

IL-4 NPs dampen T cell inflammation and increase dystrophic muscle strength; such treatment may benefit diverse dystrophies.
Abstract
Chronic inflammation contributes to the pathogenesis of all muscular dystrophies. Inflammatory T cells damage muscle, while regulatory T cells (Tregs) promote regeneration. We hypothesized that providing anti-inflammatory cytokines in dystrophic muscle would promote proregenerative immune phenotypes and improve function. Primary T cells from dystrophic (mdx) mice responded appropriately to inflammatory or suppressive cytokines. Subsequently, interleukin-4 (IL-4)– or IL-10–conjugated gold nanoparticles (PA4, PA10) were injected into chronically injured, aged, mdx muscle. PA4 and PA10 increased T cell recruitment, with PA4 doubling CD4+/CD8− T cells versus controls. Further, 50% of CD4+/CD8− T cells were immunosuppressive Tregs following PA4, versus 20% in controls. Concomitant with Treg recruitment, muscles exhibited increased fiber area and fourfold increases in contraction force and velocity versus controls. The ability of PA4 to shift immune responses, and improve dystrophic muscle function, suggests that immunomodulatory treatment may benefit many genetically diverse muscular dystrophies, all of which share inflammatory pathology.
INTRODUCTION
Inflammation is a cause of disease progression and chronic tissue degeneration (1). Increasing evidence suggests that inflammation plays a pathogenic role in several prevalent diseases including cardiovascular, metabolic, and neurodegenerative (2), as well as in genetic disease (3, 4). Historically, medical treatments focused on pathogenic factors other than inflammation have failed to cure any of the major, noninfectious, inflammation-associated diseases (1). To effectively cure these conditions, it will be imperative to control the inflammatory response. The biological complexity of chronic inflammation, with multiple cell types, such as macrophages and T cells, and cytokines, such as tumor necrosis factor (TNF), playing divergent context-dependent roles, however, has rendered the development of anti-inflammatory therapies that promote functional tissue regeneration difficult. Most broad anti-inflammatory therapies increase the risk of infection and have potentially serious toxicities (1, 5). No anti-inflammatory therapy cures a majority of patients with a disease in which inflammation plays a major role, such as arthritis (1). Therefore, the development of therapeutics that can control chronic inflammation with minimal side effects is of substantial interest.
Muscular dystrophy is one of the many conditions in which the inflammatory response plays a central role in disease progression. Although formally a genetic disease caused by mutations in dystrophin, the resultant structural instability of myofibers causes contraction-induced myofiber damage, chronic stimulation of the immune system, age-related replacement of muscle by fibrofatty tissue, progressive muscle weakness, and, ultimately, death (6). Chronic inflammation contributes substantially to muscle damage and the progression of muscle weakness (3, 4), so much so that the adoptive transfer of immune cells and muscle extracts from a dystrophic mouse to healthy recipients results in muscle pathology (7). Specifically, an imbalance between inflammatory and anti-inflammatory immune cells has been shown to promote muscle damage. Ablation of interleukin-10 (IL-10), a cytokine involved in both anti-inflammatory (M2) macrophage polarization and regulatory T cell (Treg) activation in dystrophic mice, increases muscle damage and reduces strength (8). Depletion of CD4+ helper or CD8+ cytotoxic T cells and, conversely, an increase in Tregs both improve muscle histology in dystrophic mice (7, 9). Further, murine models with macrophage polarization skewed toward the M2 phenotype show improvements in muscle histology and strength (10). Together, existing data suggest that promoting an appropriate balance of anti-inflammatory macrophages and T cells in dystrophic muscle is critical to ultimately restoring muscle strength and function.
Gene therapy has made substantial advancements in the treatment of muscular dystrophy through both ex vivo editing (11–13) and in vivo strategies. Transplantation of ex vivo edited stem cells has the potential to generate new healthy myofibers, while in vivo gene therapy, although most effective before extensive loss of muscle mass, has demonstrated the most dramatic and widespread effects in animal models (6). Multiple in vivo gene therapy human clinical trials are currently underway (14). In vivo CRISPR-Cas9–mediated exon skipping, to ultimately yield truncated yet functional dystrophin, ameliorated pathology in both mouse and canine models of Duchenne muscular dystrophy (DMD) (12, 15–18). Although dystrophin protein expression was sustained for 1 year in mice [2 months in dogs (18)], the adeno-associated virus (AAV) CRISPR delivery vector was immunogenic when administered to adult mice (19). Recently developed lipid nanoparticles (LNPs) that deliver the Cas9 ribonucleoprotein complex have shown exon skipping and dystrophin protein expression to 4.2% of normal after repeated intramuscular (IM) injections of the LNPs (20), and such lipid delivery vectors may provide significant advantages over AAV vectors. Gold nanoparticles (AuNPs) delivering the ribonucleoprotein complex, along with a DNA template to repair the dystrophin mutation, have shown gene correction in <1% of the dystrophin transcripts in adult mice after IM injection. Nevertheless, muscle function in a four-limb hanging test was significantly improved in mice treated with the AuNPs, yet was still significantly less than wild-type mice (21). To date, the only drug approved by the U.S. Food and Drug Administration (FDA) for DMD is based on exon skipping by antisense oligonucleotides (eteplirsen) and allows restoration of <1% of the normal level of dystrophin protein after extended systemic administration (22). Together, the data suggest that IM injection of gene editing therapeutics holds great potential for the treatment of muscular dystrophy, yet also serves to highlight the challenge of complete gene correction, and therefore the complete resolution of chronic inflammation likely required to halt disease progression. Toxicities associated with long-term treatment with antisense oligonucleotides (23, 24) and immunogenicity of CRISPR delivery vectors further underscore the major unmet medical need for new strategies to treat muscular dystrophy and the interplay between such treatments, disease progression, and inflammation. The most successful strategies will likely use gene editing in combination with immunomodulation to create a tissue microenvironment that both promotes tolerance to de novo dystrophin expression (25, 26) and fosters functional muscle regeneration (6). Modulating the inflammatory response such that it promotes muscle regeneration may substantially improve the use of gene therapy in patients already demonstrating loss of muscle mass.
The hypothesis underlying this study is that AuNPs can deliver anti-inflammatory cytokines, specifically IL-4 and IL-10, in dystrophic skeletal muscle, thereby promoting a proregenerative immune response and enhancing the regeneration of functional muscle tissue in aged mdx mice. Mdx mice carry a nonsense mutation in the Dmd gene encoding dystrophin, analogous to mutations found in DMD patients (27). Anti-inflammatory, or type 2, immune responses characterized by M2 macrophages and Tregs promote muscle repair (6, 9, 27). Conversely, inflammation, the type 1 response, causes muscle damage and is counter-regulated by anti-inflammatory cytokines such as IL-4 and IL-10. IL-4 is an anti-inflammatory cytokine that can induce a type 2 immune response by stimulating both M2 macrophage polarization and a CD4+ helper T cell phenotype. IL-4 has been used as a potential therapeutic in various inflammatory disease models, including autoimmune demyelinating disease, arthritis, and psoriasis (28–30). IL-10 is produced by Tregs and suppresses inflammation, in part, by activating an M2 macrophage phenotype, which can subsequently promote angiogenesis, muscle repair, and remodeling (4). Although cytokine therapy has been widely explored, its use is often limited by unfavorable pharmacokinetic profiles, requiring repeated infusions with systemic side effects (31). Further, the pleiotropic nature of cytokines, with both context-dependent beneficial and detrimental effects, often requires localized or well-targeted delivery strategies, and this has also limited the use of such treatments (31). Here, we use NPs for IL-4 and IL-10 delivery, as they are injectable and allow localized distribution throughout the target muscle. AuNPs were specifically used because they show minimal toxic or immunogenic activity in humans (32, 33). Previously, we have shown that IL-4–conjugated AuNPs (PA4s) can control the innate immune response in healthy mice after acute muscle injury and ultimately increase muscle strength (34). In the present work, the ability of these NPs to control inflammation and T cell phenotype and improve functional muscle regeneration in the context of a chronic inflammatory, muscle-wasting disease, muscular dystrophy, is explored.
RESULTS
Primary T cells from mdx mice readily adopt TH1, TH2, and Treg phenotypes ex vivo
To ensure that T cells from mdx mice could appropriately adopt muscle regenerative phenotypes, we explored the ability of T cells isolated from mdx mice aged 14 weeks to adopt the immunosuppressive regulatory (Treg) phenotype in addition to the inflammatory effector T helper cell phenotypes type 1 (TH1) and type 2 (TH2). Splenic T cells were isolated and cultured with IL-12 to induce TH1 differentiation, IL-4 for TH2 differentiation, and transforming growth factor β1 (TGFβ1) for Treg differentiation, along with supplemental antibodies required for T cell culture, as previously described (35). As expected, T cells treated with IL-12 up-regulated expression of the inflammatory transcription factor T-bet, T cells treated with IL-4 up-regulated expression of the TH2 transcription factor GATA-3 and secreted IL-4, and T cells treated with TGFβ1 up-regulated the regulatory T cell transcription factor FoxP3 (Fig. 1), suggesting that T cells in mdx mice can respond to cytokines to adopt both inflammatory and proregenerative phenotypes.
Fig. 1. Primary mdx T cells readily adopt TH1, TH2, and Treg phenotypes.
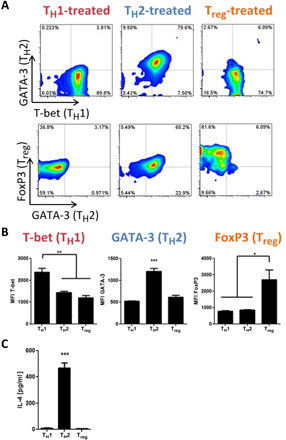
(A) Splenic naïve CD4+ T cells were isolated from mdx mice and stimulated ex vivo to induce TH1, TH2, or Treg phenotypes. Representative plots show expression of transcription factors associated with the TH1 (T-bet), TH2 (GATA-3), and Treg (FoxP3) phenotypes. (B) Median fluorescent intensity (MFI) of cells expressing each transcription factor after ex vivo stimulation to induce the TH1, TH2, or Treg phenotypes. (C) The secretion of IL-4 into cell culture medium, 22-hour culture at 106 cells/ml, was quantified with an enzyme-linked immunosorbent assay (ELISA). All data are means ± SEM; n = 4 mice (2 male, 2 female). ANOVA, with Tukey’s post hoc test: *P < 0.05, **P < 0.01, ***P < 0.001.
Generation of NPs presenting the anti-inflammatory cytokines IL-10 and IL-4
We next designed polyethylene glycol (PEG)–stabilized NPs presenting murine IL-10 or IL-4 (Fig. 2) to determine if promoting type 2 immune responses in aged mdx muscle could enhance muscle function. Monodisperse AuNPs were synthesized by hydroquinone reduction of Au onto citrate-stabilized seed particles (36) and stabilized by partial surface PEGylation with 5-kDa PEG thiol (Fig. 2A). Partial PEGylation allowed the subsequent conjugation of murine IL-10 or IL-4 to the remaining Au surface (Fig. 2B). Conjugation of both murine IL-10 and IL-4 likely occurred directly through thiol-Au bonds to the remaining Au surface and electrostatic interactions. Dynamic light scattering (DLS) showed monodisperse [polydispersity index (PDI) < 0.1] AuNPs with a core diameter of ~30 nm and a final diameter, after IL-10 or IL-4 conjugation, of ~50 nm (Table 1). A slight right shift in size distribution after PEGylation and protein conjugation confirmed the expected surface modifications (Fig. 2, C and D). ζ potential was also used to track surface modifications. As expected, the ζ potential became increasingly less negative after PEGylation and IL-10 or IL-4 conjugation (Table 1).
Fig. 2. Murine IL-4 and IL-10 conjugation to AuNPs.
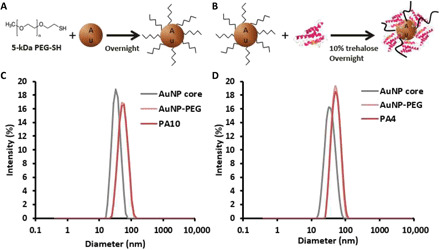
(A and B) Schematic showing partial PEGylation and subsequent IL-4 conjugation to AuNPs. IL-10 conjugation followed an identical protocol. (C) DLS size distribution of 30-nm AuNP core particles (gray) after PEGylation (light pink) and IL-10 conjugation (dark pink). (D) DLS size distribution of 30-nm AuNP core particles (gray) after PEGylation (light pink) and IL-4 conjugation (dark pink). Data are from representative syntheses.
Table 1. AuNP size and ζ potential.
|
Z average (d.nm) |
PDI |
ζ potential (mV) |
|
| AuNP core | 31.8 ± 0.5 | 0.085 ± 0.010 | −34.6 ± 0.5 |
| AuNP-PEG | 49.2 ± 0.5 | 0.069 ± 0.013 | −26.9 ± 1.3 |
| PA10 | 52.1 ± 0.7 | 0.097 ± 0.012 | −9.05 ± 0.69 |
| PA4 | 50.1 ± 0.06 | 0.073 ± 0.006 | −1.88 ± 2.2 |
IL-10 or IL-4 loading onto the AuNPs was next approximated on the basis of sphere packing, modeling IL-10 and IL-4 as spheres with diameters given by their radii of gyration, packed onto a spherical AuNP core (fig. S1). The experimentally determined conjugation efficiencies of IL-10 and IL-4 were consistent and represented 63 and 57% of the theoretical maximum, respectively (Table 2). Conjugation efficiencies less than 100% were expected, as the AuNP surface was partially PEGylated.
Table 2. IL-4 and IL-10 conjugation.
|
IL-4 or IL-10 ligands/ nm2 |
% of maximum packing |
|
| PA10 | 0.070 | 63 |
| PA4 | 0.084 | 57 |
The stability of the PEGylated, IL-4–conjugated particles has been previously assessed (34). Only 3% of murine IL-4 was released into RPMI medium containing 10% heat-inactivated fetal bovine serum (FBS) after 61 days in cell culture conditions (37°C, 5% CO2), and a similar release profile was observed for human IL-4 (34). This slow release suggests that thiol-gold bonds play an important role in the conjugation. Given the isoelectric points of IL-4 and IL-10, however, electrostatic interactions with the AuNPs cannot be ruled out (fig. S1B). Given the structural and electrostatic similarities between IL-4 and IL-10 as well as the similar packing efficiencies on AuNPs (Table 2), IL-10 conjugation and release is likely similar to IL-4.
To assess the bioactivity of PEG-stabilized NPs IL-10 (PA10) and IL-4 (PA4), the particles were used to polarize macrophages in vitro. NP-conjugated IL-10 and IL-4 up-regulated both CD163 and CD206 to the same extent as equivalent doses of soluble IL-10 and IL-4, suggesting that both cytokines retained bioactivity after conjugation (fig. S1C). PEGylated particles lacking cytokines neither are inflammatory (fig. S1D) nor promote type 2 immune responses, as indicated by macrophage CD206 expression (fig. S1E) (34).
Development of chronic muscle damage in aged mdx mice to model DMD
Although mdx mice have a point mutation in the dystrophin gene, their relative muscle degeneration is substantially slower than that observed in humans. DMD patients typically die within their first two decades of life, while mdx mice have a nearly normal murine lifespan. Mdx mice continue to gain skeletal muscle mass and show no significant loss in strength through at least 3 months of age (fig. S2, A and B). Further, mdx mice only present with minimal fibrotic lesions at very advanced ages (>18 months), while in humans fibrosis is observed at an early age and correlates with loss of ambulation. Likely due to mild clinical disease in mdx mice, treatment of 1.5- to 2.5-month-old uninjured mdx mice with PA4 did not result in any improvement in muscle force or contraction velocity (fig. S2, C and D). To more closely mimic human DMD and promote murine disease progression, microinjuries were performed in the tibialis anterior (TA) muscles of mdx mice aged 10.5 to 11 months old (37). To determine the most appropriate chronic DMD model, daily microdamage was compared to a single IM injection of Notexin Np into the TA muscle. Muscles were analyzed approximately 3 weeks after termination of the microdamage protocol or Notexin Np injection to allow acute injury and inflammation to subside and ensure that the observed effects were due to chronic injury (fig. S2E). Daily microdamage resulted in a sustained decrease in muscle mass–normalized force, while Notexin Np injection only resulted in an acute, self-resolving, injury in mdx mice with TA strength recovering to levels observed in younger uninjured mdx mice (fig. S2F). To study chronic muscle damage representative of DMD, further in vivo experiments were performed in mdx mice subjected to repeated microdamage.
PA4 increases the presence of Tregs in chronically injured mdx muscle
To determine if PA4 and/or PA10 could control the inflammatory response in aged (10.5 to 11 months) mdx mice, the particles were injected intramuscularly into the TA muscle. Two micrograms of either IL-4 or IL-10, delivered as PA4 or PA10, or phosphate-buffered saline (PBS)–only lacking particles as a negative control were administered 1 week after termination of the daily microdamage protocol to allow acute inflammation to subside before treatment (Fig. 3A). This dose was chosen on the basis of our previous work showing its ability to shift macrophage polarization and improve muscle function ~2 weeks after acute muscle injury and a single treatment in wild-type mice (34). This dose is also on the order of other local-injection, interleukin treatments used in clinical trials that have been well tolerated (30). The red/purple hue of AuNPs allows their gross biodistribution to be assessed visually. Visual inspection upon dissection revealed that AuNPs distributed throughout the TA, but not surrounding tissue, and were retained in the muscle for at least ~2 weeks after injection (Fig. 3B).
Fig. 3. PA4 increases Tregs in chronically injured aged mdx muscle.

(A) Timeline showing microdamage to induce chronic injury, treatment, and analysis. (B) Representative image showing purple hue of AuNPs throughout the TA ~2 weeks after injection. Black dashed line indicates outline of TA muscle, with the purple PA4 hue visually isolated within the TA. TA muscles removed from the leg showing PA4 throughout the TA tissue (bottom right) and no treatment (bottom left). (C) Representative flow cytometry scatterplots and quantification, showing the expression of CD45, a marker of immune cells, on cells isolated from the TA muscle. Data are means ± SEM; n = 6. (D) Immune cells were clustered based on phenotypic similarities and plotted in dimensionally reduced space using tSNE analysis in FlowJo. Left: Relative expression of CD3, a T cell marker, is visualized by color (blue, low expression, to red, high expression). T cell populations are indicated by high CD3 expression (red) and the red arrow. Right: tSNE contour plots from n = 6 mice per treatment group are overlaid to visualize differences in immune cell distribution. T cell population is indicated by the red arrow. (E) Representative flow cytometry scatterplots and quantification, showing the expression of CD3 on cells isolated from the TA muscle. Data are means ± SEM; n = 6. *P < 0.05. (F) Distribution of CD45+ cells in the TA: T cells (CD3+, pink), lymphoid DCs (CD3−/CD11b−/CD11c+, light gray), myeloid DCs (CD3−/CD11b+/CD11c+, dark gray), neutrophils (CD3−/CD11b+/CD11c−/Ly6G+, blue), NK cells (CD3−/CD11b+/CD11c−/Ly6G−/Ly6C−/F4/80−, tan), monocytes (CD3−/CD11b+/CD11c−/Ly6G−/Ly6C+/F4/80−, black), lineage negative (CD3−/CD11b−/CD11c−, white). (G) Percentage of T cells isolated from TA muscles that are CD4+/CD8− helper T cells. Data are means ± SEM; n = 6. (H) Percentage of CD4+/CD8− helper T cells isolated from TA muscles that are FoxP3+ Tregs. Data are means ± SEM; n = 6. *P < 0.05. For all, Dunnett’s comparison versus PBS control.
Both PA10 and PA4 treatments tended to increase immune cell recruitment (Fig. 3C). Automated clustering of the flow cytometry data based on dimensionality reduction [t-distributed stochastic neighbor embedding (tSNE)] was used to identify unique immune cell populations in the TA in an unbiased manner (38). The expression of cell lineage markers was visualized as a heatmap overlaid on the tSNE clustering, to identify immune cell populations (fig. S3A), and data from six mice per treatment group overlaid (fig. S3B) to visually identify differences in immune cell recruitment. Noticeably, PA10 and PA4 treatments increased T cell recruitment (Fig. 3D). A majority of the infiltrating immune cells were T cells. PA10 and PA4 treatments significantly increased both the percentage of cells in the muscle that were T cells, assessed by CD3 expression (Fig. 3E), and the percentage of immune cells that were T cells (Fig. 3F). No significant changes in the recruitment of neutrophils, dendritic cells (DCs), natural killer (NK) cells, monocytes, or macrophages were observed after PA10 or PA4 treatment (fig. S4).
To further probe T cell phenotype, automated tSNE clustering of CD3+ T cells was performed on the basis of expression of surface (CD4 and CD8) and intracellular (RORγT, T-bet, GATA-3, and FoxP3) markers of T cell phenotype (fig. S5A). Merged data from each treatment group indicated that PA4 treatment resulted in a more broad range of T cell phenotypes and in a particular increase in FoxP3+ Tregs (fig. S5B). Surface markers (CD4 and CD8) were used to assess the presence of helper and cytotoxic T cells, respectively. After treatment with PA4, the percentage of CD4+/CD8− helper T cells approximately doubled (Fig. 3G), and there was a concomitant decrease in the percentage of double-negative (CD4−/CD8−) T cells (fig. S6A). Although the percentage of CD4−/CD8+ cytotoxic T cells tended to increase as well, this change was not statistically significant (fig. S6B). Intracellular flow cytometry for transcription factors revealed that PA4 significantly increased the percentage of CD4+/CD8− helper T cells that were Tregs, identified by FoxP3 expression (Fig. 3H). While the percentage of CD4+/CD8− helper T cells expressing T-bet (identifying TH1 cells), GATA-3 (identifying TH2 cells), and RORγT (identifying TH17 cells) also tended to increase after PA4 treatment, these changes were not statistically significant (fig. S6, C to E). Approximately 50% of the CD4+/CD8− helper T cells identified in TA muscles after PA4 treatment were Tregs, compared to only ~20% after treatment with PA10 or PBS (Fig. 3H).
PA4 improves muscle histology and strength in aged mdx mice
To determine if the shift in immune cell infiltration toward Tregs after PA4 treatment was sufficient to improve muscle regeneration and function in aged and chronically injured mdx mice, TA muscles were assessed ~2 weeks after treatment. No change was observed in the TA muscle weight, or mouse weight, in any group at this time point (fig. S7). Histologic immune cell infiltration was observed in all groups, with no experimental groups demonstrating substantial fibrosis in the hematoxylin and eosin (H&E) histology (Fig. 4A and fig. S8). Cross-sectional muscle fiber area, however, was substantial increased in TA muscles treated with PA4 compared to PBS controls (Fig. 4, A and B). While PA10 treatment increased muscle fiber area, the increase was not statistically significant.
Fig. 4. PA4 increases muscle fiber area and improves strength in aged mice with muscular dystrophy.
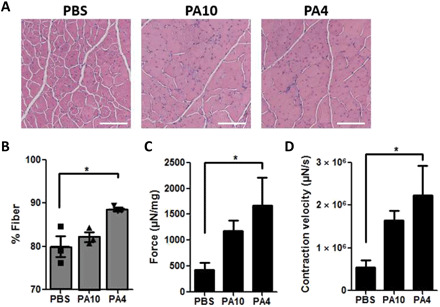
(A) Representative H&E of TA muscles, approximately 3 weeks after termination of daily microdamage and 2 weeks after treatment. Magnification, ×10. Scale bar, 300 μm. (B) Percentage of the TA cross section associated with muscle fibers. ANOVA with Tukey’s post hoc test; data are means ± SEM, n = 3. *P < 0.05. (C and D) Maximum TA muscle contraction force and velocity after ex vivo stimulation at 250 Hz, 25 V. Force was normalized to TA mass. ANOVA with Tukey’s post hoc test; data are means ± SEM; n = 5 to 6. *P < 0.05.
To determine if the increased muscle fiber area corresponded to improvements in muscle function, TA muscle contraction force and velocity were measured ex vivo after tetanic stimulation. TA muscles treated with PA4 showed fourfold increases in both muscle mass–normalized TA contraction force and velocity as compared to PBS treatment (Fig. 4, C and D). This significant increase in the ability of the TA muscle to generate force and rapidly respond to a tetanic stimulus are indicative of improved muscle function in mdx mice after treatment with PA4.
Macrophages play a minimal role in the mechanism of PA4 treatment in mdx mice
The significant Treg infiltration into PA4-treated chronically injured mdx muscles (Fig. 3, D to H) and minimal macrophage recruitment (fig. S4F) suggest that T cells play a central role in the mechanism of PA4 treatment. However, M2 macrophage polarization has previously been shown to significantly improve muscle function after acute injury in healthy mice (34). Therefore, to assess the role of macrophages in mdx muscle function, primary satellite cells isolated from mdx or wild-type mice were cocultured with RAW 264.7 cells, a murine macrophage cell line. While coculture with either wild-type or mdx-derived satellite cells promoted macrophage activation, coculture with mdx-derived satellite cells promoted significantly less macrophage activation than wild-type satellite cells. Macrophages cocultured with mdx satellite cells demonstrated lower levels of CD206 (an M2a polarization marker) (fig. S9, A and B) and CD80 (an M1 marker) expression (fig. S9, C and D), suggesting that mdx satellite cells do not inherently promote as much macrophage activation as wild-type satellite cells.
To assess the ability of primary mdx macrophages to respond to polarization cytokines, bone marrow–derived macrophages (BMDMs) were isolated from mdx or wild-type mice (fig. S10A) and polarized ex vivo with IL-4, IL-10, IL-4 + IL-10, or no cytokines as a negative control. Unlike wild-type macrophages, mdx macrophages neither down-regulated CD80 (fig. S10B) nor up-regulated CD206 (fig. S10C) in response to IL-4 and/or IL-10, suggesting that mdx macrophages are less responsive than wild-type macrophages to polarization cytokines. To assess in vivo macrophage polarization in mdx mice, PA4 was injected intramuscularly and flow cytometry was used to assess macrophage phenotype ~2 weeks after treatment, as above. Consistent with the in vitro results, PA4 treatment did not shift macrophage polarization as compared to the PBS group. There was no change in the levels of CD206 (M2a), CD86 (M1), or CD80 (M1) expression (fig. S11, A to C) or in the percentage of macrophages adopting the M1 phenotype (fig. S11D). There was a small, yet significant, increase in the percentage of M2a macrophages with PA4 treatment (fig. S11E), but the overall macrophage presence in the mdx TA muscle was still only a small fraction of the immune cells (~≤10%) and macrophage recruitment was not significantly increased by PA4 treatment (fig. S11F). Together, these data suggest that while NP IL-4 delivery can help promote an anti-inflammatory microenvironment in the muscle that supports M2a macrophage polarization, macrophages likely play a small role in the mechanism of PA4 treatment in mdx mice.
DISCUSSION
Inflammation is a driver of disease progression for many prevalent conditions ranging from cardiovascular to genetic disease. DMD is an incurable genetic disease initiated by a lack of dystrophin and structural instability in muscle tissue. All 43 genetically divergent varieties of muscular dystrophy share chronic inflammation (6). Chronic inflammation contributes substantially to DMD pathogenesis, and immune cell infiltration into skeletal muscle is strongly associated with disease severity (39). Here, we show that IL-4–conjugated AuNPs promote muscle Treg infiltration after IM injection into chronically injured aged mdx mice, a condition representative of clinically advanced DMD. PA4-driven Treg accumulation ultimately led to improved mdx muscle fiber regeneration and fourfold improvements in muscle contraction force and velocity 2 weeks after treatment.
The findings of these studies demonstrate that T cells from mdx mice can appropriately adopt both inflammatory and proregenerative phenotypes. In response to inflammatory or immunosuppressive cytokine stimulation, T cells isolated from mdx spleens up-regulated transcription factor expression associated with either inflammatory effector T cell phenotypes, TH1 and TH2, or the proregenerative Treg phenotype (Fig. 1). Further, both inflammatory and proregenerative T cells were found in mdx TA muscles (Fig. 3, G and H, and fig. S6). This is in accord with previous reports that show that muscles in both mdx mice and DMD patients undergo continuous cycles of injury and myofiber regeneration, concomitant with both inflammatory and proregenerative immune cell infiltration, respectively (6). Further, there is evidence to suggest that both branches of T cell activation contribute to DMD. Antibody depletion of either CD4+ helper T cells or CD8+ cytotoxic T cells from mdx mice significantly reduced histologically discernible muscle pathology (7). Conversely, in vivo modification of inflammatory pathways that resulted in an increase in Tregs enhanced muscle strength (40).
This study demonstrates that AuNPs can be used to deliver bioactive immunomodulatory cytokines murine IL-10 and IL-4 intramuscularly in mdx mice. We have previously shown that PEGylated AuNPs are stable, biologically inert, and nonimmunogenic (34). Further, we have shown that human and murine IL-4 conjugated to these partially PEGylated AuNPs is stable, with only ~3% released after 61 days in medium containing 10% serum proteins, and that the conjugated IL-4 retains its bioactivity (34). Here, we extend this cytokine delivery platform to murine IL-10. As expected, the size distribution of the NPs shifted to the right, and the ζ potential became less negative after partial PEGylation and IL-10 or IL-4 conjugation (Fig. 2 and Tables 1 and 2). Increased recruitment of T cells into the treated muscles and somewhat improved muscle function after both PA10 and PA4 treatments indicated that the conjugated cytokines retained bioactivity after IM injection, as expected (Figs. 3, D to F, and 4, C and D). Therapeutic efficacy observed 2 weeks after a single PA4 injection also provides some indication of the time scale and in vivo durability of PA4 therapy. Various methods of cytokine delivery have been explored previously, including continuous intravenous infusion (phase 1 clinical trial) in cancer patients (41), conjugation to decellularized scaffolds (42), and temporally controlled release from hydrogels for muscle delivery (43). However, several of these approaches have been limited by toxicity and dose limitations (especially in the case of infusions) and their ability to evenly distribute cytokine throughout the target tissue. Strategies for muscle targeting, including adeno-associated viral (AAV) vector delivery of CRISPR components or other genetic information (19, 44), and the identification of muscle targeting peptides have also been explored (45). However, gene transfer efficiency, unfavorable immune responses to the AAV capsid protein, and peptide ligand–targeted drug delivery remain challenging (46). Here, IL-10 and IL-4 conjugation to AuNPs allowed nonimmunogenic distribution of the cytokines throughout the targeted muscle, providing a model to probe their ability to control inflammation in the context of chronic DMD, and the effects of such immunomodulation on muscle function, with a clinically relevant delivery vector. Formulations of colloidal gold have been FDA-approved for the treatment of arthritis, and AuNP delivery of TNF-α has shown promising results in cancer clinical trials (47, 48).
NP delivery allows more potent and sustained immunomodulation than soluble cytokines. Previously, we have shown that PA4 macrophage polarization results in higher and more stable levels of CD206 expression than soluble IL-4 and that increased IL-4 valency on the NPs results in increased M2a polarization (34). Here, we show that AuNP-conjugated IL-10 also retains its bioactivity and ability to direct macrophage phenotype in vitro (fig. S1C). AuNPs were retained in mdx muscle for at least 2 weeks after IM injection and promoted Treg accumulation over that time period (Fig. 3). Previously, we have shown that soluble IL-4 was unable to improve muscle function after acute injury in healthy mice versus 40% improvement in strength observed after treatment with an equivalent dose of IL-4 delivered as PA4 (34). This finding suggests that NP delivery is critical. The more severe, and chronic, inflammatory nature of DMD suggests that soluble IL-4 would be even less effective at therapeutically shifting the immune response in this context; the ability of PA4 to provide a sustained polarization cytokine was likely necessary here. PEGylation has also been shown to protect interleukins from degradation in vivo, thereby improving their therapeutic efficacy (49). AuNP PEGylation likely also provided benefit in the context of acute muscle injury (34) and dystrophy studied here. An emphasis was placed on IL-4/PA4, as PA4 was found to be the most promising therapeutic. However, it is worth noting that soluble IL-10 has failed in multiple clinical trials due to its in vivo instability (50). For these reasons, in addition to the inability of PA10 to improve muscle function in mdx mice (Fig. 4), soluble IL-10 was excluded from the current study. Future studies to assess how long PA4 is retained in the muscle, to understand cellular internalization of the NPs, and determine the duration of the anti-inflammatory immune response will be important, however, as such parameters will be necessary to develop dosing regimens required to effectively treat DMD.
PA4 treatment significantly increased the presence of immunosuppressive/proregenerative T cells, Tregs, in chronically injured mdx muscle. IM PA4 injection increased T cell infiltration into the muscle and approximately doubled the percentage of CD4+/CD8− helper T cells (Fig. 3, D, E, and G). Nearly half of the CD4+/CD8− helper T cells were FoxP3-expressing Tregs after PA4 treatment, compared to only ~20% in the negative control group (Fig. 3H). This is consistent with previous findings that IL-4 can induce the formation of Tregs from naïve CD4+ T cells (51). IL-4 signaling through signal transducer and activator of transcription 6 (STAT6) is required for expression and maintenance of the Treg transcription factor FoxP3, Treg proliferation, and survival (52, 53). IL-4 also improves the ability of Tregs to inhibit inflammatory interferon-γ (IFN-γ) production by CD4+ T cells in a STAT6-independent manner (52). Increased numbers of Tregs have been observed in the skeletal muscles of mdx mice compared to wild-type mice (27), suggesting that the immune environment in dystrophic muscles may promote Treg infiltration or activation. Treatment of mdx muscles with IL-2 complexes has been shown to increase the frequency of muscle Tregs by ~10 percentage points (~15% of CD4+ T cells are Tregs in negative control versus ~25% Tregs in the treatment group) and increase muscle IL-10 nearly twofold (9). Here, PA4 treatment resulted in Tregs representing nearly half of total CD4+ T cells in the muscle. Elevated levels of IL-10 have also been observed in mdx muscles (8), and biological IL-10 saturation may have limited the therapeutic advantage of further IL-10 delivery by PA10 here. Modulation of the dystrophic microenvironment with PA4, however, further increased the presence of Tregs in chronically injured mdx muscles; therefore, PA4 may have a particular ability to direct Treg recruitment in the context of DMD, perhaps ultimately leading to dampened inflammation and improved muscle regeneration.
PA4 treatment enhanced mdx muscle regeneration and improved function. Two weeks after PA4 injection, TA muscles demonstrated increased muscle fiber area and fourfold increases in both contraction force and velocity compared to negative controls (Fig. 4). This is consistent with previous studies showing that IL-4 promotes muscle regeneration both directly, by controlling myoblast fusion and the maturation of functional myotubes (54), and indirectly via the promotion of a type 2 immune response. AAV delivery of CRISPR gene editing components (Cas9 and single-guide RNA) increased dystrophin expression and increased muscle force twofold in a murine exon 44 deletion DMD model compared to untreated controls 4 weeks after treatment (12). AuNPs delivering CRISPR components along with a DNA template to repair the dystrophin mutation yielded about twofold increase in wire-hang time 2 weeks after treatment (21). Previously, we have shown that promotion of M2 macrophage polarization by PA4 treatment improved muscle strength by 40% compared to negative control groups, in wild-type mice, after acute injury (34). Skewing M2 macrophage polarization in mdx mice, by ablation of osteopontin, increased hindlimb muscle–specific force by 1.2- to 1.3-fold (10). Here, we show a fourfold improvement in muscle force after PA4 treatment and Treg recruitment, suggesting that T cells may also play a central role in mdx muscle function.
The increased presence of Tregs in TA muscles after PA4 treatment likely played a critical role in promoting muscle regeneration and function. Conversely, the minimal therapeutic effects observed after PA10 treatment may be partially explained by its inability to increase muscle Treg infiltration (Fig. 3G, H). Ample evidence supports the role of Tregs in muscle regeneration, as these cells both modulate inflammation and directly affect muscle growth. The secretion of IL-10 by Tregs dampens muscle-damaging inflammatory processes (9). IL-10 secretion by Tregs recruited after PA4 treatment may have contributed to the improved muscle strength observed here, perhaps providing a similar benefit to combined delivery of both IL-4 and IL-10. Conversely, PA10 treatment provided minimal Treg recruitment and perhaps minimal stimulation of additional regenerative cytokines. Tregs also have a direct effect on muscle progenitors and nascent myofibers via amphiregulin (Areg). Whole muscle transcriptional analysis has shown that Treg depletion in mdx mice was associated with a down-regulation of genes associated with muscle homeostasis and function; an increase in serum levels of creatine kinase, an enzyme associated with muscle damage, was also observed with Treg depletion (27). Further, various treatments that promote Treg accumulation in mdx muscles have shown promise. Rapamycin improved mdx muscle phenotype, largely by reducing the infiltration of effector CD4+ and CD8+ T cells, while preserving Tregs (55). IL-2/anti–IL-2 complexes also decreased creatine kinase and improved muscle histology, concomitant with increasing Tregs (9). However, these studies did not measure muscle strength or function. Here, a significant increase in muscle contraction force and velocity was observed, concomitant with an increase in Tregs, after PA4 treatment in mdx mice.
Macrophage polarization likely played a small role in the mechanism of PA4 treatment of mdx muscles. Satellite cells from mdx mice did not support as much macrophage activation as wild-type satellite cells (fig. S9), and mdx macrophages were less responsive than wild-type macrophages to polarization cytokines both in vitro and in vivo (figs. S10 and S11). Although macrophages have been shown to play a central role in muscle regeneration and function in healthy mice (34), the low macrophage numbers in mdx mice observed here, the inability of PA4 treatment to promote macrophage recruitment (figs. S4F and S11F), and the relative inability of mdx macrophages to react as potently to IL-4 and IL-10 likely limited their therapeutic potential in dystrophic muscle. Together, these data indicate that the mdx mutation limits the ability of dystrophic muscle satellite cells to stimulate macrophage activation and the ability of mdx macrophages to fully respond to IL-4 and/or IL-10. Direct comparison of muscle function and PA4 therapeutic efficacy in mdx versus wild-type mice is challenging, however. Treatment of uninjured mdx mice did not result in any improvement in muscle force (fig. S2, C and D), likely due to mild clinical disease, suggesting that treating wild-type mice would also not show any change in muscle function. The microdamage used here to advance disease progression in mdx mice allowed us to study a severe chronic model of DMD and observe the therapeutic efficacy of PA4. Such microdamage, however, does not result in injury or inflammation in wild-type mice (56).
The results of these studies indicate that IL-4–conjugated AuNPs can shift the immune response in chronically inflamed dystrophic muscle and ultimately promote improved muscle regeneration and function. While more than 7000 unique genetic mutations have been identified in DMD patients (59), chronic inflammation plays a critical role in the pathogenesis of all forms of DMD. This study suggests that PA4 modulation of the immune response may slow disease progression and improve muscle function for patients suffering from a broad range of muscular dystrophies.
MATERIALS AND METHODS
For additional methods, see the Supplementary Materials.
Experimental design
The hypothesis of this investigation was that AuNPs delivering anti-inflammatory cytokines would promote a proregenerative immune response in dystrophic muscle and ultimately result in improved muscle regeneration and function. Experiments were designed to evaluate the impact of IL-4– and IL-10–conjugated AuNPs compared to PBS controls in chronically injured aged mdx mice to model clinically advanced dystrophy. Experimental conditions were chosen on the basis of previous work showing that soluble IL-4 fails to improve muscle function after acute injury (34), and are built on the expectation that soluble cytokine delivery would not provide significant benefit in the context of chronic, advanced muscular dystrophy. All treatments were administered at least 3 weeks after initiation of the chronic injury in mdx mice to ensure that acute injury and inflammation had subsided. In vivo studies were carried out on male and female mdx mice of the same generation bred at Harvard University to minimize variances between groups. All animal work was in compliance with the National Institutes of Health and institutional guidelines. The number of animals and statistical parameters are denoted in the figure legends.
T cell culture
Naïve CD4+ T cells were isolated from the spleens of mdx mice after euthanasia in compliance with the National Institutes of Health and institutional guidelines. T cells were isolated by depletion of off-target cells using magnetic-activated cell sorting and cultured (35), and medium was supplemented to activate various phenotypes (table S1).
AuNP synthesis and conjugation
AuNPs were synthesized by the hydroquinone reduction of Au onto citrate-stabilized seed particles (36). After PEGylation with 5-kDa PEG-thiol, murine IL-10 or IL-4 was added at a concentration of 0.5 ligands/nm2, with 10% (w/v) trehalose for overnight conjugation, as previously described (34).
Animal models
All animal work was in compliance with National Institutes of Health and institutional guidelines, with male and female mdx mice bred at Harvard University, aged 10.5 to 11 months old. Chronic muscle injury was induced by daily microdamage (37) and compared to a model of toxin injury induced by IM injection of Notexin Np (Supplementary Materials). To ensure treatments distributed throughout the TA, the total dose was administered via two 10-μl IM injections.
TA force
TA muscles were mounted between two electrodes and connected to a force transducer (58). Tetanic muscle contraction was evoked at 250 Hz, 25 V for 1 s. Contraction force was normalized to TA mass. Contraction velocity was measured as the slope of the force curve at the time of stimulation (34).
Histology analysis
H&E staining was performed, and 10× images were tiled across the entire muscle cross section. Color deconvolution in ImageJ was used to assess the percentage of the muscle cross-sectional area composed of muscle fibers (pink, eosin) or cell nuclei (blue, hematoxylin).
Statistical analysis
All analyses were performed with GraphPad Prism 5. Analysis of variance (ANOVA) tests with Tukey’s post hoc test were used for multiple comparisons throughout. Dunnett’s comparison versus a control condition was used as noted. Dimensionality reduction of high-parameter flow cytometry was performed using the tSNE algorithm in FlowJo v10.6.2 (TreeStar, USA) (Supplementary Materials) (38).
Acknowledgments
Funding: This work was supported by NIH grant DP3DK108224 (D.J.M.), NSF Graduate Research Fellowship Program (T.M.R.), and NIH grant R01 DE013349. Author contributions: Conceptualization: T.M.R. and D.J.M.; methodology: T.M.R.; formal analysis: T.M.R.; investigation: T.M.R.; supervision: D.J.M.; writing—original draft: T.M.R. and D.J.M.; writing—review and editing: T.M.R. and D.J.M. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabh3693/DC1
REFERENCES AND NOTES
- 1.Nathan C., Ding A., Nonresolving inflammation. Cell 140, 871–882 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Kamaly N., Fredman G., Subramanian M., Gadde S., Pesic A., Cheung L., Fayad Z. A., Langer R., Tabas I., Farokhzad O. C., Development and in vivo efficacy of targeted polymeric inflammation-resolving nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 110, 6506–6511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spencer M. J., Tidball J. G., Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul. Disord. 11, 556–564 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Paepe B. D., De Bleecker J. L., Cytokines and chemokines as regulators of skeletal muscle inflammation: Presenting the case of Duchenne muscular dystrophy. Mediators Inflamm. 2013, 540370–540380 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyatake S., Shimizu-Motohashi Y., Takeda S., Aoki Y., Anti-inflammatory drugs for Duchenne muscular dystrophy: Focus on skeletal muscle-releasing factors. Drug Des. Devel. Ther. 10, 2745–2758 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg A. S., Puig M., Nagaraju K., Hoffman E. P., Villalta S. A., Rao V. A., Wakefield L. M., Woodcock J., Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 7, 299rv4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spencer M. J., Montecino-Rodriguez E., Dorshkind K., Tidball J. G., Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin. Immunol. 98, 235–243 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Villalta S. A., Rinaldi C., Deng B., Liu G., Fedor B., Tidball J. G., Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum. Mol. Genet. 20, 790–805 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Villalta S. A., Rosenthal W., Martinez L., Kaur A., Sparwasser T., Tidball J. G., Margeta M., Spencer M. J., Bluestone J. A., Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci. Transl. Med. 6, 258ra142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capote J., Kramerova I., Martinez L., Vetrone S., Barton E. R., Sweeney H. L., Miceli M. C., Spencer M. J., Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro-regenerative phenotype. J. Cell Biol. 213, 275–288 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu P., Wu F., Mosenson J., Zhang H., He T. C., Wu W. S., CRISPR/Cas9-mediated genome editing corrects dystrophin mutation in skeletal muscle stem cells in a mouse model of muscle dystrophy. Mol. Ther. Nucleic Acids 7, 31–41 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Min Y.-L., Li H., Rodriguez-Caycedo C., Mireault A. A., Huang J., Shelton J. M., McAnally J. R., Amoasii L., Mammen P. P. A., Bassel-Duby R., Olson E. N., CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 5, eaav4324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumont N. A., Rudnicki M. A., Targeting muscle stem cell intrinsic defects to treat Duchenne muscular dystrophy. npj Regen. Med. 1, 16006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abreu N. J., Waldrop M. A., Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr. Pulmonol. 56, 710–720 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Tabebordbar M., Zhu K., Cheng J. K. W., Chew W. L., Widrick J. J., Yan W. X., Maesner C., Wu E. Y., Xiao R., Ran F. A., Cong L., Zhang F., Vandenberghe L. H., Church G. M., Wagers A. J., In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson C. E., Hakim C. H., Ousterout D. G., Thakore P. I., Moreb E. A., Castellanos Rivera R. M., Madhavan S., Pan X., Ran F. A., Yan W. X., Asokan A., Zhang F., Duan D., Gersbach C. A., In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long C., Amoasii L., Mireault A. A., McAnally J. R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J. M., Bassel-Duby R., Olson E. N., Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amoasii L., Hildyard J. C. W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T., Massey C., Shelton J. M., Bassel-Duby R., Piercy R. J., Olson E. N., Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 362, 86–91 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelson C. E., Wu Y., Gemberling M. P., Oliver M. L., Waller M. A., Bohning J. D., Robinson-Hamm J. N., Bulaklak K., Castellanos Rivera R. M., Collier J. H., Asokan A., Gersbach C. A., Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 25, 427–432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei T., Cheng Q., Min Y., Olson E. N., Siegwart D. J., Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 11, 3232 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee K., Conboy M., Park H. M., Jiang F., Kim H. J., Dewitt M. A., Mackley V. A., Chang K., Rao A., Skinner C., Shobha T., Mehdipour M., Liu H., Huang W., Lan F., Bray N. L., Li S., Corn J. E., Kataoka K., Doudna J. A., Conboy I., Murthy N., Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 1, 889–901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charleston J. S., Schnell F. J., Dworzak J., Donoghue C., Lewis S., Chen L., Young G. D., Milici A. J., Voss J., DeAlwis U., Wentworth B., Rodino-Klapac L. R., Sahenk Z., Frank D., Mendell J. R., Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production. Neurology 90, e2146–e2154 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Alfano L. N., Charleston J. S., Connolly A. M., Cripe L., Donoghue C., Dracker R., Dworzak J., Eliopoulos H., Frank D. E., Lewis S., Lucas K., Lynch J., Milici A. J., Flynt A., Naughton E., Rodino-Klapac L. R., Sahenk Z., Schnell F. J., Young G. D., Mendell J. R., Lowes L. P., Long-term treatment with eteplirsen in nonambulatory patients with Duchenne muscular dystrophy. Medicine 98, e15858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mollanoori H., Rahmati Y., Hassani B., Mehr M. H., Teimourian S., Promising therapeutic approaches using CRISPR/Cas9 genome editing technology in the treatment of Duchenne muscular dystrophy. Genes Dis. 8, 146–156 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braun S., Thioudellet C., Rodriguez P., Ali-Hadji D., Perraud F., Accart N., Balloul J. M., Halluard C., Acres B., Cavallini B., Pavirani A., Immune rejection of human dystrophin following intramuscular injections of naked DNA in mdx mice. Gene Ther. 7, 1447–1457 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Flanigan K. M., Campbell K., Viollet L., Wang W., Gomez A. M., Walker C. M., Mendell J. R., Anti-dystrophin T cell responses in Duchenne muscular dystrophy: Prevalence and a glucocorticoid treatment effect. Hum. Gene Ther. 24, 797–806 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burzyn D., Kuswanto W., Kolodin D., Shadrach J. L., Cerletti M., Jang Y., Sefik E., Tan T. G., Wagers A. J., Benoist C., Mathis D., A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Racke M. K., Bonomo A., Scott D. E., Cannella B., Levine A., Raine C. S., Shevach E. M., Röcken M., Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J. Exp. Med. 180, 1961–1966 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horsfall A. C., Butler D. M., Marinova L., Warden P. J., Williams R. O., Maini R. N., Feldmann M., Suppression of collagen-induced arthritis by continuous administration of IL-4. J. Immunol. 159, 5687–5696 (1997). [PubMed] [Google Scholar]
- 30.Ghoreschi K., Thomas P., Breit S., Dugas M., Mailhammer R., van Eden W., van der Zee R., Biedermann T., Prinz J., Mack M., Mrowietz U., Christophers E., Schlöndorff D., Plewig G., Sander C. A., Röcken M., Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat. Med. 9, 40–46 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Zidek Z., Anzenbacher P., Kmonickova E., Current status and challenges of cytokine pharmacology. Br. J. Pharmacol. 157, 342–361 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merchant B., Gold, the noble metal and the paradoxes of its toxicology. Biologicals 26, 49–59 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Root S. W., Andrews G. A., Kniseley R. M., Tyor M. P., The distribution and radiation effects of intravenously administered colloidal Au198 in man. Cancer 7, 856–866 (1954). [DOI] [PubMed] [Google Scholar]
- 34.Raimondo T. M., Mooney D. J., Functional muscle recovery with nanoparticle-directed M2 macrophage polarization in mice. Proc. Natl. Acad. Sci. U.S.A. 115, 10648–10653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwee B. J., Budina E., Najibi A. J., Mooney D. J., CD4 T-cells regulate angiogenesis and myogenesis. Biomaterials 178, 109–121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perrault S. D., Chan W. C. W., Synthesis and surface modification of highly monodispersed, spherical gold nanoparticles of 50-200 nm. J. Am. Chem. Soc. 131, 17042–17043 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Desguerre I., Arnold L., Vignaud A., Cuvellier S., Yacoub-Youssef H., Gherardi R. K., Chelly J., Chretien F., Mounier R., Ferry A., Chazaud B., A new model of experimental fibrosis in hindlimb skeletal muscle of adult mdx mouse mimicking muscular dystrophy. Muscle Nerve 45, 803–814 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Belkina A. C., Ciccolella C. O., Anno R., Halpert R., Spidlen J., Snyder-Cappions J. E., Automated optimized parameters for T-distributed stochastic neighbor embedding improve visualization and analysis of large datasets. Nat. Commun. 10, 5415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nitahara-Kasahara Y., Takeda S., Okada T., Inflammatory predisposition predicts disease phenotypes in muscular dystrophy. Inflamm. Regener. 36, 14–18 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gazzerro E., Baldassari S., Assereto S., Frusscione F., Pistorio A., Panicucci C., Volpi S., Perruzza L., Fiorillo C., Minetti C., Traggiai E., Grassi F., Bruno C., Enhancement of muscle T regulatory cells and improvement of muscular dystrophic process in mdx mice by blockade of extracellular ATP/P2X Axis. Am. J. Pathol. 185, 3349–3360 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Sosman J. A., Fisher S. G., Kefer C., Fisher R. I., Ellis T. M., A phase I trial of continuous infusion interleukin-4 (IL-4) alone and following interleukin-2 (IL-2) in cancer patients. Ann. Oncol. 5, 447–452 (1994). [DOI] [PubMed] [Google Scholar]
- 42.Spiller K. L., Nassiri S., Witherel C. E., Anfang R. R., Ng J., Nakazawa K. R., Yu T., Vunjak-Novakovic G., Sequential delivery of immunomodulatory cytokines to facilitate the M1-to-M2 transition of macrophages and enhance vascularization of bone scaffolds. Biomaterials 37, 194–207 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borselli C., Storrie H., Benesch-Lee F., Shvartsman D., Cezar C., Lichtman J. W., Vandenburgh H. H., Mooney D. J., Functional muscle regeneration with combined delivery of angiogenesis and myogenesis factors. Proc. Natl. Acad. Sci. U.S.A. 107, 3287–3292 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang Y., Cummins J., Huard J., Wang B., AAV-directed muscular dystrophy gene therapy. Expert Opin. Biol. Ther. 10, 395–408 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Seow Y., Yin H., Wood M. J. A., Identification of a novel muscle targeting peptide in mdx mice. Peptides 31, 1873–1877 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Wang Z., Tapscott S. J., Chamberlain J. S., Storb R., Immunity and AAV-mediated gene therapy for muscular dystrophies in large animal models and human trials. Front. Microbiol. 2, 201 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ward J. R., Williams H. J., Egger M. J., Reading J. C., Boyce E., Altz-Smith M., Samuelson C. O. Jr., Willkens R. F., Solsky M. A., Hayes S. P., Blocka K. L., Weinstein A., Meenan R. F., Guttadauria M., Kaplan S. B., Klippel J., Comparison of auranofin, gold sodium thiomalate, and placebo in the treatment of rheumatoid arthritis. A controlled clinical trial. Arthritis Rheum. 26, 1303–1315 (1983). [DOI] [PubMed] [Google Scholar]
- 48.Nilubol N., Oarr D., Tamarkin L., Preclinical studies evaluate pivotal TNFα nanomedicine clinical trial design. J. Clin. Oncol. 37, e14646 (2019). [Google Scholar]
- 49.Naing A., Papadopoulos K. P., Autio K. A., Ott P. A., Patel M. R., Wong D. J., Falchook G. S., Pant S., Whiteside M., Rasco D. R., Mumm J. B., Chan I. H., Bendell J. C., Bauer T. M., Colen R. R., Hong D. S., van Vlasselaer P., Tannir N. M., Oft M., Infante J. R., Safety, antitumor activity, and immune activation of pegylated recombinant human interleukin-10 (AM0010) in patients with advanced solid tumors. J. Clin. Oncol. 34, 3562–3569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mosser D. M., Zhang X., Interleukin-10: New perspectives on an old cytokine. Immunol. Rev. 226, 205–218 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Skapenko A., Kalden J. R., Lipsky P. E., Schulze-Koops H., The IL-4 receptor α-chain-binding cytokines, IL-4 and IL-13, induce forkhead box P3-expressing CD25+CD4+ regulatory T cells from CD25−CD4+ precursors. J. Immunol. 175, 6107–6116 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Maerten P., Shen C., Bullens D. M., Van Assche G., Van Gool S., Geboes K., Rutgeerts P., Ceuppens J. L., Effects of interleukin 4 on CD25+CD4+ regulatory T cell function. J. Autoimmun. 25, 112–120 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Pillemer B. B. L., Qi Z., Melgert B., Oriss T. B., Ray P., Ray A., STAT6 activation confers upon T helper cells resistance to suppression by regulatory T cells. J. Immunol. 183, 155–163 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Horsley V., Jansen K. M., Mills S. T., Pavlath G. K., IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell 113, 483–494 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Eghtesad S., Jhunjhunwala S., Little S. R., Clemens P. R., Rapamycin ameliorates dystrophic phenotype in mdx mouse skeletal muscle. Mol. Med. 17, 917–924 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lemos D. R., Babaeijandaghi F., Low M., Chang C.-K., Lee S. T., Fiore D., Zhang R., Natarajan A., Nedospasov S. A., Rossi F. M. V., Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 21, 786–794 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Bladen C. L., Salgado D., Monges S., Foncuberta M. E., Kekou K., Kosma K., Dawkins H., Lamont L., Roy A. J., Chamova T., Guergueltcheva V., Chan S., Korngut L., Campbell C., Dai Y., Wang J., Barišić N., Brabec P., Lahdetie J., Walter M. C., Schreiber-Katz O., Karcagi V., Garami M., Viswanathan V., Bayat F., Buccella F., Kimura E., Koeks Z., van den Bergen J. C., Rodrigues M., Roxburgh R., Lusakowska A., Kostera-Pruszczyk A., Zimowski J., Santos R., Neagu E., Artemieva S., Rasic V. M., Vojinovic D., Posada M., Bloetzer C., Jeannet P. Y., Joncourt F., Díaz-Manera J., Gallardo E., Karaduman A. A., Topaloğlu H., El Sherif R., Stringer A., Shatillo A. V., Martin A. S., Peay H. L., Bellgard M. I., Kirschner J., Flanigan K. M., Straub V., Bushby K., Verschuuren J., Aartsma-Rus A., Béroud C., Lochmüller H., The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 36, 395–402 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cezar C. A., Roche E. T., Vandenburgh H. H., Duda G. N., Walsh C. J., Mooney D. J., Biologic-free mechanically induced muscle regeneration. Proc. Natl. Acad. Sci. U.S.A. 113, 1534–1539 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walkey C. D., Olsen J. B., Guo H., Emili A., Chan W. C. W., Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J. Am. Chem. Soc. 134, 2139–2147 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Amend S. R., Valkenburg K. C., Pienta K. J., Murine hind limb long bone dissection and bone marrow isolation. J. Vis. Exp. 110, 53936 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Erickson H. P., Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 11, 32–51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., Skrzypek E., PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabh3693/DC1