
FIG 2.
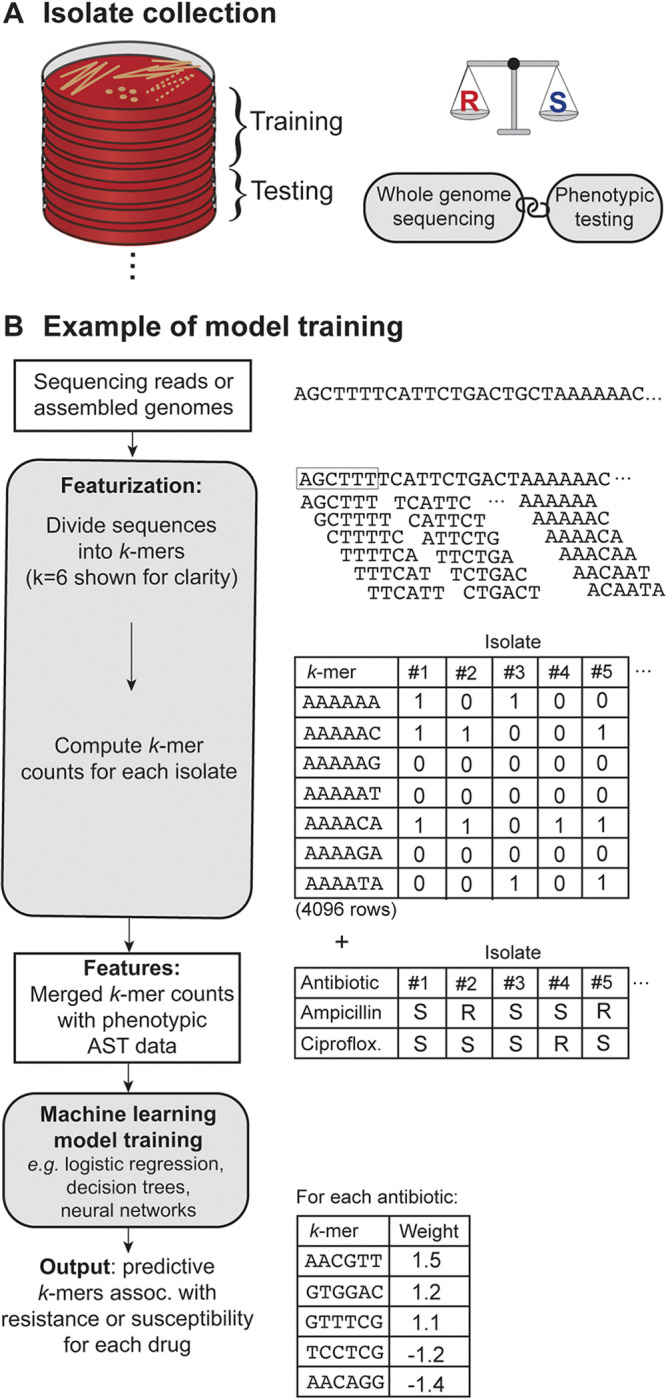
Schematic overview of the process of training a machine learning (ML) model to predict antimicrobial susceptibility testing (AST) results from whole-genome sequences. (A) ML models rely on data sets containing tens to thousands of isolates with paired whole-genome sequences and phenotypic AST results. Data sets are divided into training and test sets, where the training set is used to fit the parameters of the model and the test set evaluates the model accuracy. An optimal data set contains a balance of resistant and susceptible examples for each organism-drug combination. (B) The data inputs are usually quality-controlled sequencing reads or assembled genomes, which are transformed into overlapping subsequences of length k, referred to as a k-mer. A typical length for a k-mer feature is 13 to 31 nucleotides, but a length of 6 nucleotides is shown here for clarity. There are 4k k-mer possibilities (e.g., 4,096 when k = 6), and the counts of each k-mer present in a given sequence are tallied for each isolate in the data set. The selected features are then merged with the phenotypic AST data, and this matrix is used as the input for a supervised machine learning model. The model analyzes the matrix to find the features, e.g., k-mers, that best predict resistance or susceptibility to a given antibiotic.