of MDSCs in tumor‐bearing mice. | CRC and lymphoma (mice) | Inhibition of the immunosuppression of MDSCs. | Capuano et al., 2009. [179] |
| Decitabine | Small molecule | Decitabine promoted CD11b cells differentiation into mature F4/80/ CD11c/MHC class II‐positive APCs, with reduced IL‐13, IL‐10, PGE2, VEGF, MMP‐9, IL‐1β, IL‐6 and MIP‐2 secretion. | CRC (mice) | Differentiation of MDSCs into mature cells. | Daurkin et al., 2010. [180] |
| Triterpenoid | Small molecule | Triterpenoid mediated inhibition of ROS reduction of MDSCs. | CRC, Thymoma, LLC (mice) | Inhibition of the immunosuppression of MDSCs. | Nagaraj et al., 2010. [162] |
| Dimethyl amiloride | Small molecule | Inhibition MDSCs suppressive capacity via reduced exosome secretion. | CRC and lymphoma (mice); metastatic CRC and LA (human) | Inhibition of the immunosuppression of MDSCs. | Chalmin et al., 2010. [181] |
| Sorafenib | Small molecule | Sorafenib downregulates the population of both MDSCs and Treg. | HCC (mice) | Depletion of MDSCs. | Cao et al., 2011. [182] |
| Curcumin | Small molecule | Stimulation of differentiation into M1 macrophages through inhibition of JAK2/ STAT3. | GC, CRC(mice) | Differentiation of MDSCs into mature cells. | Tu et al., 2012. [140] |
| Zoledronic acid | Small molecule | Zoledronic acid impairs the production and recruitment of MDSCs into the periphery and tumor site. | PA (mice) | Inhibition of MDSCs population and recruitment. | Porembka et al., 2012. [129] |
| Rosiglitazone | Small molecule | Rosiglitazone limits TGFβ and COX‐2 expression in the tumor microenvironment, in turn reducing the expansion of MDSCs. | PA (mice) | Depletion of MDSCs. | Bunt et al., 2013. [183] |
| Sunitinib | Small molecule | Sunitinib treatment increased the efficacy of stereotactic body radiotherapy in patients by reversing the population and function of MDSCs. | Multiple cancer types, including HCC and CRC etc. (human) | Inhibition of the immunosuppression and accumulation of MDSCs. | Chen et al., 2015. [184] |
| PLX647 | Small molecule | Inhibition of CSF1R signaling functionally blocked tumor‐infiltrating MDSCs. | CRC and melanoma (mice) | Inhibition of MDSCs recruitment. | Holmgaard et al., 2016. [144] |
| SB225002 | Small molecule | Blockade of CXCR2 and CSF1R profound decreased PMN‐MDSC recruitment into tumor site. | CRC and other cancer types (mice and human) | Inhibition of MDSCs recruitment. | Kumar et al., 2017. [145] |
| 5‐FU | Small molecule | 5‐FU therapy selectively eliminates the sensitive MDSCs, not resistant MDSCs in a cytotoxic pathway. | CRC (mice) | Depletion of MDSCs. | Kelment et al., 2017. [185] |
| Tadalafil | Small molecule | Blockade of ARG1 and iNOS activity and accumulation of MDSCs in tumor microenvironment. | HCC (mice) | Inhibition of the immunosuppression and accumulation of MDSCs. | Yu et al., 2019. [158] |
| AMD3100 | Small molecule | AMD3100 inhibited MDSCs migration to the spleen and liver of the tumor‐bearing mice. | HCC (mice) | Inhibition of MDSCs recruitment. | Xu et al., 2019. [150] |
| SX‐682 | Small molecule | KRAS*‐mediated repression of IRF2 results in high expression of CXCL3. SX‐682 could inhibit CXCL3 binding to CXCR2 on MDSCs and promotes their migration to the tumor microenvironment. | CRC (mice) | Inhibition of MDSCs recruitment. | Liao et al., 2019. [186] |
| ATRA | Active metabolite of vitamin | ATRA promoted MDSCs converted into immunogenic APCs by IFN‐mediated activation and GSS‐mediated differentiation signals. | CRC (mice) | Differentiation of MDSCs into mature cells. | Lee et al.,2012. [187] |
| 1α,25‐Dihydroxyvitamin D3 | Vitamin D | Vitamin D could inhibit the IL‐6 signaling, reduce the MDSCs recruitment. | ESCC (mice and human) | Inhibition of MDSCs recruitment. | Chen et al., 2015. [139] |
| Bevacizumab | Antibody | Decreased accumulation of MDSCs in the peripheral blood of patients. | CRC, LA, and BC (Human) | Depletion of MDSCs. | Osada et al., 2008. [188] |
| Anti‐ENO1 antibodies | Antibody | Blockade of ENO1 on MDSCs surface could decrease the activity of Arginase. | PA (mice) | Inhibition of the immunosuppression of MDSCs. | Cappello et al., 2016. [163] |
| DS‐8273a | Antibody | Reduction of the elevated numbers of MDSCs in the peripheral blood. | CRC, HCC and other types of cancers (human) | Depletion of MDSCs. | Dominguez et al., 2017. [131] |
| KTN0158 | Antibody | Selectively reduces the immunosuppressive M‐MDSC population. | CRC and PA (mice) | Depletion of MDSCs. | Garton et al., 2017. [133] |
| Anti‐CCR2 mAb | Antibody | Blockade of CCR2 reduced radiation‐induced infiltration of M‐MDSC. | CRC and LA (mice) | Inhibition of MDSCs recruitment. | Liang et al., 2017. [148] |
| Anti S100A9 antibody | Antibody | Blockade of S100A9 could Inhibit MDSCs recruitment by down‐regulation of RAGE‐mediated p38 MAPK and TLR4‐mediated NF‐κB signalling pathway. | CRC (human) | Inhibition of MDSCs recruitment. | Huang et al., 2019. [152] |
| Gemtuzumab ozogamicin | Conjugates | Treatment of human MDSCs with gemtuzumab ozogamicin could deplete MDSCs by increasing p‐ATM and cell death. | CRC and PA (human) | Depletion of MDSCs. | Fultang et al., 2019. [134] |
| CpG | Nucleotide | CpG blocks immunosuppression by myeloid‐derived suppressor cells in tumor‐bearing mice | CRC and GC (mice) | Depletion of MDSCs. | Zoglmeier et al., 2011. [141] |
Information listed in the table are arranged in ascending chronological order in the different classifications of drug type.
Abbreviations: GI cancer, gastrointestinal cancer; MDSCs, myeloid‐derived suppressor cells; M‐MDSC, monocytic MDSCs; G‐MDSC, granulocytic MDSCs; CRC, colorectal cancer; GC, gastric cancer; HCC, hepatocellular carcinoma; BC, breast cancer; PA, pancreatic adenocarcinoma; ESCC, esophageal squamous cell carcinoma; LA, lung adenocarcinoma; MA, mammary adenocarcinoma; LLC, Lewis lung carcinoma; IL‐10, interleukin 10; IL‐6, interleukin 6; iNOS, inducible nitric oxide synthase; NO, nitric oxide; ARG1, arginase I; IL‐4a, Interleukin 4a; L‐NAME, L‐NG‐Nitroarginine methyl ester; APCs, antigen presenting cells; IL‐13, interleukin 13; PGE2, prostaglandin E2; VEGF, vascular endothelial growth factor; MMP‐9, Matrix metallopeptidase 9; IL‐1, interleukin 1; MIP‐2, macrophage inflammatory protein 2; ROS, reactive oxygen species; Treg, regulator T‐cell; JAK2, Janus kinase 2; TGFβ, transforming growth factor β; COX‐2, cyclooxygenase‐2; CSF1R, colony stimulating factor 1 receptor; CXCR2, C‐X‐C chemokine receptor 2; 5‐FU, 5‐fluorouracil; KRAS, Kirsten RAt Sarcoma; IRF2, interferon regulatory transcription factor; CXCL3, C‐X‐C motif chemokine ligand 3; CXCR2, C‐X‐C motif chemokine receptor 2; ATRA, all‐trans retinoic acid; IFN, Interferon; GSS, glutathione synthase; ENO1, α‐enolase; CCR2, C‐C motif chemokine receptor 2; S100A9, S100 calcium‐binding protein A9; MAPK, mitogen‐activated protein kinase; TLR4, Toll Like Receptor 4; NF‐κB, nuclear factor kappa‐B; ATM, Ataxia telangiectasia mutated kinase; TLR9, Toll Like Receptor 9.
6.1. Depletion of MDSCs
6.1.1. Gemcitabine, 5‐fluorouracil, and oxaliplatin
Cytotoxic agents are widely used for cancer treatment. In addition to their direct cytotoxic effects, these agents harness the host's immune system, which contributes to their antitumor activity. In preclinical models, the two chemotherapy drugs which have been demonstrated to directly reduce the number of MDSCs are gemcitabine and 5‐FU [126]. Recent studies demonstrated that gemcitabine and 5‐FU did not directly reduce the number of MDSCs in cancer patients. For instance, Annels et al. [126] reported that in advanced PC patients receiving gemcitabine and capecitabine (a 5‐FU pro‐drug), only 42% of patients (n = 19) were found with reduced MDSCs. However, when treated with combined gemcitabine and capecitabine together with administered GV1001 vaccine and adjuvant GM‐CSF, the percentage of MDSCs was significantly reduced in 18 of 21 advanced PC patients (86%).
A similar study from the Wang's group showed that the effect of combination of 5‐FU and oxaliplatin with anti‐PD1 antibody could significantly reduce the population of MDSCs and increase intra‐tumor CD8+ T cells, which is better than the treatment of anti‐PD1 antibody alone. However, these cytotoxic agents also induce expression of PD‐L1 by gastric epithelial cells, and those expressing PD‐L1 cells were more susceptible to tumorigenesis in mice, with accumulation of MDSCs [127]. Like other combination strategies, the choice of combination of cytotoxic agents with immunotherapy should be considered in the designe of therapeutic regimens to eliminate of MDSCs.
The effect of cytotoxic agents on MDSCs is more complex than simple induction of MDSC apoptosis or death. Recently, one study from François Bruchard et al. [128] demonstrated chemoresistance mediated by 5‐FU and gemcitabine and the relative mechanism of action. Evidence showed that gemcitabine and 5‐FU could deplete immunosuppressive MDSCs but also induce the release of cathepsin B from lysosomes and cause IL‐1β secretion from MDSCs via the activation of the NOD‐like receptor family pyrin domain containing‐3 protein (Nlrp3)‐dependent caspase‐1, resulting in IL‐17 production by T cells that curtails anticancer immunity. This result may explain the limits of using these cytotoxic agents such as 5‐FU and gemcitabine in antitumor treatment.
6.1.2. Zoledronic acid
MDSCs are important mediators of tumor‐induced immunosuppression in PC. Porembka et al. [129] showed that PC patients demonstrated increased frequency of MDSCs in the bone marrow and peripheral circulation. In the murine pancreatic tumor model, the evidence revealed that treatment with zoledronic acid reduced the expansion and recruitment of MDSCs in the tumor site and improved the host anti‐tumor response by increasing recruitment of T cells to the tumor with increased secretion of IFN‐γ and decreased levels of IL‐10.
6.1.3. Antibody and conjugated drugs
Importantly, Condamine et al. [130] demonstrated that compared with neutrophils and monocytes, MDSCs displayed a shorter lifespan with an increased apoptosis rate in the periphery, which was correlated with the expression of TNF‐related apoptosis‐induced ligand receptors (TRAIL‐Rs) on their surface. It seems that targeting TRAIL‐Rs by selective agonists would be an option for cancer therapy by reducing the population of MDSCs. Later, Dominguez et al. [131] tested the TRAIL receptor 2 agonist (DS‐8273a), which targeted elimination of MDSCs selectively in 16 advanced cancer patients including colorectal, pancreatic, and liver cancer patients. The treatment with DS‐8273a resulted in reduction of the number of MDSCs in the peripheral blood of most patients without affecting the numbers of neutrophils, monocytes, and other populations of myeloid and lymphoid cells. The author also showed that the decreased number of MDSCs was inversely correlated with the length of progression‐free survival. In tumor tissues, DS‐8273a treatment resulted in a decrease of MDSCs in 50% of the patients who provided pre‐ and on‐treatment biopsies. However, in several patients, MDSCs rebounded back to the pretreatment level by day 42.
It is reported that tumor‐derived stem‐cell factor (SCF) plays an important role in the expansion and accumulation of MDSCs. A study by Pan et al. [132] showed that in a colon tumor model, knockdown SCF or blockade of SCF receptor (c‐kit) could significantly reduce the MDSC expansion, leading to restored proliferative response of tumor‐infiltrating T cells, suppressed Treg development, and inhibited tumor angiogenesis. Evidence has shown that further prevention of the MDSC accumulation in conjunction with immune activation therapy showed synergistic therapeutic effect in a colon tumor model.
Another study from Garton et al. [133] targeted the stem cell factor receptor (KIT), which is an established oncogenic driver of tumor growth in gastrointestinal stromal tumors (GIST). Evidence showed that anti‐KIT mAb treatment could selectively reduce the immunosuppressive M‐MDSC population and restore the population of CD8+ and CD4+ T cells in the CT26 tumor‐bearing mice. Also anti‐KIT mAb treatment enhanced the anti‐tumor effect of anti‐CTLA‐4 and anti‐PD‐1 mAbs by promoting immune responses, which showed a rationale for investigation of the KIT‐specific mAb combined with immune checkpoint inhibitors and other immunotherapeutic agents in clinic.
More recently, Fultung et al. [134] reported the antibody‐drug conjugate, named Gemtuzumab ozogamicin, on the depletion of MDSCs in GI cancer (pancreas and colon cancers). Treatment of human MDSCs with Gemtuzumab ozogamicin could deplete MDSCs by increasing the activity of p‐ataxia telangiectasia mutated kinase (ATM) and cell death, leading to restoring T‐cell proliferation and chimeric antigen receptor (CAR)‐T‐cell activity.
6.2. Differentiation of MDSCs into mature cells
6.2.1. ATRA
All‐trans retinoic acid (ATRA) is a metabolite of vitamin A, which differentiates MDSCs into DCs, granulocytes, and monocytes [135]. ATRA treatment also reduces ROS production to improve the CTL‐mediated immune response in both cancer patients and tumor‐bearing mice through the activation of extracellular signal‐regulated kinase [136]. For instance, Nefedova et al. [136] showed that ATRA could specifically up‐regulate gene expression and protein level of glutathione synthase (GSS) in MDSCs, leading to the MDSC differentiation in CT26 and MC38 tumor‐bearing mice.
6.2.2. Vitamin D3
It is generally believed that, vitamin D3, similar to ATRA, also differentiates MDSCs and improves the antitumor immune response [137]. GI cancer research on vitamin D was mainly related to its role in inhibiting cancer cell proliferation, metastasis, and invasion or killing cancer cells by cell cycle arrest and apoptosis [138]. For example, a clinical trial using the combination of vitamin D with bevacizumab in patients with advanced or metastatic CRC is recruiting (clinical trail: NCT04094688). It was reported that vitamin D could reduce the recruitment of MDSCs in ESCC. In a 4‐nitroquinoline 1‐oxide (4‐NQO)‐induced esophageal tumor animal model, Chen et al. [139] found that vitamin D could inhibit the IL‐6 signaling, reduce MDSCs recruitment, and decrease the incidence of invasive esophageal tumors. Little is known about vitamin D on the regulation of MDSC differentiation in GI cancer, except their role on MDSC recruitment. More researches should be done to demonstrate the role vitamin D plays on the differentiation of MDSCs into mature cells in GI cancer.
6.2.3. Curcumin
Curcumin was also reported to inhibit the accumulation and promote the differentiation of MDSCs in some cancer types. A research from Wang's group examined the effects of curcumin on the activation and differentiation of MDSCs and their interaction with tumor growth. They found that treatment with curcumin could significantly reduce the percentage of MDSCs in the tissues of spleen, blood, and tumor in a human GC xenograft model and a mouse CRC allograft model, as well as decrease the secretion of IL‐6 by MDSCs in a co‐culture system. Furthermore curcumin treatment differentiated MDSCs toward M1‐like macrophages with an increased expression of CCR7 in vivo and in vitro. The evidence showed that the mechanism of curumin‐related MDSC differentiation was due to the activation of STAT3 and NF‐κB in MDSCs [140].
6.2.4. TLR9 ligands
Previously, Bourquin investigated the effect of TLR9 ligand, CpG motif‐containing oligodeoxynucleotides (CpG ODN) treatment on the proliferation and function of MDSCs in CT26 tumor‐bearing mice and in CEA424‐TAg mice bearing autochthonous gastric tumor. Evidence showed that activation of TLR9 receptor by CpG ODN significantly promoted maturation and differentiation of MDSCs and decreased the proportion of G‐MDSCs in both tumor‐bearing and tumor‐free mice. Furthermore, MDSC maturation and suppressive function inhibition were induced by IFN‐α secreted by plasmacytoid dendritic cells (pDCs) in vitro; treatment of mice with recombinant IFN‐α is sufficient to block MDSCs’ suppressive function in vivo [141]. However, the regulation of IFN‐α on the differentiation and function of MDSCs needs to be further elucidated.
6.3. Inhibition of MDSC recruitment
6.3.1. Anti‐CSF1R antibody
A well‐documented molecule that regulates the maturation and recruitment of MDSCs is the colony‐stimulating factor 1 receptor (CSF1R), whose expression is restricted to monocytes and macrophages. Various CSF1R inhibitors have shown the inhibition of the trafficking of M‐MDSC and TAMs and also have been tested in combination with chemo‐ or immunotherapies both in mice and patients [142, 143]. For instance, Holmgaard et al. [144] tested the inhibitor of CSF1R, PLX647, in a CT26 xenograft model and found that inhibition of CSF1R signaling could functionally block MDSCs trafficking to the tumor site, leading to an enhanced anti‐tumor T‐cell response. Furthermore, evidence showed that inhibition of CSF1R improved the combined CTL‐4 and PD‐1 antibodies therapy for the IDO‐expressing tumors.
Another example from Gabrilovich's group showed that combination of CSF1R inhibitor with CXCR2 inhibitor demonstrated a strong antitumor effect in a CT26 xenograft model and patients with stage III‐IV colon adenocarcinoma. Evidence showed that treatment with CSF1R inhibitor, JNJ‐40346527, disrupted cross talk between cancer cells and CAFs. Further treatment combined with CXCR2 antagonist triggered a profound decrease in PMN‐MDSC recruitment to tumor. Based on these studies, it seems that combination of CSF1R blockage with immunotherapy should be considered a treatment regimen for eliciting tumor regression [145].
6.3.2. Anti‐CCR2 antibody
CCR2 receptor is commonly expressed on the surface of MDSCs, particularly for the subset of M‐MDSC. CCR2+ M‐MDSCs are commonly found infiltrating various types of cancers and facilitating tumor cell progression. CCR2 ligands (CCL2, CCL7, and CCL12) are produced by various types of cancer cells, including GI cancer [146]. Targeting CCR2 ligand by a neutralized antibody reduced the trafficking of MDSCs into tumor sites in many studies [108, 147]. Recently, a study from Weichselbaum's group showed that treatment with an anti‐CCR2 antibody or germ‐line knockout of CCR2 could significantly block radiation‐induced infiltration of M‐MDSC in a MC38 tumor model. The combination treatment with anti‐CCR2 antibody could abrogate the immunosuppressive effect of radiotherapy or STING agonists by elimination of M‐MDSC infiltration [148].
6.3.3. CXCR2 and CXCR4 antagonists
Except for CCR2 expression on MDSCs, other chemoattractant proteins, such as CXCR2 and CXCR4, are expressed on the surface of MDSCs. Targeting these two receptors could also lead to an inhibition of MDSC chemotaxis. For instance, Katoh et al. [149] presented the genetic evidence that loss of CXCR2 dramatically inhibited the infiltration of MDSCs into colonic mucosa and tumors in a colitis‐associated cancer mouse model, suppressing chronic colonic inflammation and colitis‐associated tumorigenesis. The evidence also showed that reduced trafficking of MDSCs by loss of CXCR2 enhanced the CD8+ T‐cell cytotoxic activity. Another study from Xu et al. [150] found that hepatic stellate cells (HSCs) could induce MDSCs migration through the stromal cell‐derived factor 1 (SDF‐1)/CXCR4 axis both in vivo and in vitro. Further evidence showed that in an orthotopic mouse liver tumor model, pre‐treatment of MDSCs with AMD3100 (CXCR4 inhibitor) significantly inhibited MDSC migration to the spleen and liver in the tumor‐bearing mice, revealing an effective approach for modulating the tumor microenvironment by targeting CXCR4.
6.3.4. S100A8 and S100A9 inhibitors
S100A8/A9 are pro‐inflammatory mediators, which are abundant at inflammatory or tumor sites. Their levels in the blood were correlated with the frequency, function, and migration of MDSCs in GI cancer patients [52, 119]. Originally, Qin et al. [151] employed a competitive peptide phage display platform to identify candidate peptides binding MDSCs specifically and generated peptide‐Fc fusion proteins (peptibodies). Treatment with peptibody in multiple tumor models showed an inhibition of tumor growth in vivo by completely depleting blood, splenic, and intratumoral MDSCs in tumor‐bearing mice, instead of affecting other immune cells. Later evidence of immunoprecipitation showed that S100 family proteins were the candidate targets of peptibody among the other MDSC surface proteins. A recent study from Duan's group further confirmed that S100A9 protein could stimulate activation and chemotaxis of MDSCs, instead of their viability, through an activation of RAGE‐mediated p38 MAPK and TLR4‐mediated NF‐κB signaling pathways. Targeting S100A9 protein with an antibody could significantly reduce the immunosuppressive TME by decreasing MDSC chemotaxis and activation, thereby showing an optimal therapeutic effect [152].
6.4. Targeting the immunosuppression of MDSCs
6.4.1. PDE‐5 inhibitors
Traditionally, phosphodiesterase‐5 (PDE‐5) inhibitors such as sildenafil (Viagra), vardenafil (Levitra), and tadalafil (Cialis) have been used therapeutically to treat erectile dysfunction [153], pulmonary hypertension [154], and cardiac hypertrophy [155] and induce apoptosis in different human cancers [156, 157]. For example, Serafini et al. [154] reported that the use of PDE‐5 inhibitors (sildenafil and tadalafil) could modulate the antitumor immune response in a murine colon tumor model. They found that the restored immune response was induced by the down‐regulation of the ARG1 and NOS2 expression as well as recruitment of MDSCs by tumors, which substantially delayed tumor progression.
As PDE‐5 inhibitors have a significantly effect on modulating the immunosuppressive function of MDSCs, some studies employed the PDE‐5 inhibitors as a combination agent for cancer therapy. Recently, Yu et al. [158] reported that after adoptive cell transfer of cytokine‐induced killers (CIKs) into hepatocellular tumor‐bearing mice, the population of MDSCs was significantly increased, which impaired the antitumor efficacy by their immunosuppressive function. Evidence showed that treatment with a PDE‐5 inhibitor, tadalafil, rescued the efficacy of CIK cell therapy by reversing the immunosuppressive function via ARG1 and iNOS blockade and accumulation of MDSCs in the tumor microenvironment. Further comparison between the ARG1 inhibitor L‐NOHA [159] or iNOS inhibitor L‐NMMA [160] and tadalafil in CIK cell therapy showed a similar effect of L‐NOHA plus L‐NMMA to tadalafil alone, which further indicates tadalafil functions through the blockade of ARG1 and iNOS together [158].
6.4.2. Nitroaspirin
A similar study from De Santo et al. [161] reported that nitroaspirin corrected immune dysfunction in CT26 tumor‐bearing mice and promoted tumor eradication, which relied on the interference of the immunosuppression of MDSCs to T lymphocyte proliferation and function. The decreased MDSC immunosuppressive function was due to the administration of nitroaspirin, which could inhibit the activity of ARG1 and iNOS.
6.4.3. Triterpenoid
In a murine MC38 tumor host, Nagaraj et al. [162] reported that administration of triterpenoid could eliminate the immune suppressive effect of MDSCs and improve immune responses in tumor‐bearing mice as well as PC patients. The mechanism of triterpenoid‐mediated inhibition of immunosuppressive effect of MDSCs relies on the reduction of ROS, rather than the MDSCs viability or level of ARG1 and NO.
6.4.4. Anti‐ENO1 antibody
As reported before, α‐enolase (ENO1) was expressed on the surface of MDSCs, which increased after LPS stimulation. Cappello et al. [163] found that in the pancreatic ductal adenocarcinoma model, ENO1 mAb treatment could decrease the ARG1 activity of MDSCs, while activated T cells in the anti‐ENO1 mAb‐treated group increased IFN‐γ and IL‐17 secretion and decreased IL‐10 and TGFβ secretion compared to those in the control group. Therefore, anti‐ENO1 antibodies may not only inhibit MDSCs infiltrating into the tumor microenvironment but also attenuate their restraining of effector T‐cell response.
6.5. Potential molecules which could regulate the accumulation, recruitment, and immunosuppression of MDSCs
Apart from existing molecules that have been designed as drugs for GI cancer, we have also reviewed related studies in the past decade and summarized potential molecules, which could affect the accumulation, recruitment, and immunosuppression of MDSCs in Table 3.
TABLE 3.
Factors implicated in the expansion and recruitment of MDSCs in GI cancer
| Target | Cancer Model | Source | Mechanism | Reference a |
|---|---|---|---|---|
| IL‐10 | CRC | Tumor microenvironment and spleen | IL‐10 deficiency increases MDSCs accumulation in the spleen and tumor. | Tanikawa et al., 2012. [189] |
| CEACAM1 | CRC | Liver | Ceacam1 deficiency diminished CD11b+Gr1+MDSCs recruitment to the metastatic liver. | Arabzadeh et al., 2013. [190] |
| IL‐6 | ESCC | Peripheral blood | MDSCs recruitment was associated with invasive esophageal tumors and with increased IL‐6 levels. | Chen et al., 2014. [125] |
| CD38 | EC | Spleen | CD38 could promote monocytic MDSCs population expansion and regulate iNOS expression. | Karakashera et al., 2015. [24] |
| CCL2 | CRC | Colon adenocarcinoma tissue | CCL2 regulates G‐MDSC accumulation and T‐cell suppressive activity via STAT3. | Chun et al., 2015. [191] |
| CD40 | GC | Spleen and tumor tissue | CD40 expression upregulates the chemokine receptor CXCR5 and promotes MDSCs migration and accumulation. | Ding et al., 2015. [192] |
| G‐CSF | CAC | Colon tissues | G‐CSF could promote MDSCs survival and activation through the STAT3 signaling pathway. | Li et al., 2016. [62] |
| CCL15 | CRC | Tumor tissue | CCL15‐CCR1+ axis promotes MDSCs accumulation in the tumor microenvironment. | Inamoto et al., 2016. [114] |
| S1pr3 | CRC | Peripheral blood, spleen and bone marrow | GM‐CSF promotes MDSCs via S1pr3 through Rho kinase and the extracellular signal‐regulated kinase‐dependent pathway. | Li et al., 2017. [193] |
| STAT6 | Intestinal tumorigenesis | Spleen and lamina propria | STAT6 promoted expansion of MDSCs in the spleen and lamina propria of ApcMin/+ mice, implying regulation of antitumor T‐cell response. | Jayakumar et al., 2017. [194] |
| VEGF‐A/CXCL1 | CRC | Liver | VEGF‐A ‐CXCL1‐CXCR2 recruits MDSCs to form a pre‐metastatic niche. | Wang et al., 2017. [109] |
| GM‐CSF | CRC | Colon tissues | GM‐CSF was sufficient to differentiate hematopoietic precursors into MDSCs. | Ma et al., 2017. [195] |
| CCR5 | GC | Periphery and tumor | CCL5‐CCR5 axis recruits MDSCs, and blocks CCR5 to reduce the accumulation of MDSCs and enhances anti‐PD1 efficacy. | Yang et al., 2018. [196] |
| Acid ceramidase | CAC | Tumor tissue | Acid ceramidase protects from tumor incidence in colitis‐associated cancer and inhibits the expansion of neutrophils and G‐MDSC in the tumor microenvironment. | Espaillat et al., 2018. [197] |
| RIPK3 | CRC | Colorectal tumor tissues | In MDSCs, PGE2 suppressed RIPK3 expression and enhanced NF‐κB and COX‐2 expression, which catalyzed PGE2 synthesis. | Yan et al., 2018. [198] |
| CXCL4 | CRC | Tumor tissues and peritoneal cavity | Surgical trauma contributes to colon cancer progression by downregulating CXCL4 and hence promoting MDSCs recruitment, which leads to an immunosuppressive environment. | Xu et al., 2018. [199] |
| CXCR4 | CAC | Colon tissue | CXCR4 overexpression promotes the infiltration of bone marrow‐derived MDSCs. | Yu et al., 2019. [200] |
| DCHLL | CRC | Tumor, blood and bone marrow | Blocking DC‐HIL function is a potentially useful treatment for at least colorectal cancer with high blood levels of DC‐HIL+MDSCs. | Kobayashi et al., 2019. [164] |
| STAT3 | HCC | Liver | Inhibition of STAT3, p‐STAT3, upregulation of the pro‐apoptotic proteins Bax, cleaved caspase‐3, and downregulation of the anti‐apoptotic protein Bcl‐2. | Guha et al., 2019. [201] |
| PAR2 | CAC | Tumor tissue | Absence of PAR2 in MDSCs directly enhanced their immunosuppressive activity by promoting STAT3‐mediated ROS production. | Ke et al., 2020. [202] |
Information listed in the table are arranged in ascending chronological order.
Abbreviations: GI cancer, gastrointestinal cancer; MDSCs, myeloid‐derived suppressor cells; G‐MDSCs, granulocytic MDSCs; CRC, colorectal cancer; GC, gastric cancer; HCC, hepatocellular carcinoma; ESCC, esophageal squamous cell carcinoma; CAC, colitis‐associated colorectal cancer; IL‐10, interleukin 10; CEACAM1, carcinoembryonic antigen‐related cell adhesion molecule 1;IL‐6, interleukin 6; iNOS, inducible nitric oxide synthase; CCL2, C–C motif chemokine ligand 2; STAT3, signal transducer and activator of transcription 3; CXCR5, C–X–C chemokine receptor 5; G‐CSF, granulocyte colony‐stimulating factor;CCL15, C–C motif chemokine ligand 15; CCR1, C–C motif chemokine receptor 1; S1pr3, S1P receptor 3; GM‐CSF, granulocyte macrophage colony‐stimulating factor; STAT6, signal transducer and activator of transcription 6; VEGFA, vascular endothelial growth factor A; CXCL1, C–X–C motif chemokine ligand 1; CXCR2, C–X–C motif chemokine receptor 2; GM‐CSF, granulocyte macrophage colony‐stimulating factor;CCR5, C–C motif chemokine receptor 5; RIPK3, receptor‐interacting protein kinase 3; PGE2, prostaglandin E2; NF‐κB, nuclear factor kappa‐B; COX‐2, cyclooxygenase‐2; CXCL4, C–X–C motif chemokine ligand 4; CXCR4, C–X–C motif chemokine receptor 4; DC‐HIL, dendritic cell‐associated heparan sulfate proteoglycan‐dependent integrin ligand; Bax, Bcl‐2‐associated X; Bcl‐2, B‐cell lymphoma‐2; PAR2, protease activated receptor 2.
For instance, in 2019, Kobayashi et al. [164] studied metastatic CRC patients and found that the frequency of DC‐HIL+ MDSCs was highly elevated in the peripheral blood. Treatment with anti‐DC‐HIL mAb reduced MDSC population and increased IFN‐γ‐secreting T cells in the tumor microenvironment (with similar outcomes to anti‐PD‐L1 mAb), achieving the effect of attenuated pre‐established colon metastasis. Apart from direct regulation of MDSCs with antibody or small molecules in GI cancer, the indirectly mechanism that regulated MDSCs was also recorded. Previously, Zhuang et al. [165] found that CD8+ T cells with the production of IL‐17 (Tc17 cells) promoted MDSC migration by inducing CXCL12 production of tumor cells, resulting in impaired antitumor CD8+ cytotoxic T cells. The development of Tc17 cells was induced by the cytokines (IL‐6, IL‐1β and IL‐23) secreted from tumor‐activated monocytes, and it showed that percentages of Tc17 cells and MDSCs in gastric tumors were associated with survival time of patients.
Based on current studies that clearly record the mechanism of MDSC regulation in GI cancer models, clinical studies for GI cancer patients that target MDSC depletion, recruitment, and immunosuppression are underway or have been completed. Tested drugs include small molecule, antibody, vaccine, biotech, and oligonucleotides and are summarized in the following section.
7. CURRENT CLINICAL TRIALS IN GI CANCER TARGETING MDSCS
Based on targeting MDSCs, several therapies in GI cancer have been undergone or completed clinical trials. Here, we reviewed the website of clinicaltrials.gov and selectively summarized the therapeutic approaches involving MDSCs by different treatment regiments for GI cancer in Table 4.
TABLE 4.
Current clinical trials in GI cancer targeting MDSCs
| Compound | Type of compound | Intervention | Therapeutic target | Tumor type (Only showed GI Cancer types) | Clinical phase | Trial status | Clinical trial No. |
|---|---|---|---|---|---|---|---|
| Vicriviroc | Small molecule | Pembrolizumab | CCR5 | CRC | Phase 2 | Active, not recruiting | NCT03631407 |
| SX‐682 | Small molecule | Nivolumab | CXCR1/2 | Metastatic CRC | Phase 1 | Recruiting | NCT04599140 |
| INCB001158 | Small molecule | Pembrolizumab | Arginase | CRC and GC | Phase 1 | Active, not recruiting | NCT02903914 |
| L‐BLP25 | Vaccine | CPA+Chemoradiotherapy | MUC1 | RC | Phase 2 | Completed | NCT01507103 |
| ARG1 peptides | Vaccine | N/A | Arginase | CRC | Phase 1 | Recruiting | NCT03689192 |
| VXM01 | Vaccine | N/A | VEGFR2 | Metastatic CRC | Phase 1 | Completed | NCT02718430 |
| DS‐8273a | Antibody | Nivolumab | TRAIL‐R2 | CRC | Phase 1 | Terminated | NCT02991196 |
| Anakinra | Biotech | LV5FU2 + Bevacizumab | VEGF‐A | Metastatic CRC | Phase 2 | Completed | NCT02090101 |
| AZD9150 | Antisense Oligonucleotide | N/A | STAT3 | GI cancer | Phase 2 | Terminated | NCT02417753 |
Abbreviations: GI cancer, gastrointestinal cancer; CRC, colorectal cancer; GC, gastric cancer; RC, rectal cancer; CCR5, C‐C chemokine receptor 5; CXCR1/2, C–X–C motif chemokine receptor 1/2; MUC1, mucin 1; VEGFR2, vascular endothelial growth factor receptor 2; TRAIL‐R2, TNF‐related apoptosis‐inducing ligand receptor 2; VEGF‐A, vascular endothelial growth factor A; STAT3, signal transducer and activator of transcription 3.
For instance, a completed phase II clinical trial (NCT02090101) involved 32 participants to evaluate the influence of Anakinra on the vascularization of liver metastases by targeting VEGFA in metastatic CRC patients. Similarly, six participants were enrolled into a phase I clinical trial of VXM01 (NCT02718430), which is targeting vascular endothelial growth factor receptor 2 (VEGFR2). This completed trial was set up to examine safety, efficacy, and immune biomarkers in patients with metastatic CRC. Both the two clinical trials employed MDSCs as a secondary outcome measure. Unfortunately, neither result of these two trials were posted on the website. Another completed phase II clinical trial (NCT01507103) enrolled 124 participants to determine the impact of tecemotide (L‐BLP25) administration on the mucinous glycoprotein 1 (MUC1)‐specific immune response in subjects with newly diagnosed rectal cancer who are eligible for neoadjuvant therapy. In this clinical trial, the population of MDSCs (CD33+CD14–) and other immune cells such as NK cells, macrophages, and DCs were set as secondary outcome measures. The treatment showed a total risk of 25.64%, but the population change of MDSCs after treatment was not found from their posted results. The limitation of this study was that not all efficacy data were analyzed as no acceptable ELISpot assay is available, which led to the discontinuation of the development of tecemotide (L‐BLP25) in September 2014. Currently, there are not enough completed clinical trials that specifically target MDSCs in GI cancer, evidencing that more work should be done around this field.
8. CONCLUSIONS AND FUTURE PROSPECTS
As described in this review, MDSCs modulate several different aspects of tumor growth and metastasis, including immunosuppression, local invasion, and PMN formation. This broad range of functions suggests that these cells could be used as targets to develop both innovative liquid biopsy‐based cancer diagnostics and novel anti‐cancer therapeutic approaches. In future, researches investigating the regulatory functions of MDSCs in GI cancer should perhaps focus on their interactions with stromal cells from the tumor microenvironment, such as CAFs, because their role in the MDSC lifecycle remains to be fully elucidated. Furthermore, one of the most promising treatment approaches appears to be the promotion of MDSC differentiation into mature myeloid cells, as these lose their immunosuppressive functions. High‐throughput technologies will be of particular use as these allow the full characterization of the genetic, epigenetic, and metabolic pathways underlying the cross‐talk between MDSCs and cancer cells, as well as identifying relevant molecules. This will likely clarify some unsolved aspects of the interaction between MDSCs and GI cancer cells, laying the groundwork for more effective therapeutic strategies in the future. Finally, unlike in malignancies such as lung, renal, and skin cancers, the efficacy of immunotherapeutic agents on GI cancer has, on the whole, been much less remarkable and do not apply to the majority [166]. This could be explained by the permeated MDSCs in the tumor microenvironment, which eliminated the response of activated T cells generated by PD1/PD‐L1 blockade. Therefore, the possibility of combining checkpoint‐based immunotherapy with approaches targeting MDSCs may also represent a promising avenue in the development of more personalized treatments for GI cancer.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS' CONTRIBUTIONS
Conception/design: CC and LF. Manuscript preparing and writing: CC. Tables preparation: CC and PL. Manuscript writing and editing: LF. All authors read and approved the final manuscript.
ACKNOWLEDGEMENTS
This work was supported by grants from the National Natural Science Foundation of China (81772957, 82002546), the China Postdoctoral Science Foundation (2020M672815), the National Key R&D Program of China (2017YFA0503900), the Science and Technology Program of Guangdong Province in China (2019B030301009), the Industry and Information Technology Foundation of Shenzhen (20180309100135860), and the SZU Top Ranking Project (86000000210).
Cui C, Lan P, Fu L. The role of myeloid‐derived suppressor cells in gastrointestinal cancer. Cancer Commun. 2021;41:442–471. 10.1002/cac2.12156
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018;68(6):394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Ahmad A, Reha J, Saied A, Espat NJ, Somasundar P, Katz SC. Association of primary tumor lymph node ratio with burden of liver metastases and survival in stage IV colorectal cancer. Hepatobiliary surgery and nutrition. 2017;6(3):154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quante M, Varga J, Wang TC, Greten FR. The gastrointestinal tumor microenvironment. Gastroenterology. 2013;145(1):63‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hirbod‐Mobarakeh A, Mirghorbani M, Hajiju F, Marvi M, Bashiri K, Rezaei N. Myeloid‐derived suppressor cells in gastrointestinal cancers: A systematic review. Journal of gastroenterology and hepatology. 2016;31(7):1246‐56. [DOI] [PubMed] [Google Scholar]
- 5. Feng R‐M, Zong Y‐N, Cao S‐M, Xu R‐H. Current cancer situation in China: good or bad news from the 2018 Global Cancer Statistics? Cancer Communications. 2019;39(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim JH, Kim BJ, Kim HS, Kim JH. Current status and perspective of immunotherapy in gastrointestinal cancers. Journal of Cancer. 2016;7(12):1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao K, Wu J. National trend of gastric cancer mortality in China (2003‐2015): a population‐based study. Cancer Communications. 2019;39(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Waldron TJ, Quatromoni JG, Karakasheva TA, Singhal S, Rustgi AK. Myeloid derived suppressor cells: targets for therapy. Oncoimmunology. 2013;2(4):e24117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raghavan S, Quiding‐Järbrink M. Regulatory T cells in gastrointestinal tumors. Expert review of gastroenterology & hepatology. 2011;5(4):489‐501. [DOI] [PubMed] [Google Scholar]
- 10. Noy R, Pollard JW. Tumor‐associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gabrilovich DI, Nagaraj S. Myeloid‐derived suppressor cells as regulators of the immune system. Nature reviews immunology. 2009;9(3):162‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pak AS, Wright MA, Matthews JP, Collins SL, Petruzzelli GJ, Young M. Mechanisms of immune suppression in patients with head and neck cancer: presence of CD34 (+) cells which suppress immune functions within cancers that secrete granulocyte‐macrophage colony‐stimulating factor. Clinical Cancer Research. 1995;1(1):95‐103. [PubMed] [Google Scholar]
- 13. Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid‐derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin‐13. Cancer Immunology, Immunotherapy. 2011;60(10):1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmielau J, Finn OJ. Activated granulocytes and granulocyte‐derived hydrogen peroxide are the underlying mechanism of suppression of t‐cell function in advanced cancer patients. Cancer research. 2001;61(12):4756‐60. [PubMed] [Google Scholar]
- 15. Huang T‐X, Fu L. The immune landscape of esophageal cancer. Cancer Communications. 2019;39(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer‐expanded myeloid‐derived suppressor cells induce anergy of NK cells through membrane‐bound TGF‐β1. The Journal of Immunology. 2009;182(1):240‐9. [DOI] [PubMed] [Google Scholar]
- 17. Huang B, Pan P‐Y, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr‐1+ CD115+ immature myeloid suppressor cells mediate the development of tumor‐induced T regulatory cells and T‐cell anergy in tumor‐bearing host. Cancer research. 2006;66(2):1123‐31. [DOI] [PubMed] [Google Scholar]
- 18. Condamine T, Ramachandran I, Youn J‐I, Gabrilovich DI. Regulation of tumor metastasis by myeloid‐derived suppressor cells. Annual review of medicine. 2015;66:97‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889:571‐3. [PubMed] [Google Scholar]
- 20. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature. 2005;438(7069):820‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The role of myeloid‐derived suppressor cells in patients with solid tumors: a meta‐analysis. PloS one. 2016;11(10):e0164514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Limagne E, Euvrard R, Thibaudin M, Rébé C, Derangère V, Chevriaux A, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX–bevacizumab drug treatment regimen. Cancer research. 2016;76(18):5241‐52. [DOI] [PubMed] [Google Scholar]
- 23. Markowitz J, Brooks TR, Duggan MC, Paul BK, Pan X, Wei L, et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid‐derived suppressor cells upon progression of disease. Cancer Immunology, Immunotherapy. 2015;64(2):149‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karakasheva TA, Waldron TJ, Eruslanov E, Kim S‐B, Lee J‐S, O'Brien S, et al. CD38‐expressing myeloid‐derived suppressor cells promote tumor growth in a murine model of esophageal cancer. Cancer research. 2015;75(19):4074‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen M‐F, Chen P‐T, Kuan F‐C, Chen W‐C. The predictive value of pretreatment neutrophil‐to‐lymphocyte ratio in esophageal squamous cell carcinoma. Annals of Surgical Oncology. 2019;26(1):190‐9. [DOI] [PubMed] [Google Scholar]
- 26. Tcyganov E, Mastio J, Chen E, Gabrilovich DI. Plasticity of myeloid‐derived suppressor cells in cancer. Current opinion in immunology. 2018;51:76‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rossi J‐F, Céballos P, Lu Z‐Y. Immune precision medicine for cancer: a novel insight based on the efficiency of immune effector cells. Cancer Communications. 2019;39(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu T, Wu X, Wang H‐Y, Chen L. Immune contexture defined by single cell technology for prognosis prediction and immunotherapy guidance in cancer. Cancer Communications. 2019;39(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Terrazas LI, Walsh KL, Piskorska D, McGuire E, Harn DA. The schistosome oligosaccharide lacto‐N‐neotetraose expands Gr1+ cells that secrete anti‐inflammatory cytokines and inhibit proliferation of naive CD4+ cells: a potential mechanism for immune polarization in helminth infections. The Journal of Immunology. 2001;167(9):5294‐303. [DOI] [PubMed] [Google Scholar]
- 30. Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly‐Scumpia KM, et al. MyD88‐dependent expansion of an immature GR‐1+ CD11b+ population induces T cell suppression and Th2 polarization in sepsis. The Journal of experimental medicine. 2007;204(6):1463‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr‐1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. The Journal of Immunology. 2006;176(4):2085‐94. [DOI] [PubMed] [Google Scholar]
- 32. Zhu B, Bando Y, Xiao S, Yang K, Anderson AC, Kuchroo VK, et al. CD11b+ Ly‐6Chi suppressive monocytes in experimental autoimmune encephalomyelitis. The Journal of Immunology. 2007;179(8):5228‐37. [DOI] [PubMed] [Google Scholar]
- 33. Talmadge JE, Gabrilovich DI. History of myeloid‐derived suppressor cells. Nature Reviews Cancer. 2013;13(10):739‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gabrilovich DI. Myeloid‐derived suppressor cells. Cancer immunology research. 2017;5(1):3‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mandruzzato S, Brandau S, Britten CM, Bronte V, Damuzzo V, Gouttefangeas C, et al. Toward harmonized phenotyping of human myeloid‐derived suppressor cells by flow cytometry: results from an interim study. Cancer Immunology, Immunotherapy. 2016;65(2):161‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu T, Daniels CK, Cao S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacology & therapeutics. 2012;136(3):354‐74. [DOI] [PubMed] [Google Scholar]
- 37. Bronte V, Brandau S, Chen S‐H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid‐derived suppressor cell nomenclature and characterization standards. Nature communications. 2016;7(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor‐induced myeloid‐derived suppressor cell subpopulations with distinct T cell–suppressive activity. Blood. 2008;111(8):4233‐44. [DOI] [PubMed] [Google Scholar]
- 39. Dietlin TA, Hofman FM, Lund BT, Gilmore W, Stohlman SA, Van der Veen RC. Mycobacteria‐induced Gr‐1+ subsets from distinct myeloid lineages have opposite effects on T cell expansion. Journal of leukocyte biology. 2007;81(5):1205‐12. [DOI] [PubMed] [Google Scholar]
- 40. Youn J‐I, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid‐derived suppressor cells in tumor‐bearing mice. The Journal of Immunology. 2008;181(8):5791‐802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ding L, Hayes MM, Photenhauer A, Eaton KA, Li Q, Ocadiz‐Ruiz R, et al. Schlafen 4–expressing myeloid‐derived suppressor cells are induced during murine gastric metaplasia. The Journal of clinical investigation. 2016;126(8):2867‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ding L, Li Q, Chakrabarti J, Munoz A, Faure‐Kumar E, Ocadiz‐Ruiz R, et al. MiR130b from Schlafen4+ MDSCs stimulates epithelial proliferation and correlates with preneoplastic changes prior to gastric cancer. Gut. 2020;69(10):1750‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karakasheva TA, Dominguez GA, Hashimoto A, Lin EW, Chiu C, Sasser K, et al. CD38+ M‐MDSC expansion characterizes a subset of advanced colorectal cancer patients. JCI insight. 2018;3(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. T‐t Wang, Y‐l Zhao, L‐s Peng, Chen N, Chen W, Y‐p Lv, et al. Tumour‐activated neutrophils in gastric cancer foster immune suppression and disease progression through GM‐CSF‐PD‐L1 pathway. Gut. 2017;66(11):1900‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Peng L‐s, Zhang J‐y, Teng Y‐s, Zhao Y‐l, Wang T‐t, Mao F‐y, et al. Tumor‐associated monocytes/macrophages impair NK‐cell function via TGFβ1 in human gastric cancer. Cancer immunology research. 2017;5(3):248‐56. [DOI] [PubMed] [Google Scholar]
- 46. Mao F‐y, Zhao Y‐l, Lv Y‐p, Teng Y‐s, Kong H, Liu Y‐g, et al. CD45+ CD33 low CD11b dim myeloid‐derived suppressor cells suppress CD8+ T cell activity via the IL‐6/IL‐8‐arginase I axis in human gastric cancer. Cell death & disease. 2018;9(7):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. The Journal of clinical investigation. 2006;116(10):2777‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mills CD, Shearer J, Evans R, Caldwell MD. Macrophage arginine metabolism and the inhibition or stimulation of cancer. The Journal of Immunology. 1992;149(8):2709‐14. [PubMed] [Google Scholar]
- 49. Kusmartsev SA, Li Y, Chen S‐H. Gr‐1+ myeloid cells derived from tumor‐bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. The Journal of Immunology. 2000;165(2):779‐85. [DOI] [PubMed] [Google Scholar]
- 50. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T‐cell receptor expression and antigen‐specific T‐cell responses. Cancer research. 2004;64(16):5839‐49. [DOI] [PubMed] [Google Scholar]
- 51. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of t cell receptor cd3ζ chain expression byl‐arginine. Journal of Biological Chemistry. 2002;277(24):21123‐9. [DOI] [PubMed] [Google Scholar]
- 52. Wang L, Chang EW, Wong SC, Ong S‐M, Chong DQ, Ling KL. Increased myeloid‐derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. The Journal of Immunology. 2013;190(2):794‐804. [DOI] [PubMed] [Google Scholar]
- 53. Rodriguez PC, Quiceno DG, Ochoa AC. L‐arginine availability regulates T‐lymphocyte cell‐cycle progression. Blood. 2007;109(4):1568‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang TT, Zhao YL, Peng LS, Chen N, Chen W, Lv YP, et al. Tumour‐activated neutrophils in gastric cancer foster immune suppression and disease progression through GM‐CSF‐PD‐L1 pathway. Gut. 2017;66(11):1900‐11. 10.1136/gutjnl-2016-313075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ren W, Zhang X, Li W, Feng Q, Feng H, Tong Y, et al. Circulating and tumor‐infiltrating arginase 1‐expressing cells in gastric adenocarcinoma patients were mainly immature and monocytic Myeloid‐derived suppressor cells. Scientific reports. 2020;10(1):8056. 10.1038/s41598-020-64841-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Oberlies J, Watzl C, Giese T, Luckner C, Kropf P, Müller I, et al. Regulation of NK cell function by human granulocyte arginase. The Journal of Immunology. 2009;182(9):5259‐67. [DOI] [PubMed] [Google Scholar]
- 57. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB‐1158 blocks myeloid cell‐mediated immune suppression in the tumor microenvironment. Journal for immunotherapy of cancer. 2017;5(1):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid‐derived suppressor cells impair T cell responses through independent nitric oxide‐related pathways. International journal of cancer. 2014;134(12):2853‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen‐specific T cells. Journal of Experimental Medicine. 2011;208(10):1949‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Markowitz J, Wang J, Vangundy Z, You J, Yildiz V, Yu L, et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4+ T cells in cancer and measurement of STAT1 nitration. Scientific reports. 2017;7(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stiff A, Trikha P, Mundy‐Bosse B, McMichael E, Mace TA, Benner B, et al. Nitric oxide production by myeloid‐derived suppressor cells plays a role in impairing Fc receptor–mediated natural killer cell function. Clinical Cancer Research. 2018;24(8):1891‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li W, Zhang X, Chen Y, Xie Y, Liu J, Feng Q, et al. G‐CSF is a key modulator of MDSC and could be a potential therapeutic target in colitis‐associated colorectal cancers. Protein & cell. 2016;7(2):130‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid‐derived suppressor cells in tumor‐bearing mice. Journal of immunology. 2008;181(8):5791‐802. 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mantovani G, Maccio A, Madeddu C, Mura L, Gramignano G, Lusso MR, et al. Antioxidant agents are effective in inducing lymphocyte progression through cell cycle in advanced cancer patients: assessment of the most important laboratory indexes of cachexia and oxidative stress. Journal of molecular medicine. 2003;81(10):664‐73. 10.1007/s00109-003-0476-1. [DOI] [PubMed] [Google Scholar]
- 65. Szuster‐Ciesielska A, Hryciuk‐Umer E, Stepulak A, Kupisz K, Kandefer‐Szerszen M. Reactive oxygen species production by blood neutrophils of patients with laryngeal carcinoma and antioxidative enzyme activity in their blood. Acta oncologica. 2004;43(3):252‐8. 10.1080/02841860410029708. [DOI] [PubMed] [Google Scholar]
- 66. Agostinelli E, Seiler N. Non‐irradiation‐derived reactive oxygen species (ROS) and cancer: therapeutic implications. Amino acids. 2006;31(3):341‐55. 10.1007/s00726-005-0271-8. [DOI] [PubMed] [Google Scholar]
- 67. Gabrilovich DI, Nagaraj S. Myeloid‐derived suppressor cells as regulators of the immune system. Nature reviews Immunology. 2009;9(3):162‐74. 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2001;11(4):173‐86. 10.1159/000047804. [DOI] [PubMed] [Google Scholar]
- 69. Otsuji M, Kimura Y, Aoe T, Okamoto Y, Saito T. Oxidative stress by tumor‐derived macrophages suppresses the expression of CD3 ζ chain of T‐cell receptor complex and antigen‐specific T‐cell responses. Proceedings of the National Academy of Sciences. 1996;93(23):13119‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schmielau J, Nalesnik MA, Finn OJ. Suppressed T‐cell receptor ζ chain expression and cytokine production in pancreatic cancer patients. Clinical cancer research. 2001;7(3):933s‐9s. [PubMed] [Google Scholar]
- 71. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nature medicine. 2007;13(7):828‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. OuYang L‐Y, Wu X‐J, Ye S‐B, R‐x Zhang, Li Z‐L, Liao W, et al. Tumor‐induced myeloid‐derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. Journal of translational medicine. 2015;13(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor‐induced myeloid‐derived suppressor cells. The Journal of Immunology. 2009;182(9):5693‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Neurath M, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, et al. The transcription factor T‐bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. The Journal of experimental medicine. 2002;195(9):1129‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lúdvíksson BR, Seegers D, Resnick AS, Strober W. The effect of TGF‐β1 on immune responses of naïve versus memory CD4+ Th1/Th2 T cells. European journal of immunology. 2000;30(7):2101‐11. [DOI] [PubMed] [Google Scholar]
- 76. Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF‐β inhibits Th type 2 development through inhibition of GATA‐3 expression. The Journal of Immunology. 2000;165(9):4773‐7. [DOI] [PubMed] [Google Scholar]
- 77. Takaku S, Terabe M, Ambrosino E, Peng J, Lonning S, McPherson JM, et al. Blockade of TGF‐β enhances tumor vaccine efficacy mediated by CD8+ T cells. International journal of cancer. 2010;126(7):1666‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jayakumar A, Bothwell AL. Functional diversity of myeloid‐derived suppressor cells: the multitasking hydra of cancer. The Journal of Immunology. 2019;203(5):1095‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor‐expressed IDO recruits and activates MDSCs in a Treg‐dependent manner. Cell reports. 2015;13(2):412‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand‐Rosenberg S. Cross‐talk between myeloid‐derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. The Journal of Immunology. 2007;179(2):977‐83. [DOI] [PubMed] [Google Scholar]
- 81. Hu C‐E, Gan J, Zhang R‐D, Cheng Y‐R, Huang G‐J. Up‐regulated myeloid‐derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scandinavian journal of gastroenterology. 2011;46(2):156‐64. [DOI] [PubMed] [Google Scholar]
- 82. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD‐L1 is a novel direct target of HIF‐1α, and its blockade under hypoxia enhanced MDSC‐mediated T cell activation. Journal of Experimental Medicine. 2014;211(5):781‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jingjing, Zhu , Céline, G. , Powis, de , et al. Resistance to cancer immunotherapy mediated by apoptosis of tumor‐infiltrating lymphocytes. Nature Communications. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Trovato R, Canè S, Petrova V, Sartoris S, Ugel S, De Sanctis F. The engagement between MDSCs and metastases: partners in crime. Frontiers in Oncology. 2020;10:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jayaraman P, Parikh F, Newton JM, Hanoteau A, Rivas C, Krupar R, et al. TGF‐β1 programmed myeloid‐derived suppressor cells (MDSC) acquire immune‐stimulating and tumor killing activity capable of rejecting established tumors in combination with radiotherapy. Oncoimmunology. 2018;7(10):e1490853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Iwata T, Kondo Y, Kimura O, Morosawa T, Fujisaka Y, Umetsu T, et al. PD‐L1+MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Scientific Reports. 2016;6(1):39296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD‐L1 in tumor‐induced myeloid‐derived suppressor cells. Oncoimmunology. 2016;5(12):e1247135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chiu DK‐C, Tse AP‐W, Xu IM‐J, Di Cui J, Lai RK‐H, Li LL, et al. Hypoxia inducible factor HIF‐1 promotes myeloid‐derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nature communications. 2017;8(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. The Journal of Immunology. 2010;185(6):3190‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nagaraj S, Gabrilovich DI. Tumor escape mechanism governed by myeloid‐derived suppressor cells. Cancer research. 2008;68(8):2561‐3. [DOI] [PubMed] [Google Scholar]
- 92. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid‐derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138(2):105‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shibata M, Gonda K, Takenoshita S. MDSC: Myeloid‐derived suppressor cells. Springer, Japan; 2016. [Google Scholar]
- 94. Raskovalova T, Lokshin A, Huang X, Jackson EK, Gorelik E. Adenosine‐mediated inhibition of cytotoxic activity and cytokine production by IL‐2/NKp46‐activated NK cells. Immunologic research. 2006;36(1‐3):91‐9. [DOI] [PubMed] [Google Scholar]
- 95. Jacob A, Prekeris R. The regulation of MMP targeting to invadopodia during cancer metastasis. Frontiers in cell and developmental biology. 2015;3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Huh SJ, Liang S, Sharma A, Dong C, Robertson GP. Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer research. 2010;70(14):6071‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Spicer JD, McDonald B, Cools‐Lartigue JJ, Chow SC, Giannias B, Kubes P, et al. Neutrophils promote liver metastasis via Mac‐1–mediated interactions with circulating tumor cells. Cancer research. 2012;72(16):3919‐27. [DOI] [PubMed] [Google Scholar]
- 98. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGFβ signaling in mammary carcinomas recruits Gr‐1+ CD11b+ myeloid cells that promote metastasis. Cancer cell. 2008;13(1):23‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Heeke S, Mograbi B, Alix‐Panabières C, Hofman P. Never travel alone: the crosstalk of circulating tumor cells and the blood microenvironment. Cells. 2019;8(7):714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361(6409). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sprouse ML, Welte T, Boral D, Liu HN, Yin W, Vishnoi M, et al. PMN‐MDSCs enhance CTC metastatic properties through reciprocal interactions via ROS/Notch/Nodal signaling. International journal of molecular sciences. 2019;20(8):1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, et al. Myeloid‐derived suppressor cells endow stem‐like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross‐talk signaling. Cancer research. 2016;76(11):3156‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Trovato R, Cane S, Petrova V, Sartoris S, Ugel S, De Sanctis F. The Engagement Between MDSCs and Metastases: Partners in Crime. Frontiers in oncology. 2020;10:165. 10.3389/fonc.2020.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yang C, Chen F, Wang S, Xiong B. Circulating Tumor Cells in Gastrointestinal Cancers: Current Status and Future Perspectives. Frontiers in oncology. 2019;9:1427. 10.3389/fonc.2019.01427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Peinado H, Lavotshkin S, Lyden D, editors. The secreted factors responsible for pre‐metastatic niche formation: old sayings and new thoughts. Seminars in cancer biology; 2011: Elsevier. [DOI] [PubMed] [Google Scholar]
- 106. Giles AJ, Reid CM, Evans JD, Murgai M, Vicioso Y, Highfill SL, et al. Activation of hematopoietic stem/progenitor cells promotes immunosuppression within the pre–metastatic niche. Cancer research. 2016;76(6):1335‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Qian B‐Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast‐tumour metastasis. Nature. 2011;475(7355):222‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer‐associated fibroblasts in the tumor microenvironment via STAT3–CCL2 signaling. Cancer research. 2016;76(14):4124‐35. [DOI] [PubMed] [Google Scholar]
- 109. Wang D, Sun H, Wei J, Cen B, DuBois RN. CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer research. 2017;77(13):3655‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sceneay J, Parker BS, Smyth MJ, Möller A. Hypoxia‐driven immunosuppression contributes to the pre‐metastatic niche. Oncoimmunology. 2013;2(1):e22355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Shi H, Zhang J, Han X, Li H, Xie M, Sun Y, et al. Recruited monocytic myeloid‐derived suppressor cells promote the arrest of tumor cells in the premetastatic niche through an IL‐1beta‐mediated increase in E‐selectin expression. International journal of cancer. 2017;140(6):1370‐83. 10.1002/ijc.30538. [DOI] [PubMed] [Google Scholar]
- 112. Shi H, Zhang J, Han X, Li H, Xie M, Sun Y, et al. Recruited monocytic myeloid‐derived suppressor cells promote the arrest of tumor cells in the premetastatic niche through an IL‐1β‐mediated increase in E‐selectin expression. International journal of cancer. 2017;140(6):1370‐83. [DOI] [PubMed] [Google Scholar]
- 113. Kitamura T, Fujishita T, Loetscher P, Revesz L, Hashida H, Kizaka‐Kondoh S, et al. Inactivation of chemokine (CC motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proceedings of the National Academy of Sciences. 2010;107(29):13063‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Inamoto S, Itatani Y, Yamamoto T, Minamiguchi S, Hirai H, Iwamoto M, et al. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid‐Derived Suppressor Cells through the CCL15–CCR1 Chemokine Axis. Clinical Cancer Research. 2016;22(2):492‐501. [DOI] [PubMed] [Google Scholar]
- 115. Hsu Y‐L, Yen M‐C, Chang W‐A, Tsai P‐H, Pan Y‐C, Liao S‐H, et al. CXCL17‐derived CD11b+ Gr‐1+ myeloid‐derived suppressor cells contribute to lung metastasis of breast cancer through platelet‐derived growth factor‐BB. Breast Cancer Research. 2019;21(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G‐CSF‐initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti‐VEGF therapy in mouse models. Proceedings of the National Academy of Sciences. 2009;106(16):6742‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP‐9 supplied by bone marrow–derived cells contributes to skin carcinogenesis. Cell. 2000;103(3):481‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Shojaei F, Ferrara N. Refractoriness to antivascular endothelial growth factor treatment: role of myeloid cells. Cancer research. 2008;68(14):5501‐4. [DOI] [PubMed] [Google Scholar]
- 119. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand‐Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid‐derived suppressor cells. The Journal of Immunology. 2008;181(7):4666‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ichikawa M, Williams R, Wang L, Vogl T, Srikrishna G. S100A8/A9 activate key genes and pathways in colon tumor progression. Molecular cancer research. 2011;9(2):133‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zhang H, Maric I, DiPrima MJ, Khan J, Orentas RJ, Kaplan RN, et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood. 2013;122(7):1105‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zoso A, Mazza EM, Bicciato S, Mandruzzato S, Bronte V, Serafini P, et al. Human fibrocytic myeloid‐derived suppressor cells express IDO and promote tolerance via Treg‐cell expansion. European journal of immunology. 2014;44(11):3307‐19. [DOI] [PubMed] [Google Scholar]
- 123. Gao XH, Tian L, Wu J, Ma XL, Zhang CY, Zhou Y, et al. Circulating CD14+ HLA‐DR−/low myeloid‐derived suppressor cells predicted early recurrence of hepatocellular carcinoma after surgery. Hepatology Research. 2017;47(10):1061‐71. [DOI] [PubMed] [Google Scholar]
- 124. Solito S, Falisi E, Diaz‐Montero CM, Doni A, Pinton L, Rosato A, et al. A human promyelocytic‐like population is responsible for the immune suppression mediated by myeloid‐derived suppressor cells. Blood, The Journal of the American Society of Hematology. 2011;118(8):2254‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Chen M‐F, Kuan F‐C, Yen T‐C, Lu M‐S, Lin P‐Y, Chung Y‐H, et al. IL‐6‐stimulated CD11b+ CD14+ HLA‐DR− myeloid‐derived suppressor cells, are associated with progression and poor prognosis in squamous cell carcinoma of the esophagus. Oncotarget. 2014;5(18):8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Annels NE, Shaw VE, Gabitass RF, Billingham L, Corrie P, Eatock M, et al. The effects of gemcitabine and capecitabine combination chemotherapy and of low‐dose adjuvant GM‐CSF on the levels of myeloid‐derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunology, Immunotherapy. 2014;63(2):175‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kim W, Chu TH, Nienhüser H, Jiang Z, Del Portillo A, Remotti HE, et al. PD‐1 Signaling Promotes Tumor‐Infiltrating Myeloid‐Derived Suppressor Cells and Gastric Tumorigenesis in Mice. Gastroenterology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Bruchard M, Mignot G, Derangère V, Chalmin F, Chevriaux A, Végran F, et al. Chemotherapy‐triggered cathepsin B release in myeloid‐derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nature medicine. 2013;19(1):57‐64. [DOI] [PubMed] [Google Scholar]
- 129. Porembka MR, Mitchem JB, Belt BA, Hsieh C‐S, Lee H‐M, Herndon J, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid‐derived suppressor cells which promote primary tumor growth. Cancer Immunology, Immunotherapy. 2012;61(9):1373‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Condamine T, Kumar V, Ramachandran IR, Youn JI, Gabrilovich DI. ER stress regulates myeloid‐derived suppressor cell fate through TRAIL‐R–mediated apoptosis. The Journal of clinical investigation. 2014;124(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Dominguez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, et al. Selective targeting of myeloid‐derived suppressor cells in cancer patients using DS‐8273a, an agonistic TRAIL‐R2 antibody. Clinical Cancer Research. 2017;23(12):2942‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Pan P‐Y, Wang GX, Yin B, Ozao J, Ku T, Divino CM, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid‐derived suppressor cell development by blockade of stem‐cell factor function. Blood. 2008;111(1):219‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Garton AJ, Seibel S, Lopresti‐Morrow L, Crew L, Janson N, Mandiyan S, et al. Anti‐KIT monoclonal antibody treatment enhances the antitumor activity of immune checkpoint inhibitors by reversing tumor‐induced immunosuppression. Molecular cancer therapeutics. 2017;16(4):671‐80. [DOI] [PubMed] [Google Scholar]
- 134. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine. 2019;47:235‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Liu Y, Wei G, Cheng WA, Dong Z, Sun H, Lee VY, et al. Targeting myeloid‐derived suppressor cells for cancer immunotherapy. Cancer Immunology, Immunotherapy. 2018;67(8):1181‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all‐trans retinoic acid effect on tumor‐associated myeloid‐derived suppressor cells. Cancer research. 2007;67(22):11021‐8. [DOI] [PubMed] [Google Scholar]
- 137. Tobin RP, Davis D, Jordan KR, McCarter MD. The clinical evidence for targeting human myeloid‐derived suppressor cells in cancer patients. Journal of leukocyte biology. 2017;102(2):381‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Mahendra A, Karishma BKC, Sharma T, Bansal N, Bansal R, Gupta S. Vitamin D and gastrointestinal cancer. Journal of laboratory physicians. 2018;10(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Chen P‐T, Hsieh C‐C, Wu C‐T, Yen T‐C, Lin P‐Y, Chen W‐C, et al. 1α, 25‐dihydroxyvitamin D3 inhibits esophageal squamous cell carcinoma progression by reducing IL6 signaling. Molecular cancer therapeutics. 2015;14(6):1365‐75. [DOI] [PubMed] [Google Scholar]
- 140. Tu SP, Jin H, Shi JD, Zhu LM, Suo Y, Lu G, et al. Curcumin induces the differentiation of myeloid‐derived suppressor cells and inhibits their interaction with cancer cells and related tumor growth. Cancer prevention research. 2012;5(2):205‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Zoglmeier C, Bauer H, Nörenberg D, Wedekind G, Bittner P, Sandholzer N, et al. CpG blocks immunosuppression by myeloid‐derived suppressor cells in tumor‐bearing mice. Clinical Cancer Research. 2011;17(7):1765‐75. [DOI] [PubMed] [Google Scholar]
- 142. De Cicco P, Ercolano G, Ianaro A. The new era of cancer immunotherapy: targeting myeloid‐derived suppressor cells to overcome immune evasion. Frontiers in Immunology. 2020;11:1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Cannarile MA, Weisser M, Jacob W, Jegg A‐M, Ries CH, Rüttinger D. Colony‐stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. Journal for immunotherapy of cancer. 2017;5(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid‐derived suppressor cells with colony stimulating factor‐1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2, 3‐dioxygenase‐expressing tumors. EBioMedicine. 2016;6:50‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla‐Alonso S, et al. Cancer‐associated fibroblasts neutralize the anti‐tumor effect of CSF1 receptor blockade by inducing PMN‐MDSC infiltration of tumors. Cancer cell. 2017;32(5):654‐68. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wolf MJ, Hoos A, Bauer J, Boettcher S, Knust M, Weber A, et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2‐Stat5 and p38MAPK pathway. Cancer cell. 2012;22(1):91‐105. [DOI] [PubMed] [Google Scholar]
- 147. Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. Journal of neuro‐oncology. 2011;104(1):83‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING‐dependent MDSC mobilization drives extrinsic radiation resistance. Nature communications. 2017;8(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Katoh H, Wang D, Daikoku T, Sun H, Dey SK, DuBois RN. CXCR2‐expressing myeloid‐derived suppressor cells are essential to promote colitis‐associated tumorigenesis. Cancer cell. 2013;24(5):631‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Xu Y, Fang F, Jiao H, Zheng X, Huang L, Yi X, et al. Activated hepatic stellate cells regulate MDSC migration through the SDF‐1/CXCR4 axis in an orthotopic mouse model of hepatocellular carcinoma. Cancer Immunology, Immunotherapy. 2019;68(12):1959‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Qin H, Lerman B, Sakamaki I, Wei G, Cha SC, Rao SS, et al. Generation of a new therapeutic peptide that depletes myeloid‐derived suppressor cells in tumor‐bearing mice. Nature medicine. 2014;20(6):676‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Huang M, Wu R, Chen L, Peng Q, Li S, Zhang Y, et al. S100A9 regulates MDSCs‐mediated immune suppression via the RAGE and TLR4 signaling pathways in colorectal carcinoma. Frontiers in immunology. 2019;10:2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Setter SM, Iltz JL, Fincham JE, Campbell RK, Baker DE. Phosphodiesterase 5 inhibitors for erectile dysfunction. Annals of Pharmacotherapy. 2005;39(7‐8):1286‐95. [DOI] [PubMed] [Google Scholar]
- 154. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase‐5 inhibition augments endogenous antitumor immunity by reducing myeloid‐derived suppressor cell function. The Journal of experimental medicine. 2006;203(12):2691‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nature medicine. 2005;11(2):214‐22. [DOI] [PubMed] [Google Scholar]
- 156. Liu L, Li H, Underwood T, Lloyd M, David M, Sperl G, et al. Cyclic GMP‐dependent protein kinase activation and induction by exisulind and CP461 in colon tumor cells. Journal of Pharmacology and Experimental Therapeutics. 2001;299(2):583‐92. [PubMed] [Google Scholar]
- 157. Sarfati M, Mateo V, Baudet S, Rubio M, Fernandez C, Davi F, et al. Sildenafil and vardenafil, types 5 and 6 phosphodiesterase inhibitors, induce caspase‐dependent apoptosis of B‐chronic lymphocytic leukemia cells. Blood. 2003;101(1):265‐9. [DOI] [PubMed] [Google Scholar]
- 158. Yu SJ, Ma C, Heinrich B, Brown ZJ, Sandhu M, Zhang Q, et al. Targeting the crosstalk between cytokine‐induced killer cells and myeloid‐derived suppressor cells in hepatocellular carcinoma. Journal of hepatology. 2019;70(3):449‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Miret JJ, Kirschmeier P, Koyama S, Zhu M, Li YY, Naito Y, et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a NSCLC mouse model by activating anti‐tumor immunity. Journal for immunotherapy of cancer. 2019;7(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Zhao Y, Shen X‐F, Cao K, Ding J, Kang X, Guan W‐x, et al. Dexamethasone‐induced myeloid‐derived suppressor cells prolong allo cardiac graft survival through iNOS‐and glucocorticoid receptor‐dependent mechanism. Frontiers in immunology. 2018;9:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. De Santo C, Serafini P, Marigo I, Dolcetti L, Bolla M, Del Soldato P, et al. Nitroaspirin corrects immune dysfunction in tumor‐bearing hosts and promotes tumor eradication by cancer vaccination. Proceedings of the National Academy of Sciences. 2005;102(11):4185‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Nagaraj S, Youn J‐I, Weber H, Iclozan C, Lu L, Cotter MJ, et al. Anti‐inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clinical cancer research. 2010;16(6):1812‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Cappello P, Tonoli E, Curto R, Giordano D, Giovarelli M, Novelli F. Anti‐α‐enolase antibody limits the invasion of myeloid‐derived suppressor cells and attenuates their restraining effector T cell response. Oncoimmunology. 2016;5(5):e1112940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Kobayashi M, Chung J‐S, Beg M, Arriaga Y, Verma U, Courtney K, et al. Blocking monocytic myeloid‐derived suppressor cell function via anti‐DC‐HIL/GPNMB antibody restores the in vitro integrity of T cells from cancer patients. Clinical Cancer Research. 2019;25(2):828‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Zhuang Y, Peng LS, Zhao YL, Shi Y, Mao XH, Chen W, et al. CD8+ T cells that produce interleukin‐17 regulate myeloid‐derived suppressor cells and are associated with survival time of patients with gastric cancer. Gastroenterology. 2012;143(4):951‐62. e8. [DOI] [PubMed] [Google Scholar]
- 166. Turkes F, Mencel J, Starling N. Targeting the immune milieu in gastrointestinal cancers. Journal of gastroenterology. 2020:1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Dong J, Li J, Liu S‐M, Feng X‐Y, Chen S, Chen Y‐B, et al. CD33+/p‐STAT1+ double‐positive cell as a prognostic factor for stage IIIa gastric cancer. Medical oncology. 2013;30(1):442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Sun H‐L, Zhou X, Xue Y‐F, Wang K, Shen Y‐F, Mao J‐J, et al. Increased frequency and clinical significance of myeloid‐derived suppressor cells in human colorectal carcinoma. World journal of gastroenterology: WJG. 2012;18(25):3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, et al. Circulating and tumor‐infiltrating myeloid‐derived suppressor cells in patients with colorectal carcinoma. PloS one. 2013;8(2):e57114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Arihara F, Mizukoshi E, Kitahara M, Takata Y, Arai K, Yamashita T, et al. Increase in CD14+ HLA‐DR−/low myeloid‐derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunology, Immunotherapy. 2013;62(8):1421‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Teng F, Meng X, Kong L, Mu D, Zhu H, Liu S, et al. Tumor‐infiltrating lymphocytes, forkhead box P3, programmed death ligand‐1, and cytotoxic T lymphocyte–associated antigen‐4 expressions before and after neoadjuvant chemoradiation in rectal cancer. Translational research. 2015;166(6):721‐32. e1. [DOI] [PubMed] [Google Scholar]
- 172. Ni Y, Chen W. Relationship between Immunosuppressive Cells Treg, MDSCs and Clinicopathological Characteristics of Gastric Cancer. Chinese Journal of Gastroenterology. 2015(4):210‐3. [Google Scholar]
- 173. Huang H, Zhang G, Li G, Ma H, Zhang X. Circulating CD14+ HLA‐DR−/low myeloid‐derived suppressor cell is an indicator of poor prognosis in patients with ESCC. Tumor Biology. 2015;36(10):7987‐96. [DOI] [PubMed] [Google Scholar]
- 174. Shimura T, Shibata M, Gonda K, Hayase S, Sakamoto W, Okayama H, et al. Prognostic impact of preoperative lymphocyte‐to‐monocyte ratio in patients with colorectal cancer with special reference to myeloid‐derived suppressor cells. Fukushima Journal of Medical Science. 2018;64(2):64‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Fědorová L, Pilátová K, Selingerová I, Bencsiková B, Budinská E, Zwinsová B, et al. Circulating Myeloid‐Derived Suppressor Cell Subsets in Patients with Colorectal Cancer–Exploratory Analysis of Their Biomarker Potential. Klinicka onkologie: casopis Ceske a Slovenske onkologicke spolecnosti. 2018;31(Suppl 2):88‐92. [PubMed] [Google Scholar]
- 176. Urakawa S, Yamasaki M, Goto K, Haruna M, Hirata M, Morimoto‐Okazawa A, et al. Peri‐operative monocyte count is a marker of poor prognosis in gastric cancer: increased monocytes are a characteristic of myeloid‐derived suppressor cells. Cancer Immunology, Immunotherapy. 2019;68(8):1341‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Talmadge JE, Hood KC, Zobel LC, Shafer LR, Coles M, Toth B. Chemoprevention by cyclooxygenase‐2 inhibition reduces immature myeloid suppressor cell expansion. International immunopharmacology. 2007;7(2):140‐51. [DOI] [PubMed] [Google Scholar]
- 178. Ko H‐J, Kim Y‐J, Kim Y‐S, Chang W‐S, Ko S‐Y, Chang S‐Y, et al. A combination of chemoimmunotherapies can efficiently break self‐tolerance and induce antitumor immunity in a tolerogenic murine tumor model. Cancer research. 2007;67(15):7477‐86. [DOI] [PubMed] [Google Scholar]
- 179. Capuano G, Rigamonti N, Grioni M, Freschi M, Bellone M. Modulators of arginine metabolism support cancer immunosurveillance. BMC immunology. 2009;10(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Daurkin I, Eruslanov E, Vieweg J, Kusmartsev S. Generation of antigen‐presenting cells from tumor‐infiltrated CD11b myeloid cells with DNA demethylating agent 5‐aza‐2′‐deoxycytidine. Cancer immunology, immunotherapy. 2010;59(5):697‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy‐Martin J‐P, et al. Membrane‐associated Hsp72 from tumor‐derived exosomes mediates STAT3‐dependent immunosuppressive function of mouse and human myeloid‐derived suppressor cells. The Journal of clinical investigation. 2010;120(2):457‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Cao M, Xu Y, Youn J‐i, Cabrera R, Zhang X, Gabrilovich D, et al. Kinase inhibitor Sorafenib modulates immunosuppressive cell populations in a murine liver cancer model. Laboratory Investigation. 2011;91(4):598‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Bunt SK, Mohr AM, Bailey JM, Grandgenett PM, Hollingsworth MA. Rosiglitazone and Gemcitabine in combination reduces immune suppression and modulates T cell populations in pancreatic cancer. Cancer Immunology, Immunotherapy. 2013;62(2):225‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Chen H‐M, Ma G, Gildener‐Leapman N, Eisenstein S, Coakley BA, Ozao J, et al. Myeloid‐derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clinical Cancer Research. 2015;21(18):4073‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Klement JD, Paschall AV, Savage NM, Nayak‐Kapoor A, Liu K. 5‐Fluorouracil regulation of myeloid‐derived suppressor cell differentiation in vitro and in vivo. Am Assoc Immnol; 2017. [Google Scholar]
- 186. Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS‐IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer cell. 2019;35(4):559‐72. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Lee JM, Seo JH, Kim YJ, Kim YS, Ko HJ, Kang CY. The restoration of myeloid‐derived suppressor cells as functional antigen‐presenting cells by NKT cell help and all‐trans‐retinoic acid treatment. International journal of cancer. 2012;131(3):741‐51. [DOI] [PubMed] [Google Scholar]
- 188. Osada T, Chong G, Tansik R, Hong T, Spector N, Kumar R, et al. The effect of anti‐VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunology, Immunotherapy. 2008;57(8):1115‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Tanikawa T, Wilke CM, Kryczek I, Chen GY, Kao J, Núñez G, et al. Interleukin‐10 ablation promotes tumor development, growth, and metastasis. Cancer research. 2012;72(2):420‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Arabzadeh A, Chan C, Nouvion A, Breton V, Benlolo S, DeMarte L, et al. Host‐related carcinoembryonic antigen cell adhesion molecule 1 promotes metastasis of colorectal cancer. Oncogene. 2013;32(7):849‐60. [DOI] [PubMed] [Google Scholar]
- 191. Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, et al. CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid‐derived suppressor cell population and function. Cell reports. 2015;12(2):244‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Ding Y, Shen J, Zhang G, Chen X, Wu J, Chen W. CD40 controls CXCR5‐induced recruitment of myeloid‐derived suppressor cells to gastric cancer. Oncotarget. 2015;6(36):38901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Li Y, Zhou T, Wang Y, Ning C, Lv Z, Han G, et al. The protumorigenic potential of FTY720 by promoting extramedullary hematopoiesis and MDSC accumulation. Oncogene. 2017;36(26):3760‐71. [DOI] [PubMed] [Google Scholar]
- 194. Jayakumar A, Bothwell AL. Stat6 promotes intestinal tumorigenesis in a mouse model of adenomatous polyposis by expansion of MDSCs and inhibition of cytotoxic CD8 response. Neoplasia. 2017;19(8):595‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Ma N, Liu Q, Hou L, Wang Y, Liu Z. MDSCs are involved in the protumorigenic potentials of GM‐CSF in colitis‐associated cancer. International journal of immunopathology and pharmacology. 2017;30(2):152‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Yang L, Wang B, Qin J, Zhou H, Majumdar AP, Peng F. Blockade of CCR5‐mediated myeloid derived suppressor cell accumulation enhances anti‐PD1 efficacy in gastric cancer. Immunopharmacology and Immunotoxicology. 2018;40(1):91‐7. [DOI] [PubMed] [Google Scholar]
- 197. Espaillat MP, Snider AJ, Qiu Z, Channer B, Coant N, Schuchman EH, et al. Loss of acid ceramidase in myeloid cells suppresses intestinal neutrophil recruitment. The FASEB Journal. 2018;32(5):2339‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J, et al. A RIPK3‐PGE2 Circuit Mediates Myeloid‐Derived Suppressor Cell–Potentiated Colorectal Carcinogenesis. Cancer research. 2018;78(19):5586‐99. [DOI] [PubMed] [Google Scholar]
- 199. Xu P, He H, Gu Y, Wang Y, Sun Z, Yang L, et al. Surgical trauma contributes to progression of colon cancer by downregulating CXCL4 and recruiting MDSCs. Experimental cell research. 2018;370(2):692‐8. [DOI] [PubMed] [Google Scholar]
- 200. Yu X, Wang D, Wang X, Sun S, Zhang Y, Wang S, et al. CXCL12/CXCR4 promotes inflammation‐driven colorectal cancer progression through activation of RhoA signaling by sponging miR‐133a‐3p. Journal of Experimental & Clinical Cancer Research. 2019;38(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Guha P, Gardell J, Darpolor J, Cunetta M, Lima M, Miller G, et al. STAT3 inhibition induces Bax‐dependent apoptosis in liver tumor myeloid‐derived suppressor cells. Oncogene. 2019;38(4):533‐48. [DOI] [PubMed] [Google Scholar]
- 202. Ke Z, Wang C, Wu T, Wang W, Yang Y, Dai Y. PAR2 deficiency enhances myeloid cell‐mediated immunosuppression and promotes colitis‐associated tumorigenesis. Cancer Letters. 2020;469:437‐46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.