

SUMMARY
Alveolar epithelial type 2 (AT2) cells integrate signals from multiple molecular pathways to proliferate and differentiate to drive regeneration of the lung alveolus. Utilizing in vivo genetic and ex vivo organoid models, we investigated the role of Fgfr2 signaling in AT2 cells across the lifespan and during adult regeneration after influenza infection. We show that, although dispensable for adult homeostasis, Fgfr2 restricts AT2 cell fate during postnatal lung development. Using an unbiased computational imaging approach, we demonstrate that Fgfr2 promotes AT2 cell proliferation and restrains differentiation in actively regenerating areas after injury. Organoid assays reveal that Fgfr2-deficient AT2 cells remain competent to respond to multiple parallel proliferative inputs. Moreover, genetic blockade of AT2 cell cytokinesis demonstrates that cell division and differentiation are uncoupled during alveolar regeneration. These data reveal that Fgfr2 maintains AT2 cell fate, balancing proliferation and differentiation during lung alveolar regeneration.
In brief
Using an unbiased computational imaging approach, Liberti et al. demonstrate that alveolar epithelial type 2 (AT2) cells actively maintain their fate independent of their ability to divide. AT2 cells balance the decision to divide versus differentiate to drive lung regeneration after acute injury.
Graphical Abstract
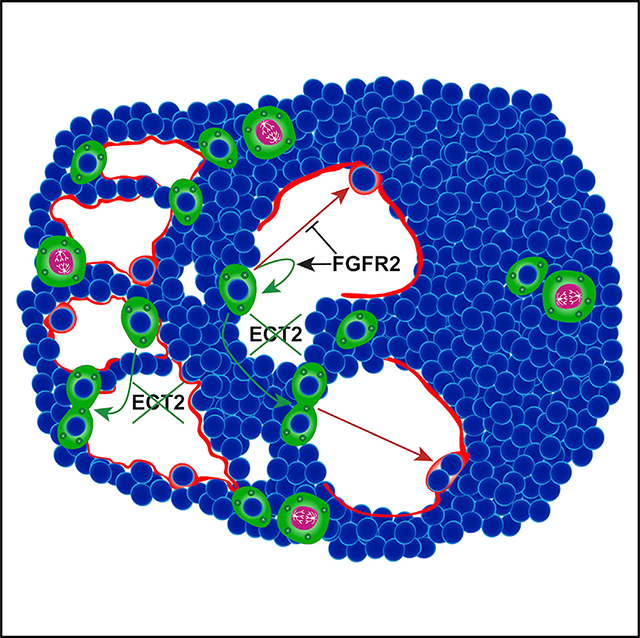
INTRODUCTION
The distal compartment of the lungs contains the alveoli, where the majority of gas exchange with the external environment occurs in mammals. The predominant epithelial cell lineages within this niche are the alveolar type 1 (AT1) and type 2 (AT2) cells (Zepp and Morrisey, 2019). AT1 cells form a thin, gas diffusible interface with the pulmonary capillary plexus, which is essential for respiration. AT2 cells generate and secrete pulmonary surfactant, which reduces surface tension to prevent alveolar collapse. During normal adult homeostasis, the lungs are quiescent, exhibiting a very low rate of cellular turnover. However, acute injury damages alveoli by destroying resident AT1 and AT2 cells, depleting the lung of respiratory capacity.
Injury to the lungs is often heterogeneous and can elicit a varied regenerative response depending on the type of damage and its severity (Basil et al., 2020; Sivakumar and Frank, 2019; Whitsett et al., 2019; Xi et al., 2017; Zepp and Morrisey, 2019; Alysandratos et al., 2021). These varying responses are highlighted by distinct alterations in tissue architecture (Zacharias et al., 2018). Appropriate alveolar architecture must be reconstructed to restore gas exchange, which requires the activation of the lung’s resident stem/progenitors: AT2 cells. In response to lung damage, AT2 cells both rapidly proliferate and differentiate into AT1 cells to regenerate alveoli (Barkauskas et al., 2013). Yet, the cellular and molecular processes that regulate these AT2 cell behaviors are poorly understood, due in part to the heterogeneous nature of injury and tissue repair, which makes the investigation of these processes complicated to address empirically. Understanding the molecular mechanisms involved in activating AT2 cell proliferation and driving differentiation into AT1 cells is critical to develop methods to promote alveolar regeneration after lung damage.
Fibroblast growth factor (Fgf) signaling is a potent mitogenic pathway that drives lung development and injury regeneration. Fgfr2 signaling is required for lung branching morphogenesis and for maintenance of prenatal AT2 cell fate (Abler et al., 2009; Chang et al., 2013; Lebeche et al., 1999; Li et al., 2018; Weaver et al., 2000; Sekine et al., 1999; De Moerlooze et al., 2000; Mori et al., 2019). Moreover, loss of Fgf signaling during the postnatal period of lung development, alveologenesis, increases the vulnerability of the lung to injury (Hokuto et al., 2004). In adults, Fgf signaling has been implicated as essential for AT2 cell homeostasis (Dorry et al., 2020; Yuan et al., 2019). A recent study suggests that the combined loss of Fgfr1, Fgfr2, and Fgfr3 in AT2 cells results in decreased survival and proliferation of AT2 cells during adult homeostasis and after bleomycin injury (Dorry et al., 2020). Fgf ligands, Fgf7 and Fgf10, stimulate AT2 cell proliferation through Fgfr2 after injury, and exogenous stimulation with or overexpression of these ligands may aid in lung regeneration (MacKenzie et al., 2015; Gupte et al., 2009; Quantius et al., 2016; Yuan et al., 2019; Zacharias et al., 2018; Zepp et al., 2017). However, this stimulation may also cause AT2 cells to be more susceptible to viral infection (Nikolaidis et al., 2017). Together, these findings suggest that Fgfr2 has other functions beyond regulating cell proliferation during injury repair and regeneration in the lung.
We assessed the role of Fgfr2 in AT2 cells across the lifespan to determine how it may balance proliferation versus differentiation both at homeostasis and after acute injury to the lung. Loss of Fgfr2 during alveologenesis reveals that this signaling pathway acts to restrict AT2 cell fate. In the adult lung, AT2 cell-specific inactivation of Fgfr2 did not disrupt AT2 cell homeostasis up to 1 year. Given the heterogenous nature of lung injury and regeneration, we leveraged an unbiased machine learning approach to classify regenerating regions into distinct zones for analysis. Using this technique, we show that Fgfr2-deficient AT2 cells exhibit both a decreased level of proliferation and a greater ability to differentiate into AT1 cells exclusively in regions of active alveolar remodeling, indicating the necessity of Fgf signaling in a spatially specific manner in tissue regeneration. Organoid assays reveal that although inactivation of Fgfr2 eliminates the ability of AT2 cells to respond to Fgfr2 ligand stimulation, Fgfr2 is not necessary for overall AT2 cell proliferation. This result is due in part to Fgfr2-deficient AT2 cells maintaining their competence to respond to parallel mitogenic signals, including immune cytokines. Moreover, blockade of AT2 cell division through inactivation of Ect2 uncouples division from differentiation in these cells, demonstrating that AT2 cell fate decisions are made independent of proliferative competency. Thus, our studies reveal that Fgf-mediated alveolar regeneration occurs in a spatially specific manner and highlights the critical parallel inputs that control AT2 cell proliferation in an inflammatory injury and regeneration model. Importantly, our studies reveal that AT2 cell identity is actively maintained during regeneration and suggest that Fgfr2 acts not only to promote proliferation but also to restrain AT2 cell fate.
RESULTS
Fgfr2 restricts AT2 cell identity during postnatal lung development
To assess the role of Fgfr2 in AT2 cell development during alveologenesis, we generated SftpcCreERT2;Fgfr2fl/fl;R26EYFP mice to both functionally inactivate Fgfr2 and to trace the fate of the AT2 cell lineage starting at postnatal day zero (P0). We found that AT2 cell-specific loss of Fgfr2 resulted in an increase in the percentage of lineage-traced cells expressing the AT1 cell marker Hopx (Figure 1A). Although the majority of these Hopx+ lineage-traced cells also expressed the canonical AT2 cell marker Sftpc at P5, as alveologenesis progressed to P15 and P28, these cells lost expression of Sftpc, while retaining Hopx expression. This progressive loss of Sftpc and gain of Hopx expression in lineage-traced cells suggests that loss of Fgfr2 in AT2 cells leads to a transition from an AT2 to an AT1 cell fate (Figures 1A–1C). We confirmed that Fgfr2 was functionally inactivated in Fgfr2 mutant AT2 cells using a custom RNA fluorescence in situ hybridization (FISH) probe to detect the deleted Fgfr2 exons 8–10 (Figure 1D).
Figure 1. Fgfr2 maintains AT2 cell identity during alveologenesis.

(A) Immunohistochemistry (IHC) for the SftpcCreERT2 lineage trace marker EYFP, an AT1 cell marker (HOPX), and an AT2 cell marker (SFTPC) in P5, P15, and P28 lungs. This staining demonstrates a loss of AT2 and gain of AT1 cell fate in Fgfr2 mutant lungs over the course of alveologenesis. Zoomed in regions marked by dashed white boxes, dashed white circles demarcate AT2 cells, dashed yellow circles and arrows indicate HOPX+ cells.
(B) Quantification of the percentage of SFTPC+HOPX+ lineage-traced cells at P5, P15, and P28 between control and SftpcCreERT2;Fgfr2fl/fl (knockout [KO]) cells.
(C) Quantification of the percentage of HOPX+ lineage-traced cells at P5, P15, and P28 between control and KO cells.
(D) RNA fluorescence in situ hybridization (FISH) staining, using a custom probe designed to bind the region of Fgfr2 flanked by loxP sites, demonstrates the successful inactivation of Fgfr2 in lineage-traced cells in mutant lungs. Dashed white lines demarcate AT2 cells.
(E) Staining for the lineage trace marker EYFP, HOPX, and EdU at P5 shows an increase in EdU+ lineage-traced AT2 cells. Zoomed in regions marked by dashed white boxes, dashed white circles and arrows demarcate AT2 cells, yellow arrow indicates an AT1 cell.
(F) Quantification of the percentage of EdU+ control and KO AT2 cells.
(G) IHC for cleaved CASPASE 3 reveals no increase in cell death in Fgfr2-deficient AT2 cells.
(H and I) Morphometric analysis measuring mean linear intercept (MLI) on P28 H&E stained slides.
(J–L) Large images show the lineage trace and HOPX, small images also show NKX2.1. Quantification of AT1 cells as a percentage of total epithelial cells and of lineage labeled cells per mm3 of tissue. White boxes demarcate zoomed in region, white circles mark an AT2 cell, and yellow circles indicate an AT1 cell.
All quantification data are represented as mean ± SEM. Two-tailed t tests: ns, not significant; *p ≤ 0.05, **p ≤ 0.01, n = 4–7 mice per group. All scale bars, 50 μm.
AT2 cells are known to proliferate to a limited degree over the course of alveologenesis (Frank et al., 2016; Yang et al., 2016). Therefore, we compared AT2 cell proliferative potential between control and Fgfr2 mutant lungs by 5-ethynyl-2′-deoxyuridine (EdU) incorporation around the earliest peak in AT2 cell proliferation at P5 (Frank et al., 2016). These data reveal more EdU incorporation in Fgfr2-deficient AT2 cells compared to controls, suggesting there is an increase in AT2 cell proliferation on loss of Fgfr2 at this time (Figures 1E and 1F). EdU+ Fgfr2-deficient AT2-derived AT1 cells were rare, consistent with the minimal proliferation of the normal AT1 cell population during postnatal development (Frank et al., 2016; Yang et al., 2016). Additionally, there was no apparent change in markers of cell death, suggesting that the increased in the percentage of lineage-traced AT1 cells was not due to loss of AT2 cells (Figure 1G). To determine whether the loss of AT2 cell fate in Fgfr2 mutant lungs impacts adult lung architecture, we performed mean linear intercept (MLI) analysis on P28 lungs and found no significant morphometric difference between control and mutant lungs (Figures 1H and 1I). Further, although the expansion of Fgfr2-deficient AT2 cells we observed at P5 did not lead to a statistically significant increase in total lineage-traced cells at P28, it seemed to increase the AT2 cell population enough that the percentage of total AT1 cells of the total alveolar epithelium at P28 appeared unchanged (Figures 1J–1L). These results show that Fgfr2 acts to restrict AT2 cell fate during postnatal alveologenesis.
Fgfr2 is dispensable for AT2 cell homeostasis
To determine whether Fgfr2 is required for AT2 cell homeostasis in the adult lung, we inactivated Fgfr2 in AT2 cells and lineage-traced these cells in adult mice to assess lung morphometry and AT2 cell fate at three time points: 1 month, 6 months, and 1 year. We observed no obvious change in lung architecture in Fgfr2 mutant lungs compared to controls (Figure 2A). Moreover, MLI analysis revealed no difference between mutant and control lungs at any time point (Figure 2B). Although the lung is largely a quiescent organ at homeostasis, AT2 cells are known to self-renew and differentiate into AT1 cells slowly over time during normal homeostasis (Barkauskas et al., 2013). To determine whether loss of Fgfr2 would impact long term AT2 cell survival or AT2 to AT1 cell differentiation at homeostasis, we compared the number of lineage-labeled cells and the percentage of lineage-labeled AT1 cells in Fgfr2-deficient versus control lungs. We found no change in the number of lineage-labeled cells between Fgfr2 mutant and control lungs at any time point, suggesting there was no change in AT2 cell survival over time (Figures 2C and 2D). Even after 1 year, we did not observe a difference in the percentage of lineage labeled AT1 cells between mutant and control lungs, suggesting mutant AT2 cells exhibited normal homeostatic turnover into AT1 cells over time (Figures 2C and 2E). Together, our results demonstrate that Fgfr2 is not required in AT2 cells to maintain lung alveolar homeostasis.
Figure 2. Fgfr2 is not required to maintain AT2 cell identity during adult homeostasis.

(A) H&E staining of control and Fgfr2-deficient lungs 1 month, 6 months, and 1 year post tamoxifen induction.
(B) Morphometric analysis reveals no statistically significant difference in MLI between control and mutant lungs at any time point.
(C) IHC for the lineage trace marker EYFP, HOPX, and SFTPC at 1 month, 6 months, and 1 year post tamoxifen induction. Dashed white boxes demarcate zoomed in regions, dashed white circles mark AT2 cells, and dashed yellow circles demarcate AT1 cells.
(D and E) Quantification of the number of lineage-traced cells per mm3 and the percentage of lineage-traced AT1 cells shows no statistically significant difference between control and Fgfr2-deficient lungs at any time point.
All quantification data are represented as mean ± SEM. Two-tailed t tests: ns, not significant; n = 4–5 mice per group. All scale bars, 50 μm.
Zonal analysis reveals a spatially specific role for Fgfr2 in regulating AT2 cell proliferation
To examine whether Fgfr2 is required for AT2 cell-mediated lung regeneration, we investigated the behavior of adult Fgfr2-deficient AT2 cells after acute influenza infection. In control lungs 14 days post influenza infection (dpi), we found enriched expression of Fgfr2 ligands Fgf7 and Fgf10 in morphologically perturbed areas of tissue (Figures 3A and S1A). This enrichment is consistent with previous reports that both the intracellular signaling and the accompanying injury response differ across the heterogeneously damaged tissue (Xi et al., 2017; Katsura et al., 2019; Zacharias et al., 2018). Moreover, our lab previously described the heterogenous damage caused by influenza and identified distinct zones of injury (Zacharias et al., 2018). However, the distinction between these zones can often be difficult to discern, making analysis challenging. To ensure rigorous, reproducible analysis, we developed a lung damage assessment program, which leverages the heterogenous nature of lung injury and computer vision to unbiasedly bin injured regions into distinct zones. Using a computational image analysis approach that clusters image pixels based on the degree of staining, we defined three zones of injury for robust and simple analysis: “severe,” “damaged,” and “normal” (Figure 3B). Severe zones are characterized by maximal alveolar damage. A hallmark of severe influenza infection is the emergence of Keratin 5 (Krt5) expressing airway epithelial cells, which migrate into the alveolar space in an attempt to provide a rapid but dysplastic response to injury (Kumar et al., 2011; Ray et al., 2016; Zuo et al., 2015; Vaughan et al., 2015; Fernanda de Mello Costa et al., 2020). We found Krt5+ cells exclusively in severe zones, whereas markers of AT1 cells are largely absent from this zone (Figures 3C, 3E, and 3F). Although these zones are typically thought to be alveolar deserts, we find a small number of AT2 cells in some of these zones (Figures 3C and 3G). The AT2 cells in severe zones are highly proliferative and very rarely differentiate into AT1 cells in this injury context as noted by the vast majority of SftpcCreERT2-traced cells in these zones expressing Sftpc (Figure 3C). In contrast, damaged zones are defined by active morphological remodeling, are devoid of Krt5+ cells, and harbor dense collections of AT1 and AT2 cells (Figures 3D and 3E′–3G′). These zones contain both proliferating and differentiating AT2 cells (Figure 3D). Normal zones similarly lack Krt5+ cells and consist of largely homeostatic tissue architecture with both AT1 cells lining the alveolar walls and AT2 cells distributed throughout the alveoli. (Figures 3E″–G″). To determine whether Fgfr2 regulates AT2 cell-mediated lung regeneration, we compared the percentage of Ki67+ cells in control or Fgfr2-deficient AT2 cells in both damaged and severe zones on 14 dpi. Although we saw a decrease in proliferative AT2 cells in damaged zones consistent with previous reports, we did not see a statistically significant change in proliferation in AT2 cells in severe zones (Figures 3H and 3I). The lack of a significant, detectable difference in severe zones could not be explained by an inability of Fgfr2-deficient AT2 cells to survive injury as we confirmed the presence of Fgfr2-deficient AT2 cells in both severe and damaged zones at 14 dpi (Figure S1B). Moreover, although there is some AT2 cell proliferation in normal zones, it is quite variable, and no difference was observed between Fgfr2-deficient or replete AT2 cells at 14 dpi (Figures S1C and S1D). Together, our data suggest that Fgfr2 is preferentially important to promote AT2 cell proliferation in damaged zones, which are sites of active tissue regeneration (Figure 3J).
Figure 3. Fgfr2 drives AT2 cell proliferation in a spatially restricted manner.

(A) Experimental schematic indicating the lungs were harvested 14 days post influenza infection (dpi).
(B) Representative images showing the workflow of the lung damage assessment program run on an H&E stained control mouse lung section 14 dpi.
(C) Left image: zoom in from the clustered image in (B). White lines demarcate the border between the severe and damaged clusters encircling the severe zone. Right image: representative IHC for the SftpcCreERT2 lineage trace marker EYFP, KI67, and SFTPC in the severe zone. White arrows indicate KI67+ lineage-traced AT2 cells.
(D) Left image: zoom in from the clustered image in (B). White lines demarcate the border between the damaged and normal clusters encircling the damaged zone. Right image: representative IHC for the SftpcCreERT2 lineage trace marker EYFP, HOPX, and SFTPC in the damaged zone. The dashed white box demarcates zoomed in area, dashed white circles mark a lineage-traced AT2 cell, and dashed yellow circles mark a lineage-traced AT1 cell.
(E–G″) IHC for KRT5 (E), PDPN (F), and SFTPC (G) in severe zones, damaged zones (E′)–(G′), and normal zones (E″)–(G″).
(H) IHC for the lineage trace marker EYFP, KI67, and SFTPC in control and Fgfr2-deficient lungs in both damaged and severe zones. White dashed lines mark KI67+ lineage-traced AT2 cells.
(I) Quantification of the percentage of KI67+ lineage-traced AT2 cells shows a statistically significant decrease in Fgfr2-deficient lungs compared to controls specifically in damaged zones and not in severe zones.
(J) Summary diagram showing the distribution of AT2 cells (green, proliferating AT2 cells drawn with pink nuclei and segregating chromosomes) and AT1 cells (red) across severe, damaged, and normal zones. Dashed white box outlines a single AT2 cell in a damaged zone. Expanded dashed white box demonstrates that Fgfr2 drives AT2 cell proliferation in damaged zones.
All quantification data are represented as mean ± SEM. Two-tailed t tests: ns, not significant; *p ≤ 0.05, n = 5–6 mice per group. All scale bars, 50 μm.
Multiple and redundant inputs promote AT2 cell proliferation
To investigate more directly whether Fgfr2 is required for AT2 cell proliferation, we employed an ex vivo organoid model to compare the growth of isolated, lineage-traced Fgfr2-deficient versus control AT2 cells from uninjured mice (Figure S2A) (Barkauskas et al., 2013). Consistent with our previous findings in severe zones in the influenza injury and regeneration model, we observed no obvious difference in size between control and mutant organoids after 21 days of culture (Figure 4A). To verify that this lack of a difference was not due to competitive loss of Fgfr2-deficient AT2 cells from impeded survival or growth of Fgfr2-replete cells resulting from incomplete inactivation of Fgfr2, we confirmed the loss of Fgfr2 in these mutant organoid cultures (Figure 4A). Moreover, we verified expression of Sftpc and Ager, an AT1 cell marker, in these mutant organoids, suggesting that Fgfr2-deficient AT2 cells were able to expand and differentiate comparably to controls (Figure 4A). Comparing control and mutant organoid growth over the course of 21 days in culture, we found no statistically significant difference in organoid size or growth rate over time (Figures 4B and 4C). Additionally, we found no difference in colony forming efficiency, suggesting that Fgfr2-deficient AT2 cells survive and grow comparably to controls (Figure 4D). Because our findings suggest that Fgfr2 is not required for AT2 cell proliferation, we sought to determine whether loss of Fgfr2 would impact AT2 cell responsiveness to other mitogenic signals (Figure 4E). Fgfr2 is the primary receptor for Fgf7, which we have previously shown stimulates mature AT2 cell proliferation (Zacharias et al., 2018; Zepp et al., 2017). To investigate the responsiveness of control versus Fgfr2-deficient AT2 cells to Fgf signaling, we provided exogenous Fgf7 to the organoid cultures. Control AT2 cell-derived organoids exhibited robust growth on Fgf7 stimulation due to augmented AT2 cell proliferation and differentiation into AT1 cells consistent with our previous studies (Figure 4F) (Zacharias et al., 2018; Zepp et al., 2017). Conversely, organoids generated from Fgfr2-deficient AT2 cells did not respond to Fgf7 treatment (Figure 4F). Visualizing growth over time, we found that control organoids responded rapidly to exogenous Fgf7 (Figure 4G). Fgf7-treated control organoids demonstrated a statistically significant increase in growth compared to untreated control and Fgf7-treated Fgfr2-deficient organoids after 7 days of stimulation (14 days of culture) (Figures 4G–4I). However, this growth plateaued after 9 days of stimulation (16 days of culture) (Figure 4G). Thus, Fgfr2 deficient AT2 cells do not respond to exogenous Fgf7, even though the level added in these assays is likely higher than what occurs in vivo during alveolar regeneration.
Figure 4. Fgfr2 is not required for AT2 cell proliferation.

(A) Left: images using endogenous fluorescence of a representative well of AT2 cell-derived organoids from either SftpcCreERT2;R26RtdTomato (top) or SftpcCreERT2;Fgfr2fl/fl;R26RtdTomato (bottom) mice at 21 days of culture, white arrows indicate a single organoid (scale bar, 500 μm). Middle: RNA FISH for Fgfr2 on representative control (top) and Fgfr2-deficient (bottom) AT2 cell-derived organoids. Dashed white boxes mark zoomed in regions. Right: IHC for AGER and SFTPC on representative control (top) and Fgfr2-deficient (bottom) AT2 cell-derived organoids.
(B) Analysis of organoid size from 11 to 21 days of culture. Each dot represents the average of organoids derived from three individual mice. Dashed black box demarcates 21 days of culture.
(C and D) Analysis of organoid size and colony forming efficiency at 21 days of culture. Each dot represents the average of at least three replicate wells from one mouse for a total of n = 3 mice per group.
(E) Experimental schematic outlining the timing of cytokine treatment. Transforming growth factor β (TGF-β) inhibitor was added for the first week of culture. At 7 days of culture, cytokines were added for the remainder of the experiment.
(F) Left: images using endogenous fluorescence of a representative well of AT2 cell-derived organoids from either SftpcCreERT2;R26RtdTomato (top) or SftpcCreERT2;Fgfr2fl/fl;R26RtdTomato (bottom) mice at 21 days of culture after treatment with FGF7 for the last 14 days, white arrows indicate a single organoid (scale bar, 500 μm). Middle: RNA FISH for Fgfr2 on representative control (top) and Fgfr2-deficient (bottom) AT2 cell-derived organoids. Dashed white boxes mark zoomed in regions. Right: IHC for AGER and SFTPC on representative control (top) and Fgfr2-deficient (bottom) AT2 cell-derived organoids.
(G) Analysis of organoid size from 11 to 21 days of culture. Each dot on this graph represents the average from three individual mice, n = 3. Dashed black box demarcates 14 days of culture.
(H and I) Analysis of organoid size in untreated versus FGF7 treated control and Fgfr2-deficient (KO) organoids at 14 days of culture highlighted in (G). Each dot represents the average of at least three replicate wells from one mouse for a total of n = 3 mice per group.
(J) Representative images of AT2 cell-derived organoids from either SftpcCreERT2;R26RtdTomato (top) or SftpcCreERT2;Fgfr2fl/fl;R26RtdTomato (bottom) mice at 21 days of culture after treatment with IL-1α, IL-1β, or TNF-α for the last 14 days of culture (scale bar, 500 μm).
(K) Organoid size at 21 days of culture. Each dot represents the average of at least three replicate wells from one mouse for a total of n = 3 mice per group.
All quantification data are represented as mean ± SEM. Two-tailed t tests: ns, not significant; *p ≤ 0.05, **p ≤ 0.01. Scale bars, 50 μm unless otherwise noted.
Recent reports have suggested that inflammatory signaling is important for stimulating AT2 cell-mediated lung regeneration (Choi et al., 2020; Katsura et al., 2019). To examine whether loss of Fgfr2 impacts the AT2 cell response to immune cytokine stimulation, we treated Fgfr2-deficient and control AT2 cell organoids with interleukin (IL)-1α, IL-1β, and tumor necrosis factor alpha (TNF-α). At 21 days of culture, both IL-1α and IL-1β stimulated robust organoid growth, whereas TNF-α led to a slight but not statistically significant increase in organoid size, regardless of presence or absence of Fgfr2 (Figures 4J and 4K). Additionally, we found no difference between the organoid growth of Fgfr2-deficient and control organoids stimulated with any of the three cytokines (Figures S2B–S4D). Together, our results show that immune cytokines promote robust AT2 cell growth independent of Fgfr2 signaling, suggesting that Fgfr2 is not the sole regulator of AT2 cell proliferation and alveolar regeneration integrates multiple proliferative inputs.
Loss of Fgfr2 promotes AT2 to AT1 cell differentiation
To assess whether loss of Fgfr2 would alter the fate of AT2 cells during lung regeneration similar to our findings during alveologenesis, we lineage-traced Fgfr2-deficient and control AT2 cells and evaluated AT2 to AT1 cell differentiation through expression of Sftpc and Hopx in damaged zones at 14 and 28 dpi. Fgfr2-deficient AT2 cells differentiated more readily into AT1 cells in damaged zones leading to an increase in lineage-traced AT1 cells in mutant lungs at 14 dpi (Figures 5A, 5B, S3A, and S3B). This difference persisted to 28 dpi (Figures 5C and 5D). Moreover, the increase in lineage-traced AT1 cells in mutant lungs led to an overall increase in total AT1 cells as a percentage of total alveolar epithelium in damaged zones (Figures 5E and 5F). AT1 cells are known to spread dramatically during normal development, contributing to the complex architecture of the adult lung alveolus (Wang et al., 2016; Yang et al., 2016). We surmised that the differentiation of AT2 cells into large squamous AT1 cells serves not only to replace lost epithelium destroyed as a consequence of injury, but also as a mechanism to restore the homeostatic alveolar architecture and that this architectural remodeling process would become altered if the ratio of AT1:AT2 cells was perturbed. We found that the increase in AT2 to AT1 cell differentiation, which results in an overall increase in AT1 cells in Fgfr2-deficient lungs, did impact the alveolar architecture of mutant lungs (Figure 5G). Because it is difficult to know the extent of injury in normal zones where the tissue has largely regenerated by 28 dpi, we focused our morphological analysis in damaged zones. Morphologic analysis of damaged zones in Fgfr2-deficient versus control lungs at 28 dpi revealed an increase in the average alveolar area of mutant lungs resulting from large, extended air spaces due to an increase in AT1 cells (Figures 5H and 5I). This H1N1 influenza model does not commonly elicit a fibrotic response, and we confirmed there was no obvious change in collagen deposition or infiltration of Acta2 expressing myofibroblasts between control and mutant lungs (Figures S3C and S3D) (Kumar et al., 2011). Moreover, although we did note the presence of airway-derived Krt5+ pods common to severe influenza injury in both control and mutant lungs, we did not observe an obvious difference between the size or frequency of these pods between control and mutant lungs (Figure S3E) (Kumar et al., 2011; Zacharias et al., 2018; Vaughan et al., 2015). Together, these data demonstrate that Fgfr2 acts to restrain AT2 cell fate, promote expansion of the AT2 cell pool, and regulate AT2 to AT1 cell differentiation to drive restoration of the homeostatic alveolar architecture (Figure 5J).
Figure 5. Fgfr2 maintains AT2 cell identity.

(A–D) IHC for the lineage trace marker EYFP, HOPX, and SFTPC 14 and 28 dpi in damaged zones and quantification of the percentage of lineage labeled AT1 cells (marked by yellow arrows and dashed yellow circles). Zoomed in regions marked by dashed white boxes.
(E and F) IHC for HOPX and NKX2.1 at 28 dpi shows a statistically significant increase in AT1 cells (marked by yellow arrows and dashed yellow circles) as a percentage of total epithelium in damaged zones. Zoomed in regions marked by white boxes.
(G) H&E staining at 28 dpi demonstrates stark morphological differences between control and Fgfr2-deficient lungs. Zoomed in regions marked by dashed black boxes (scale bars, 500 μm and 100 μm).
(H and I) Morphological analysis of alveolar area in damaged zones using IHC for PDPN to outline alveoli shows a statistically significant increase in average alveolar area in Fgfr2-deficient versus control lungs. Zoomed in regions marked by dashed white boxes.
(J) Summary diagram showing the distribution of AT2 cells (green, proliferating AT2 cells drawn with pink nuclei and segregating chromosomes) and AT1 cells (red) across severe, damaged, and normal zones. Dashed white box outlines a single AT2 cell in a damaged zone. Expanded dashed white box demonstrates that Fgfr2 maintains AT2 cell identity in damaged zones.
All quantification data are represented as mean ± SEM. Two-tailed t tests: *p ≤ 0.05, **p ≤ 0.01, n = 5–6 mice per group. Scale bars, 50 μm unless otherwise noted.
AT2 cell division and differentiation are uncoupled processes
Because Fgfr2 regulates both AT2 cell proliferation and identity in a spatially restricted manner, we sought to determine whether these two cellular processes were linked during tissue regeneration in the lung. To investigate whether cell division and differentiation are coupled processes in the lung alveolus, we employed an Ect2 genetic deletion model to block cell division. Ect2 is essential for cytokinesis, and loss of Ect2 leads to a failure in cell division (Cook et al., 2011; Windmueller et al., 2020). We hypothesized that genetic inactivation of Ect2 would leave AT2 cells receptive to proliferative stimuli while preventing AT2 cell division, resulting in the generation of binucleated cells during lung regeneration (Figure 6A). As expected, we confirmed the presence of binucleated AT2 cells, containing lamellar bodies and indication of functional surfactant production (Figure 6B). Both binucleated AT1 and AT2 cells are present at 14 and 28 dpi, but binucleated AT1 cells are found exclusively in damaged zones at 14 dpi, consistent with AT2 cell differentiation occurring in these zones (Figures 6C, 6D, and S4A–S4F). Importantly, the presence of binucleated AT2 cell-derived AT1 cells demonstrates that cell division is not required for differentiation to occur. The presence of binucleated AT1 cells exclusively in damaged zones at 14 dpi along with the appearance of binucleated AT2 cells in normal and severe zones at 28 dpi suggests that blocking cytokinesis alone is not sufficient to drive an AT2 cell to differentiate into an AT1 cell (Figures 6C and S4C–S4F). However, Ect2-deficient AT2 cells can become binucleated at any time during regeneration in any injury zone, highlighting the importance of determining when these binucleated cells arise and whether they stably maintain their identity. To address this point and to further determine whether cytokinesis is required to maintain AT2 cell identity, we performed an EdU pulse-chase experiment to mark cells that had undergone the S phase of the cell cycle, including emerging binucleated cells, and followed their fate decisions. We pulsed mice with EdU once a day for 5 consecutive days beginning at 9 dpi when AT2 cells should be proliferating and chased for 2 weeks until 28 dpi (Figure 6E) (Katsura et al., 2019). The presence of EdU+ binucleated AT1 and AT2 cells in damaged zones at 28 dpi confirms that binucleated AT2 cells can differentiate into and maintain an AT1 cell fate, and these data show that AT2 cells can stably maintain their identity independent of their ability to divide for at least 2 weeks after injury (Figures 6F and 6G). Our data demonstrate that cell division is not required to maintain AT2 cell fate, demonstrating that loss of the ability to undergo cytokinesis does not deterministically result in differentiation of AT2 cells into an AT1 cell fate. To test this possibility further, we investigated whether blockade of AT2 cell division would increase the number of AT1 cells as a percentage of the total alveolar epithelium during regeneration. Quantification of the number of AT1 cells as a percentage of Nkx2.1+ cells in damaged zones in Ect2-deficient and control lungs at 28 dpi shows that loss of AT2 cell division does not change the overall number of AT1 cells as a percentage of the total alveolar epithelium (Figures 6H and 6I). Together, these data show that AT2 cell division and differentiation are uncoupled processes and that cell fate is maintained independent of proliferative competency.
Figure 6. Cell division is not required to maintain AT2 cell identity during regeneration.

(A) Model showing Ect2 is required for cytokinesis.
(B) Electron micrographs of a normal AT2 cell (top) and a binucleated AT2 cell (bottom) from EPCAM-sorted cells from SftpcCreERT2;Ect2fl/fl mice 34 dpi (n, nucleus; scale bars, 2 μm).
(C) IHC for NKX2.1, CDH1, and LAMP3 (left) and NKX2.1, HOPX, and AGER (right) at 14 dpi in damaged zones. Zoomed in regions marked by dashed white boxes and individual nuclei marked by dashed white circles (AT2 cells) or dashed yellow circles (AT1 cells).
(D) IHC for the lineage trace marker EYFP, CDH1, and LAMP3 (left) and the lineage trace marker EYFP, HOPX, and AGER (right) at 28 dpi in damaged zones. Zoomed in regions marked by dashed white boxes and individual nuclei marked by dashed white circles (AT2 cells) or dashed yellow circles (AT1 cells).
(E) Experimental schematic showing five EdU pulses between 9 and 13 dpi followed by a 2-week chase.
(F) Staining for AGER, HOPX, and EdU (top) and CDH1, LAMP3, and EdU (bottom) at 28 dpi in damaged zones. Zoomed in regions marked by dashed white boxes and individual nuclei marked by white arrows (AT2 cells) or yellow arrows (AT1 cells).
(G) Model showing, despite loss of the ability to divide, AT2 cells can either maintain their fate or differentiate into AT1 cells.
(H) IHC for the lineage trace marker EYFP, HOPX, and SFTPC (left) and NKX2.1, HOPX, and AGER (right) at 28 dpi in damaged zones. Zoomed in regions marked by dashed white boxes, AT2 cells marked by white arrows, AT1 cells indicated by yellow arrows, and individual AT1 cell nuclei marked by dashed yellow circles.
(I) Quantification shows no change between control and Ect2-deficient (KO) lungs in the number of AT1 cells as a percentage of total epithelium in damaged zones.
Quantification data are represented as mean ± SEM. Two-tailed t test: ns, not significant; n = 3 mice per group. Scale bars, 50 μm unless otherwise noted.
DISCUSSION
Restoration of gas exchange after lung injury requires both replenishment of alveolar epithelial cells and re-establishment of alveolar architecture. The major facultative progenitor of the lung alveolus, the AT2 cell, is thought to play a critical role in these processes through proliferating and differentiating into AT1 cells. However, it remains unclear how AT2 cells balance the decision to divide versus differentiate. Our studies reveal that multifarious parallel pathways converge to drive AT2 cell proliferation and that Fgfr2 acts to restrain AT2 cell identity in a spatially restricted manner to balance proliferation versus differentiation to restore alveolar architecture and normal lung function after lung injury.
Fgf signaling is essential for prenatal development in the lung and it serves to guide branching morphogenesis and to maintain AT2 cell fate (Abler et al., 2009; Chang et al., 2013; Lebeche et al., 1999; Li et al., 2018; Liberti et al., 2019; Weaver et al., 2000; Mori et al., 2019; Sekine et al., 1999; De Moerlooze et al., 2000). Our results demonstrate that although Fgf signaling via Fgfr2 is likely not the only regulator of AT2 cell identity, it remains critical during postnatal development to maintain AT2 cell fate long after alveolar epithelial fate specification (Frank et al., 2019). Prior work has suggested that Fgfr2 is important for adult lung epithelial homeostasis and repair after injury, but whether Fgfr2 acts purely as a mitogenic signal transducer in the lung epithelium has remained unclear (Dorry et al., 2020; Balasooriya et al., 2017; Yuan et al., 2019). During airway homeostasis, loss of a single Fgfr2 allele leads to depletion of the basal cell lineage into an immature luminal cell fate, whereas after injury overexpression of Fgf10 increases airway cell contribution to the AT2 cell lineage, suggesting a role for Fgfr2 signaling in regulating lung epithelial cell fate (Balasooriya et al., 2017; Yuan et al., 2019). Although our data show that AT2 cell-specific loss of Fgfr2 for up to 1 year did not lead to any change in lung morphometry, AT2 cell survival, or AT2 to AT1 cell differentiation, our results are consistent with Fgfr2 being critical for regulating AT2 cell fate both during development and lung regeneration.
Our results reveal that Fgfr2-deficient AT2 cells remain competent to respond to other mitogenic stimuli, in particular inflammatory cytokines. This is especially relevant in the context of an influenza injury model, where the immune response plays an important role in both injury resolution and regeneration. In particular, immune cytokines such as IL-1α and IL-1β play key roles in promoting AT2 cell-mediated alveolar regeneration (Katsura et al., 2019). A recent study revealed that IL-1 receptor-expressing AT2 cells respond to acute lung injury by proliferating, but chronic IL-1β stimulation decreased AT2 to AT1 cell differentiation (Choi et al., 2020). These results demonstrate that multiple, parallel, and possibly redundant proliferative stimuli encourage AT2 cell proliferation, highlighting the importance of AT2 cell proliferation to promote rapid tissue regeneration in the lung. Moreover, the similarity of our organoid results to the behavior of AT2 cells in severe versus damaged zones underscores the importance of comparing ex vivo experiments in the context of in vivo biology and suggests that additional advances in culturing conditions will benefit these organoid models (Katsura et al., 2020; Weiner et al., 2019; Shiraishi et al., 2019a, 2019b).
Although cell proliferation is an essential aspect of robust tissue regeneration, how this process is balanced by the need for differentiation has remained less clear in the lung. We demonstrate that although AT2 cells do not require the ability to divide to differentiate into AT1 cells, AT2 cells similarly do not require proliferative competency to maintain their identity. Indeed, the persistence of binucleated cells versus further multinucleation or death as a result of blocking cytokinesis suggests that AT2 cells actively balance the decision to divide versus differentiate. These findings suggest that not only division, but also proliferation may be uncoupled from fate choice in AT2 cells during regeneration. Moreover, the loss of the ability to divide does not perturb the balance between AT1 and AT2 cells as with the loss of Fgfr2, suggesting that Fgfr2 acts to maintain AT2 cell identity independently of its role in mitogenesis. Previous work showed that a Wnt-responsive alveolar epithelial progenitor (AEP) sublineage within the AT2 cell population contributes primarily to the proliferative response of AT2 cells (Zacharias et al., 2018; Nabhan et al., 2018). In light of our current findings, it is possible that the non-AEP fraction of the AT2 cell population more readily converts into AT1 cells versus undergoing cell division. This differential fate choice likely hinges both on the local signaling niche as well cell intrinsic factors reinforcing a more or less plastic AT2 cell identity, consistent with epithelial progenitor cell behavior in other organs like the skin and intestine (Nabhan et al., 2018; Zepp et al., 2017; Xi et al., 2017; Rompolas et al., 2012, 2013; Gehart and Clevers, 2019). Such a balance between cellular responses within the AT2 cell population may allow for rapid regeneration to occur, while maintaining the surfactant production required for alveolar homeostasis.
One of the challenges of defining the molecular mechanisms driving pro-regenerative cellular behaviors during lung repair is the heterogeneous nature of lung injury. To overcome this difficulty, we developed an unbiased computational imaging approach, which takes advantage of the heterogenous architectural distortion typical to lung injury, to bin different domains of injury and regeneration in the lung. Our analysis demonstrates that most of the morphological remodeling of the tissue due to AT2 cell differentiation into AT1 cells occurs in areas separate from the formation of dysplastic keratinized epithelium, consistent with previous reports (Zacharias et al., 2018). Our studies underscore the importance of examining lung repair and regeneration in a zonally specific manner as the AT2 cell response differs significantly across different regions of injured tissue. Moreover, utilizing this imaging approach to analyze morphological changes in specific zones allows unbiased assessment of secondary defects and promotes reproducibility in comparison between conditions by controlling for injury heterogeneity. Use of this lung damage assessment program may better define and clarify future studies on the impact of severe injury and subsequent regeneration in the lung.
Given the complex signaling milieu that occurs after severe lung injury, multiple parallel and competing pathways are likely required to provide a robust balance between cell proliferation and differentiation and to promote proper restoration of functional homeostasis. Our findings reveal that a combination of pathways and inputs are required for the proliferative response to acute injury in the lung alveolus. Moreover, we have demonstrated that cytokinesis is uncoupled from AT2 to AT1 cell differentiation, indicating a complex and flexible molecular response of the alveolar epithelium to injury.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for further information and resources related to this study may be directed to the Lead Contact, Edward Morrisey (emorrise@pennmedicine.upenn.edu).
Materials availability
The RNAscope probe Fgfr2-O1 (Cat#: 806301) was generated by and is available from Advanced Cell Diagnostics upon request.
Data and software availability
Lung damage assessment software (LungDamage) is publicly available on Github (https://github.com/WALIII/LungDamage).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse lines
Development and genotyping information for mouse lines SftpcCreERT2, Fgfr2fl/fl (Jackson Laboratory stock # 007569), R26REYFP (Jackson Laboratory stock # 007903), Ai14(RCL-tdT)-D (R26RtdTomato) (Jackson Laboratory stock # 007914), and Ect2fl/fl have been previously described (Chapman et al., 2011; Madisen et al., 2010; Windmueller et al., 2020; Yu et al., 2003). Fgfr2fl/fl, R26RtdTomato, and R26REYFP mouse lines were purchased from the Jackson Laboratory. All mice were maintained on a mixed background (C57BL/6 and CD1). No obvious defects were observed in heterozygous mice, so SftpcCreERT2;Fgfr2fl/+;R26REYFP and SftpcCreERT2;Ect2fl/+;R26REYFP littermates were used as controls for all experiments except for the organoid experiments in Figure 4 where SftpcCreERT2;R26RtdTomato mice were used and the adult one month or longer influenza experiments in Figures 5C–5I,6B, and 6D–6I where SftpcCreERT2;R26REYFP mice, including littermates, were used. Experiments were all performed with a minimum of n = 3 mice per condition of mixed gender, and unless otherwise stated each dot on a graph represents one mouse. All procedures for animal experiments were performed under the guidance of the University of Pennsylvania Institutional Animal Care and Use Committee.
Lung alveolar organoid assay
Organoid assays were performed as previously described with modification (Barkauskas et al., 2013; Frank et al., 2016; Peng et al., 2015; Zacharias et al., 2018; Zepp et al., 2017). In brief, single cell suspensions were made from SftpcCreERT2;R26RtdTomato or SftpcCreERT2;Fgfr2fl/fl; R26RtdTomato mice by physical and enzymatic dissociation and tdTomato+ cells were sorted into FACS buffer (Hanks Balanced Salt Solution (HBSS, GIBCO, catalog # 14175095), 25mM HEPES (GIBCO, catalog # 15630080), 2mM EDTA (Invitrogen, catalog # 15575020), and 2% Fetal Bovine Serum (FBS) (Deville, catalog # FB5001)). Live cell number was verified using a hemocytometer and Trypan Blue Solution, 0.4% (GIBCO, catalog # 15250061). In each technical replicate, 5×103 tdTomato+ cells were combined with 5×104 primary lung fibroblasts in 50% Matrigel (Corning, catalog # 356231) and 50% MTEC-SAGM in a Falcon Cell Culture Insert (Thermo Fisher Scientific, catalog # 08770). The MTEC-SAGM media was made as previously described. In brief, Small Airway Epithelial Cell Growth Basal Media (SABM) (Lonza, catalog # CC-3119) was mixed with selected Small Airway Epithelial Cell Growth Medium supplements (SAGM) (Lonza, catalog # CC-4124): Insulin/Transferrin, Bovine Pituitary Extract, Gentamycin, and Retinoic Acid as well as 0.1 μg/mL Cholera Toxin (Millipore Sigma, catalog # C9903), 25ng/mL EGF (Peprotech, catalog # AF-100–15), and 5% FBS (Zepp et al., 2017; Barkauskas et al., 2013). Cell/matrigel mixtures were allowed to solidify for fifteen minutes before MTEC-SAGM media was added under the transwell into the bottom of the well. Media was changed every other day. Antibiotic-Antimycotic (GIBCO, catalog # 15240062) and 10 μM rock inhibitor (Y-27632 dihydrochloride, Millpore Sigma, catalog # Y0503) were added to the media for the first two days of culture. A Tgfbr1 inhibitor (SB431542, Abcam, catalog # ab120163) was added to the media to a final concentration of 10 μM for the first seven days of culture to promote organoid formation (Katsura et al., 2019). Experiments involving IL-1α (10ng/mL, BioLegend, catalog # 575002), IL-1β (10ng/mL, BioLegend, catalog # 575102), TNFα (10ng/mL, BioLegend, catalog # 575202), or mFGF7 (25ng/mL, R&D Systems, catalog # 5028-KG-025) treatment involved cytokine addition beginning after the first seven days of culture. Concentrations may differ from endogenous levels but were consistent with previous studies (Katsura et al., 2019; Zepp et al., 2017). Treatment occurred during normal media changes and continued for the final two weeks of culture. Images were captured over the course of culture using an EVOS FL Auto 2 Imaging System. On the final day of culture, organoids were removed from transwell inserts, embedded in 4% UltraPure Low Melting Point Agarose (Invitrogen, catalog # 16520050) in HBSS, fixed in 2% paraformaldehyde solution (Thermo Fisher Scientific, catalog # AAJ19943K2) for 30 minutes at room temperature, washed in Phosphate Buffered Saline (PBS) (Quality Biological, catalog # 119–068-151), dehydrated in a graded series of ethanol washes (30%, 50%, 70%, 95%, and 100%), embedded in paraffin wax, and sectioned at a 6 μm thickness. Results are reflective of three independent experiments with at least three replicate wells per condition.
METHOD DETAILS
Tamoxifen delivery
Tamoxifen administration to P0 mice was performed by intraperitoneal injection of 20 μL of a 20mg/mL mixture of tamoxifen in ethanol (10%) and corn oil (90%). Tamoxifen delivery to adults was performed by oral gavage at a dose of 200mg/kg for three consecutive days. All adult experiments were performed on 6–12 week old mice. A two week washout period was observed after the final tamoxifen dose for all in vivo lineage tracing experiments.
EdU incorporation
EdU (Santa Cruz, catalog # sc-284628B) was dissolved in filtered H2O and given via intraperitoneal injection at a dose of 50mg/kg. P5 mice treated with EdU were harvested four hours after treatment. Adult mice were given EdU once per day for five consecutive days from 9 to 13 dpi. EdU staining was performed with the Click-iT EdU cell proliferation kit (Invitrogen, catalog # C10340) according to the manufacturer’s instructions.
Influenza infection
PR8 H1N1 influenza was a kind gift from Dr. John Wherry at the University of Pennsylvania. Virus was diluted in cold PBS and 50uL was delivered to anesthetized mice intranasally at a dose of approximately 1LD50 (determined experimentally based on delivery to six to eight week old C57BL/6 female mice).
Histology, immunohistochemistry, and RNAscope
Lungs were harvested at a constant pressure of 25cm H2O and were fixed overnight at 4°C in 2% paraformaldehyde. Lungs were washed, dehydrated, embedded, and sectioned as stated above for organoids. Hematoxylin and eosin (H&E) and Masson’s trichrome staining were performed according to standard procedures. Immunohistochemistry was performed with the following antibodies: Acta2 (mouse, Millipore Sigma, A5228, 1:200), Ager (Rage) (rat, R&D Systems, MAB1179, 1:50), cleaved Caspase 3 (rabbit, Cell Signaling, 9664s, 1:50), Lamp3 (DC-Lamp) (rat, Novus, DDX0191P-100, 1:100), Cdh1 (E-Cadherin) (rabbit, Cell Signaling, 3195, 1:100), GFP (chicken, Aves Labs, GFP-1020, 1:200), Hopx (mouse, Santa Cruz, sc-398703, 1:100), Keratin 5 (Krt5) (rabbit, Abcam, ab52635, 1:500), Ki67 (mouse, BD Biosciences, 550609, 1:200), Nkx2.1 (Ttf1) (rabbit, Santa Cruz, sc-13040, 1:50), Nkx2.1 (Ttf1) (mouse, Thermo Fisher Scientific, MS-699-P1, 1:25), Pdpn (hamster, Developmental Studies Hybridoma Bank, 8.1.1, 1:100), and Sftpc, (rabbit, Millipore Sigma, AB3786, 1:100). Slides were mounted in Vectashield Antifade Mounting Medium (Vector Laboratories catalog # H-1000) or Slowfade Diamond Antifade Mountant (Invitrogen, catalog # S36972). Slowfade Diamond Antifade Mountant was used to avoid the quenching action of Vectashield on Alexa Fluor 647 secondary antibodies. RNAscope was performed according to the manufacturer’s instructions using the following probes: Mm-Fgf7-no-XHs (Catalog # 443521), Mm-Fgf10 (Catalog # 446371), Fgfr2-O1 (custom probe designed to bind specifically within the floxed region of Fgfr2flox allele). H&E, Masson’s trichrome, and widefield fluorescence images in Figures S3C–S3E were acquired with either a Nikon Eclipse 80i or an EVOS FL Auto 2 Imaging System. All other fluorescence images, including all images used for quantification, were captured using a Zeiss LSM 710 confocal microscope.
Transmission Electron microscopy
Influenza infected lungs 34 dpi were digested as for the lung alveolar organoid assay and total Epcam+ cells were isolated using the Dynabeads FlowComp Flexi Kit according to the manufacturer’s instructions. Briefly, a purified Epcam antibody (Rat, Thermo Fisher Scientific, 14–5791-85) was biotinylated and incubated with digested lung tissue for 20 minutes at 4°C. The cells were subsequently washed with FACS buffer and incubated with FlowComp Dynabeads on a rotating platform for 15 minutes at 4°C before being placed on a magnetic stand. The cells were washed with FACS buffer, then were released from the beads by adding Release Buffer for 20 minutes at 4°C. The magnetic beads were removed using the magnetic stand, and the cells were fixed in 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1M sodium cacodylate buffer, pH 7.4 overnight at 4°C. Samples were washed, post-fixed in 2.0% osmium tetroxide with 1.5% K3Fe(CN)6 for 1 hour at room temperature, and rinsed in dH2O. After dehydration through a graded acetone series, the samples were infiltrated and embedded in EMbed-812 (Electron Microscopy Sciences, Fort Washington, PA). Thin sections were cut then stained with uranyl acetate and SATO lead and were subsequently imaged on a JEOL 1010 electron microscope fitted with a Hamamatsu digital camera using AMT Advantage NanoSprint500 software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Immunohistochemistry quantification
All image quantification was performed on confocal acquired z stack images through 6 μm thick tissue sections. For a given stain, at least 200 cells or five confocal stacks taken at 40x magnification were used for quantification for each individual mouse.
Mean linear intercept
Morphometric analysis of mean linear intercept was performed on images acquired automatically from a scan of an entire lobe using an EVOS FL Auto 2 Imaging System at 20x. Images were manually sorted to avoid large airways and vessels to concentrate analysis on alveolar regions. At least seven images were randomly selected per mouse for analysis. Quantification was performed using custom MATLAB software described previously (Paris et al., 2020; Obraztsova et al., 2020).
Organoid size and colony forming efficiency
Organoid images were captured using endogenous fluorescence on an EVOS FL Auto 2 Imaging System. Images were processed in Fiji using simple thresholding, and were then binarized, subjected to dilation and erosion to remove background. To enable proper visualization of whole organoids, images were subjected to the fill holes function followed by watershedding to prevent organoid overlap. Organoids were then quantified in Fiji using the analyze particles function with a cutoff of 1000 μm2. Organoid areas were pooled from three to four replicate wells per mouse to calculate average size per condition. Colony number was divided by the number of input tdTomato+ cells to determine colony forming efficiency.
Lung damage assessment program
Our lung damage assessment program was developed to enable the direct comparison of similarly injured areas in tissue sections from the same mouse and between different mice. We leveraged previous work from our lab where we identified and binned injured regions by the level of morphological perturbation to develop an unbiased method of mapping the severity of damaged regions across tissue sections (Zacharias et al., 2018). Our MATLAB software utilizes a K-means clustering algorithm to rank and sort points on H&E slides according to staining intensity and density which varies with injury severity (Zacharias et al., 2018). We independently sort all pixels from the tissue images into four clusters. We find that pixels are reproducibly well-sorted into clusters comprised of a background component and three clusters that highly correlate with injury zones, as compared to manual curation. To account for the possibility of bias in the relative clustering of heterogeneously injured tissue into distinct clusters, an image of a single lobe is defined as a means of comparison and each image is clustered together with that image. To further ensure reproducibility, sections from whole lobes were tile-scanned using an EVOS FL Auto 2 Imaging System at 20x, and tissue sections were only compared if they were acquired from the same slide and from a single imaging session to control for differences in acquisition settings or staining. We are able to use the pixel-wise labeling of tissue to identify locations of overlap of clusters that correlate with the two most damaged regions (false-colored in red and green, each region is displayed surrounded by a boundary line). We demarcate the areas of maximal damage (red) as Severe zones, the areas of active morphological remodeling (green) as Damaged zones, and the homeostatic appearing regions (blue) as Normal zones. Once the maps are generated, they are used to identify the corresponding zones on immediately adjacent slides, stained for various markers of interest.
Alveolar area measurement
Alveolar area analysis was performed on z stack images of Pdpn stained slides captured using a Zeiss LSM 710 confocal microscope. Maximum projections were created from these stacks in Fiji to produce 2D images with sharp alveolar boundaries. These images were further processed in Fiji using simple thresholding. Images were then binarized, inverted, and subjected to dilation and erosion to remove background. Quantification was performed in Fiji using the analyze particles function with a cutoff of 10 μm2 and a circularity cutoff of 0.2.
Statistics
All results are reported as mean ± SEM. Comparisons were made using two-tailed t tests with p ≤ 0.05 considered significant. Statistical tests were conducted in Graphpad Prism8 software.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Acta2 | Millipore Sigma | Cat#: A5228; RRID: AB_262054 |
| Rat anti-Ager (Rage) | R&D Systems | Cat#: MAB1179; RRID: AB_2289349 |
| Rabbit anti-Cleaved Caspase 3 | Cell Signaling | Cat#: 9664s; RRID: AB_2070042 |
| Rat anti-Lamp3 (DC-Lamp) | Novus | Cat#: DDX0191P-100; RRID: AB_2827532) |
| Rabbit anti-Cdh1 (E-Cadherin) | Cell Signaling | Cat#: 3195; RRID: AB_2291471 |
| Chicken anti-GFP | Aves Labs | Cat#: GFP-1020; RRID: AB_10000240 |
| Mouse anti-Hopx | Santa Cruz | Cat#: sc-398703; RRID: AB_2687966 |
| Rabbit anti-Keratin 5 | Abcam | Cat#: ab52635; RRID: AB_869890 |
| Mouse anti-Ki67 | BD Biosciences | Cat#: 550609; RRID: AB_393778 |
| Rabbit anti-Nkx2.1 (TTF1) | Santa Cruz | Cat#: sc-13040; RRID: AB_793532 |
| Mouse anti-Nkx2.1 (TTF1) | Thermo Fisher Scientific | Cat#: MS-699-P1; RRID: AB_142087 |
| Syrian hamster anti-Pdpn | Developmental Studies Hybridoma Bank | Cat#: 8.1.1; RRID: AB_531893 |
| Rabbit anti-Sftpc | Millipore Sigma | Cat#: AB3786; RRID: AB_91588 |
| Rat anti-Epcam | Thermo Fisher Scientific | Cat#: 14–5791-85; RRID: AB_953626 |
| Mm-Fgf7-no-XHs (RNA FISH probe) | Advanced Cell Diagnostics | Cat#: 443521 |
| Mm-Fgf10 (RNA FISH probe) | Advanced Cell Diagnostics | Cat#: 446371 |
| Fgfr2-O1 (RNA FISH probe) | Advanced Cell Diagnostics | Cat#: 806301 |
| Bacterial and virus strains | ||
| H1N1 Pr8 | John Wherry Lab (University of Pennsylvania) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Fgf7 | R&D Systems | Cat#: 5028-KG-025 |
| IL-1α | BioLegend | Cat#: 575002 |
| IL-1β | BioLegend | Cat#: 575102 |
| TNFα | BioLegend | Cat#: 575202 |
| Small Airway Epithelial Cell Growth Medium (SAGM) supplements | Lonza | Cat#: CC-4124 |
| Small airway epithelial cell growth basal media (SABM) | Lonza | Cat#: CC-3119 |
| Cholera toxin | Millipore Sigma | Cat#: C9903 |
| EGF | Peprotech | Cat#: AF-100–15 |
| Y-27632 | Millipore Sigma | Cat#: Y0503 |
| Antibiotic-Antimycotic | GIBCO | Cat#: 15240062 |
| Fetal Bovine Serum | Denville | Cat#: FB5001 |
| Matrigel | Corning | Cat#: 356231 |
| EdU | Santa Cruz | Cat#: sc-284628B |
| Vectashield Antifade Mounting Medium | Vector Laboratories | Cat#: H-1000 |
| Slowfade Diamond Antifade Mountant | Invitrogen | Cat#: S36972 |
| Critical commercial assays | ||
| Click-iT EdU Cell Proliferation Kit | Invitrogen | Cat#: C10340 |
| Experimental models: Organisms/strains | ||
| Sftpc-CreERT2 | PMCID: PMC3223845 | N/A |
| Fgfr2-flox | Jackson Laboratories | Cat#: 007569 |
| Ect2-Flox | PMCID: PMC7194103 | N/A |
| R26R-EYFP | Jackson Laboratories | Cat#: 007903 |
| Ai14(RCL-tdT)-D (R26R-tdTomato) | Jackson Laboratories | Cat#: 007914 |
| Software and algorithms | ||
| LungDamage program | This paper | https://github.com/WALIII/LungDamage |
| Graphpad Prism8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Fiji | PMID: 22743772 | https://fiji.sc/ |
| Other | ||
| Falcon Cell Culture Insert | Thermo Fisher Scientific | Cat#: 08770 |
Highlights.
Fgfr2 maintains AT2 cell fate during postnatal development and after acute injury
AT2 cell differentiation drives morphological remodeling during lung regeneration
Cell division is not required to maintain AT2 cell fate during regeneration
ACKNOWLEDGMENTS
The authors thank the members of the Morrisey laboratory and Dr. Aravind Sivakumar, Dr. David Frank, and particularly Rafael Fernandez for helpful discussion and technical assistance. The authors are appreciative for the assistance of the Cell and Developmental Biology Microscopy Core, in particular Dr. Andrea Stout and Jasmine Zhao, for technical assistance with confocal microscopy. The authors thank Dr. Biao Zuo and Inna Martynyuk of the Electron Microscopy Resource Laboratory at the Perelman School of Medicine for technical assistance with sample preparation and transmission electron microscopy. The authors are also grateful to the Flow Cytometry Core Laboratory at Children’s Hospital of Philadelphia for help with flow cytometry. This study was supported by funding from the NIH (R01-HL132999, R01-HL087825, and U01-HL134745 to E.E.M. and T32-HD083185 to D.C.L.). E.E.M. is funded by the BREATH Consortium in the Longfonds Foundation.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109092.
REFERENCES
- Abler LL, Mansour SL, and Sun X (2009). Conditional gene inactivation reveals roles for Fgf10 and Fgfr2 in establishing a normal pattern of epithelial branching in the mouse lung. Dev. Dyn 238, 1999–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alysandratos KD, Herriges MJ, and Kotton DN (2021). Epithelial Stem and Progenitor Cells in Lung Repair and Regeneration. Annu. Rev. Physiol 83, 529–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasooriya GI, Goschorska M, Piddini E, and Rawlins EL (2017). FGFR2 is required for airway basal cell self-renewal and terminal differentiation. Development 144, 1600–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, and Hogan BL (2013). Type 2 alveolar cells are stem cells in adult lung. J. Clin. Invest 123, 3025–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basil MC, Katzen J, Engler AE, Guo M, Herriges MJ, Kathiriya JJ, Windmueller R, Ysasi AB, Zacharias WJ, Chapman HA, et al. (2020). The Cellular and Physiological Basis for Lung Repair and Regeneration: Past, Present, and Future. Cell Stem Cell 26, 482–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DR, Martinez Alanis D, Miller RK, Ji H, Akiyama H, McCrea PD, and Chen J (2013). Lung epithelial branching program antagonizes alveolar differentiation. Proc. Natl. Acad. Sci. USA 110, 18042–18051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, Sonnenberg A, Wei Y, and Vu TH (2011). Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J. Clin. Invest 121, 2855–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Park JE, Tsagkogeorga G, Yanagita M, Koo BK, Han N, and Lee JH (2020). Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 27, 366–382.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DR, Solski PA, Bultman SJ, Kauselmann G, Schoor M, Kuehn R, Friedman LS, Cowley DO, Van Dyke T, Yeh JJ, et al. (2011). The ect2 rho Guanine nucleotide exchange factor is essential for early mouse development and normal cell cytokinesis and migration. Genes Cancer 2, 932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, and Dickson C (2000). An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 127, 483–492.10631169 [Google Scholar]
- Dorry SJ, Ansbro BO, Ornitz DM, Mutlu GM, and Guzy RD (2020). FGFR2 Is Required for AEC2 Homeostasis and Survival after Bleomycin-induced Lung Injury. Am. J. Respir. Cell Mol. Biol 62, 608–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernanda de Mello Costa M, Weiner AI, and Vaughan AE (2020). Basal-like Progenitor Cells: A Review of Dysplastic Alveolar Regeneration and Remodeling in Lung Repair. Stem Cell Reports 15, 1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DB, Peng T, Zepp JA, Snitow M, Vincent TL, Penkala IJ, Cui Z, Herriges MJ, Morley MP, Zhou S, et al. (2016). Emergence of a Wave of Wnt Signaling that Regulates Lung Alveologenesis by Controlling Epithelial Self-Renewal and Differentiation. Cell Rep 17, 2312–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DB, Penkala IJ, Zepp JA, Sivakumar A, Linares-Saldana R, Zacharias WJ, Stolz KG, Pankin J, Lu M, Wang Q, et al. (2019). Early lineage specification defines alveolar epithelial ontogeny in the murine lung. Proc. Natl. Acad. Sci. USA 116, 4362–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehart H, and Clevers H (2019). Tales from the crypt: new insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol 16, 19–34. [DOI] [PubMed] [Google Scholar]
- Gupte VV, Ramasamy SK, Reddy R, Lee J, Weinreb PH, Violette SM, Guenther A, Warburton D, Driscoll B, Minoo P, and Bellusci S (2009). Overexpression of fibroblast growth factor-10 during both inflammatory and fibrotic phases attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Crit. Care Med 180, 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokuto I, Perl AK, and Whitsett JA (2004). FGF signaling is required for pulmonary homeostasis following hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol 286, L580–L587. [DOI] [PubMed] [Google Scholar]
- Katsura H, Kobayashi Y, Tata PR, and Hogan BLM (2019). IL-1 and TNFα Contribute to the Inflammatory Niche to Enhance Alveolar Regeneration. Stem Cell Reports 12, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura H, Sontake V, Tata A, Kobayashi Y, Edwards CE, Heaton BE, Konkimalla A, Asakura T, Mikami Y, Fritch EJ, et al. (2020). Human Lung Stem Cell-Based Alveolospheres Provide Insights into SARS-CoV-2-Mediated Interferon Responses and Pneumocyte Dysfunction. Cell Stem Cell 27, 890–904.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, et al. (2011). Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147, 525–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeche D, Malpel S, and Cardoso WV (1999). Fibroblast growth factor interactions in the developing lung. Mech. Dev 86, 125–136. [DOI] [PubMed] [Google Scholar]
- Li J, Wang Z, Chu Q, Jiang K, Li J, and Tang N (2018). The Strength of Mechanical Forces Determines the Differentiation of Alveolar Epithelial Cells. Dev. Cell 44, 297–312.e5. [DOI] [PubMed] [Google Scholar]
- Liberti DC, Zepp JA, Bartoni CA, Liberti KH, Zhou S, Lu M, Morley MP, and Morrisey EE (2019). Dnmt1 is required for proximal-distal patterning of the lung endoderm and for restraining alveolar type 2 cell fate. Dev. Biol 454, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie B, Henneke I, Hezel S, Al Alam D, El Agha E, Chao CM, Quantius J, Wilhelm J, Jones M, Goth K, et al. (2015). Attenuating endogenous Fgfr2b ligands during bleomycin-induced lung fibrosis does not compromise murine lung repair. Am. J. Physiol. Lung Cell. Mol. Physiol 308, L1014–L1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Furuhashi K, Danielsson JA, Hirata Y, Kakiuchi M, Lin CS, Ohta M, Riccio P, Takahashi Y, Xu X, et al. (2019). Generation of functional lungs via conditional blastocyst complementation using pluripotent stem cells. Nat. Med 25, 1691–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, and Desai TJ (2018). Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359, 1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaidis NM, Noel JG, Pitstick LB, Gardner JC, Uehara Y, Wu H, Saito A, Lewnard KE, Liu H, White MR, et al. (2017). Mitogenic stimulation accelerates influenza-induced mortality by increasing susceptibility of alveolar type II cells to infection. Proc. Natl. Acad. Sci. USA 114, E6613–E6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obraztsova K, Basil MC, Rue R, Sivakumar A, Lin SM, Mukhitov AR, Gritsiuta AI, Evans JF, Kopp M, Katzen J, et al. (2020). mTORC1 activation in lung mesenchyme drives sex- and age-dependent pulmonary structure and function decline. Nat. Commun 11, 5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris AJ, Hayer KE, Oved JH, Avgousti DC, Toulmin SA, Zepp JA, Zacharias WJ, Katzen JB, Basil MC, Kremp MM, et al. (2020). STAT3-BDNF-TrkB signalling promotes alveolar epithelial regeneration after lung injury. Nat. Cell Biol 22, 1197–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T, Frank DB, Kadzik RS, Morley MP, Rathi KS, Wang T, Zhou S, Cheng L, Lu MM, and Morrisey EE (2015). Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature 526, 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quantius J, Schmoldt C, Vazquez-Armendariz AI, Becker C, El Agha E, Wilhelm J, Morty RE, Vadász I, Mayer K, Gattenloehner S, et al. (2016). Influenza Virus Infects Epithelial Stem/Progenitor Cells of the Distal Lung: Impact on Fgfr2b-Driven Epithelial Repair. PLoS Pathog 12, e1005544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Chiba N, Yao C, Guan X, McConnell AM, Brockway B, Que L, McQualter JL, and Stripp BR (2016). Rare SOX2+ Airway Progenitor Cells Generate KRT5+ Cells that Repopulate Damaged Alveolar Parenchyma following Influenza Virus Infection. Stem Cell Reports 7, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompolas P, Deschene ER, Zito G, Gonzalez DG, Saotome I, Haberman AM, and Greco V (2012). Live imaging of stem cell and progeny behaviour in physiological hair-follicle regeneration. Nature 487, 496–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompolas P, Mesa KR, and Greco V (2013). Spatial organization within a niche as a determinant of stem-cell fate. Nature 502, 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, and Kato S (1999). Fgf10 is essential for limb and lung formation. Nat. Genet 21, 138–141. [DOI] [PubMed] [Google Scholar]
- Shiraishi K, Nakajima T, Shichino S, Deshimaru S, Matsushima K, and Ueha S (2019a). In vitro expansion of endogenous human alveolar epithelial type II cells in fibroblast-free spheroid culture. Biochem. Biophys. Res. Commun. 515, 579–585. [DOI] [PubMed] [Google Scholar]
- Shiraishi K, Shichino S, Ueha S, Nakajima T, Hashimoto S, Yamazaki S, and Matsushima K (2019b). Mesenchymal-Epithelial Interactome Analysis Reveals Essential Factors Required for Fibroblast-Free Alveolosphere Formation. iScience 11, 318–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumar A, and Frank DB (2019). Paradigms that define lung epithelial progenitor cell fate in development and regeneration. Curr. Stem Cell Rep 5, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, Tan K, Tan V, Liu FC, Looney MR, et al. (2015). Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 517, 621–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Frank DB, Morley MP, Zhou S, Wang X, Lu MM, Lazar MA, and Morrisey EE (2016). HDAC3-Dependent Epigenetic Pathway Controls Lung Alveolar Epithelial Cell Remodeling and Spreading via miR-17–92 and TGF-β Signaling Regulation. Dev. Cell 36, 303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver M, Dunn NR, and Hogan BL (2000). Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development 127, 2695–2704. [DOI] [PubMed] [Google Scholar]
- Weiner AI, Jackson SR, Zhao G, Quansah KK, Farshchian JN, Neupauer KM, Littauer EQ, Paris AJ, Liberti DC, Scott Worthen G, et al. (2019). Mesenchyme-free expansion and transplantation of adult alveolar progenitor cells: steps toward cell-based regenerative therapies. NPJ Regen. Med 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitsett JA, Kalin TV, Xu Y, and Kalinichenko VV (2019). Building and Regenerating the Lung Cell by Cell. Physiol. Rev 99, 513–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windmueller R, Leach JP, Babu A, Zhou S, Morley MP, Wakabayashi A, Petrenko NB, Viatour P, and Morrisey EE (2020). Direct Comparison of Mononucleated and Binucleated Cardiomyocytes Reveals Molecular Mechanisms Underlying Distinct Proliferative Competencies. Cell Rep 30, 3105–3116.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Kim T, Brumwell AN, Driver IH, Wei Y, Tan V, Jackson JR, Xu J, Lee DK, Gotts JE, et al. (2017). Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat. Cell Biol 19, 904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Hernandez BJ, Martinez Alanis D, Narvaez del Pilar O, Vila-Ellis L, Akiyama H, Evans SE, Ostrin EJ, and Chen J (2016). The development and plasticity of alveolar type 1 cells. Development 143, 54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, and Ornitz DM (2003). Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063–3074. [DOI] [PubMed] [Google Scholar]
- Yuan T, Volckaert T, Redente EF, Hopkins S, Klinkhammer K, Wasnick R, Chao CM, Yuan J, Zhang JS, Yao C, et al. (2019). FGF10-FGFR2B Signaling Generates Basal Cells and Drives Alveolar Epithelial Regeneration by Bronchial Epithelial Stem Cells after Lung Injury. Stem Cell Reports 12, 1041–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, Zhou S, Cantu E, and Morrisey EE (2018). Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 555, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepp JA, and Morrisey EE (2019). Cellular crosstalk in the development and regeneration of the respiratory system. Nat. Rev. Mol. Cell Biol 20, 551–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, and Morrisey EE (2017). Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell 170, 1134–1148.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo W, Zhang T, Wu DZ, Guan SP, Liew AA, Yamamoto Y, Wang X, Lim SJ, Vincent M, Lessard M, et al. (2015). p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature 517, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Lung damage assessment software (LungDamage) is publicly available on Github (https://github.com/WALIII/LungDamage).