

Abstract
Three-dimensional fluorescence-based imaging of living cells and organisms requires the sample to be exposed to substantial excitation illumination energy, typically causing phototoxicity and photobleaching. Light sheet fluorescence microscopy dramatically reduces phototoxicity, yet most implementations are limited to objective lenses with low numerical aperture and particular sample geometries that are built for specific biological systems. To overcome these limitations, we developed a single-objective light sheet fluorescence system for biological imaging based on axial plane optical microscopy and digital confocal slit detection, using either Bessel or Gaussian beam shapes. Compared to spinning disk confocal microscopy, this system displays similar optical resolution, but a significantly reduced photobleaching at the same signal level. This single-objective light sheet technique is built as an add-on module for standard research microscopes and the technique is compatible with high-numerical aperture oil immersion objectives and standard samples mounted on coverslips. We demonstrate the performance of this technique by imaging three-dimensional dynamic processes, including bacterial biofilm dispersal, the response of biofilms to osmotic shocks, and macrophage phagocytosis of bacterial cells.
1. Introduction
Live-cell optical microscopy is a powerful tool for characterizing the dynamics of life in three dimensions (3D). Living samples are typically photosensitive and traditional 3D imaging techniques, such as confocal microscopy or two-photon microscopy, cause significant phototoxicity. This phototoxicity can interfere with biological systems, particularly when samples are imaged with high temporal resolution for extended periods. Recently, light-sheet fluorescence microscopy (LSFM) or selective plane illumination microscopy (SPIM) have re-emerged as important imaging modalities for 3D live-cell imaging, due to their strongly reduced phototoxicity compared with other 3D imaging techniques [1–5]. In LSFM, only the focal plane of an objective lens is illuminated with a thin light sheet. Therefore, only the plane in the sample that is actually imaged is also illuminated by the excitation light, which reduces phototoxicity and photobleaching to a minimum needed for imaging [1,6–12]. In contrast, the volume of the sample that is illuminated by excitation light in confocal microscopy or two-photon microscopy is significantly larger than the volume that is imaged at a given time, resulting in stronger phototoxicity and photobleaching.
Light sheet microscopy is typically implemented by uncoupling the illumination and detection beam paths using two separate objective lenses. The first objective illuminates the plane of interest with a thin light sheet and the second objective, arranged orthogonally to the illumination objective, collects the resulting fluorescence light. This scheme requires that the illumination and detection objectives have to be positioned so that their focal planes intersect, which poses constraints on both objective selection and sample preparation [2]. To enable the use of two-objective LSFM with “open top” sample geometries that are widely used in biology, such as liquid-filled multi-well plates, several microscope designs have been developed that typically rely on objectives that are mounted at a 45° angle with the cover slip [3,13–15].
Despite the advances of two-objective light sheet microscopy, which include very thin illumination designs such as optical fibres [16,17], oil immersion objectives cannot currently be used in a two-objective configuration, because their short working distance and their physical size make it impossible to fit an illumination objective next to them. Therefore, two-objective light sheet microscopy is limited to air- or water-immersion objectives with currently a maximum numerical aperture (NA) of 1.1 [18], which is substantially below the NA of oil immersion objectives. Objectives that are not mounted at right angles to each other can enable the use of oil immersion objectives for LSFM, albeit with a lower-NA illumination objective [19]. In addition to the constraints on the NA, the existing two-objective LSFM configurations are not optimized for imaging the wide variety of sample geometries for which confocal microscopy is typically used. Specifically, widely-used multi-well plates, as well as most microfluidic chip designs are difficult or impossible to handle for two-objective LSFM techniques.
To overcome the limitations of two-objective light sheet microscopy, several single-objective techniques have been developed. These techniques are based on using the same objective lens for illumination and detection, either by employing an oblique light sheet [20–22] or by using micromirrors to form a horizontal light sheet in the focal plane [23,24]. To achieve a horizontal light sheet in the focal plane, a vertical light sheet is launched from the objective and reflected from a 45° micromirror that is either incorporated into the sample chamber [23–28] or mounted on a cantilever that is placed near the sample [29,30]. To tune the position of the waist of the light sheet, an electrically tunable lens can be used [23]. While the reflected light sheet approach enables the use of high-NA oil immersion objectives on standard microscope bodies, this method still requires the samples to fit in either microchannels with in-built micromirrors, which are complex to manufacture, or the samples need to be mounted in chambers that are open to the top to enable the positioning of the cantilever mirror. Another disadvantage of the LSFM techniques that require a reflection of the illumination beam into the focal plane is that during volumetric imaging, the light sheet has to be repositioned continuously in order to match the focal plane of the detection objective, which requires precise positioning, alignment, and hardware control of the light sheet.
In single-objective oblique plane microscopy (OPM) on the other hand, the light sheet is launched from the objective lens at an angle that is oblique to the focal plane [20,22,31–33]. In order to image the oblique plane with a camera sensor, it is necessary to reimage and rotate the oblique plane. This is achieved using two additional objective lenses in which one objective forms an intermediate image of the oblique plane, which can be imaged with the other objective that must be aligned so that the intermediate image is in its focal plane, to form a horizontal image. This image rotation is based on the principle of remote focusing [34–37]. While most OPM systems rely on stage scanning for volumetric imaging, a galvo mirror can also be used for fast scanning and de-scanning of the light sheet and the image plane, to dramatically increase the imaging speed [21,38–40]. The image rotation in OPM techniques leads to a degradation in overall light collection efficiency and resolving power, due to aperture loss in the remote imaging unit, which may be anisotropic along the x- and y-directions. For example, a recent OPM system constructed around a water immersion detection objective with NA = 1.27 achieved an effective NA along the x-direction of 1.06, and an effective NA along the y-direction of 1.20, with an effective collection efficiency of the entire system that is equivalent to an NA of 1.17 [41]. Recently, this method was improved by employing a custom glass-tipped objective [42,43], which increased the collection efficiency and reduced the aperture clipping in the remote beampath. This resulted in an effective NA of 1.28 of the system, based on a primary objective NA of 1.35. An important further improvement of this system is that the secondary as well as the custom tertiary objective are color and field curvature corrected lenses (plan apochromatic), which reduces optical aberrations over a larger field of view [42,43].
An extreme version of OPM is to orient the illuminated plane orthogonally to the focal plane of the primary objective. This implementation of OPM, termed axial plane optical microscopy (APOM), simplifies the required image rotation because only a single remote focusing objective is used in conjunction with a 45° mirror placed in the focal space of the remote objective [32,44–46]. APOM therefore acquires vertical images of the sample, and is compatible with high-NA oil immersion objectives and sample mounting on coverslips and multi-well plates on standard commercial microscope bodies.
Motivated by the need for high-resolution and low phototoxicity imaging of living 3D samples on standard coverslips and standard microscope bodies, we developed an improved APOM system which we termed digital confocal light-sheet axial plane optical microscopy (DC-APOM). For this system, we combined the concept of APOM [44] with digitally scanned Gaussian and Bessel beam light-sheets [47,48] and confocal slit detection [49,50], and optimal objectives for 3D living samples, in order to improve contrast and resolution. We further implemented a two-fold increased detection efficiency of the detection beam path, by introducing a polarizing beam splitter and quarter waveplate into the DC-APOM detection beampath at the second objective [46]. We demonstrate and evaluate the capabilities of DC-APOM compared with traditional confocal microscopy, for imaging 3D dynamics of highly-scattering bacterial biofilms and macrophage phagocytosis.
2. Methods
The key components of the DC-APOM design are shown in Fig. 1(A) and the complete optical system is described in Fig. 1(B). We implemented DC-APOM on a standard commercial inverted microscope body, as indicated in Fig. 1(B). By translating the sample in the y-direction through the light sheet positioned in the xz-plane, and by optically rotating the detected vertical image into a horizontal image on the camera, 3D images of challenging biological samples, such as bacterial biofilms, can be obtained at single-cell resolution (Fig. 1(C)). Using a pair of identical objective lenses for the primary image detection and the remote image rotation, images of the xz-plane with minimal aberrations can be obtained (Fig. 1(D)). These properties make DC-APOM a suitable technique for 3D live-cell imaging in standard sample formats based on coverslips or multi-well plates on commercial microscope bodies (Fig. 1(E)).
Fig. 1.

Digital Confocal Axial Plane Optical Microscopy for high-resolution imaging of living samples. (A) DC-APOM employs the same objective lens for light sheet illumination and detection, to image a vertical xz-plane with high-NA oil-immersion objectives. The vertical image is rotated by 90° into a horizontal image that can be projected onto a camera sensor using a remote objective lens (Supplement 1 (3.8MB, pdf) , Fig. S5). Blue lines at the sample and on the camera sensor indicate the beam that is scanned across the sample to form a light sheet. A red box indicates the rows of pixels on the camera sensor that are currently read out relative to the position of the scanned beam. While the beam is moved across the sample, the virtual confocal slit follows. (B) Optical system of DC-APOM. The beam path is described briefly in the Methods, and in detail in Appendix A. The following designations are used: SMF - single mode fiber; CL - collimator; L1, L2, L3, L4, L5, L6 - lenses; GX - X-axis galvo; SL - scan lens; TL1, TL2, TL3, TL4 - tube lenses 1; O1, O2 - objectives; DM - dichroic mirror; EF - emission filter; M1, M2, M3, M4, M5, M6 - mirrors; PBS - polarizing beam splitter; QWP – quarter-wave plate. The blue and green arrows indicate the direction of light propagation for the excitation and emission, respectively. (C) Samples that are difficult to image at cellular resolution, such as densely-packed bacterial biofilms can be fully resolved and reconstructed in 3D using DC-APOM imaging. In the V. cholerae biofilm shown here, each cell is coloured according to its distance to the center of mass of the whole biofilm colony. (D) Widefield xy-image and APOM xz-image of the same nanohole array target used for alignment of the system. The holes in the chromium layer of the target have a diameter of 400 nm and a spacing of 7 µm. (E) Schematic drawings of examples for sample geometries that can be used for DC-APOM imaging. The only constraint on the sample geometry is the necessity for a coverslip.
The experimental setup consists of three main beam paths (Fig. 1(B)): excitation (Laser-CL-TL3), illumination-detection (O1-TL1), and detection (TL2-O2-TL4). The exact components and considerations for these beam paths are described in Appendix A. The alignment procedure (see Supplement 1 (3.8MB, pdf) Section S1) strongly relies on a suitable calibration target with a predefined pattern. As a target we used a knife-edge prism onto which we deposited a 160 nm thick chromium layer (OD = 2.7) into which a nanohole array pattern was inscribed using a focused ion beam, as described in detail in Supplement 1 (3.8MB, pdf) Section S2 and shown on Fig. 1(D).
In the excitation beam path, lasers are coupled into to a single-mode fiber SMF, followed by outcoupling to a collimated beam of 6 mm in diameter (measured on 1/e2 intensity level). After collimation, the light passes through a custom-made chromium mask GBM, which performs amplitude modulation of the beam to shape a Gaussian or a Bessel beam with a selectable size, resulting in a light sheet with a selectable width and length. This GBM mask and the resulting beam shapes are described in detail in Supplement 1 (3.8MB, pdf) , Section S3. The beam then falls on a galvo mirror GX, which controls the beam location in the x-axis in the sample space. The beam then passes through a scan lens SL and tube lens TL3, which form a telescope (1.28x beam magnification), to image the beam on the back focal plane (BFP) of the illumination objective O1. The beam is coupled into the commercial microscope body (Nikon Ti-E) through the epi-illumination port. This ultimately results in a beam along the z-axis in the sample space, which can be scanned along the x-axis using the galvo mirror to form a light sheet.
In DC-APOM the illumination and detection are both performed by the same objective O1. The fluorescence signal originating from the sample is therefore collected by O1 and passes through the dichroic DM. The image formed by the O1-TL1 system in the focal plane of TL1 is subsequently re-imaged and relayed by another tube lens TL2 to a polarizing beam splitter PBS, quarter waveplate QWP and a second objective O2 (identical to O1). In the objective space of O2, along the z-axis, an intermediate image is formed with the same orientation and magnification as in the O1 objective xyz-space. As the illumination of the sample is due to a light sheet, only a single vertical xz-plane (a cross-section of the sample) is present in the intermediate image formed by objective O2. In order to detect this vertical plane, a mirror M5 tilted at 45° degrees to the optical axis is employed in the space of O2, which effectively rotates the vertical intermediate image into a horizontal one. Here, it is necessary to achieve a high position precision of mirror M5 in the focal volume of O2 and we achieved this by employing a combination of fine adjustment positioners (see Appendix A). The resulting xy-plane is then detected by the O2 objective and transmitted back towards the QWP, PBS, mirror M6, and the camera. The sCMOS camera has a rolling shutter readout mode, whose speed and active pixel rows (slit width) can be controlled, creating a virtual confocal slit moving throughout the sCMOS chip. This virtual slit is synchronized with the scanned light sheet by the GX galvo to produce a confocal detection along x-axis. The hardware and software control scheme is described in detail in Supplement 1 (3.8MB, pdf) , Section S4.
The preparation of biological samples and calibration samples, as well as technical details regarding the biological assays are described in detail in Appendix B.
3. Results
The DC-APOM system was developed based on the original APOM design [44], but with important modifications to improve the system’s performance for biological samples. These changes include a confocal slit light sheet detection, for which a light sheet is generated along the optical axis of the primary objective lens by scanning a beam across the x-direction in the xz-plane using a galvo mirror. For a given beam position, only those camera pixels that are close to the image generated by the beam on the camera sensor are read out (Fig. 1(A)), which effectively rejects out-of-focus scattered light. This digital confocal slit is based on a particular readout mode of scientific CMOS cameras, which enables the rows of pixels that are exposed and those that are read out to be scanned across the camera sensor at user-defined speeds so that they can be synchronized with the scan speed of the excitation beam [49]. In order to improve the transfer efficiency of the emitted fluorescence to the camera, and thereby reduce the required excitation illumination intensity, we introduced a polarizing beam splitter and quarter waveplate into the DC-APOM detection beam path of the second objective (Fig. 1(B)), resulting in a twofold increase of the detection efficiency [46]. We further determined the optimal detection objectives, excitation illumination beam shapes, and confocal slit widths for a model biological sample, as described below.
As a model for a challenging biological sample for evaluating the capabilities of DC-APOM, we used bacterial biofilms of the species Vibrio cholerae, which consist of densely packed cells (approximately 2 µm length, 0.8 µm diameter) in a 3D structure, held together by an extracellular matrix. Biofilms are the most abundant form of microbial life on Earth [51], yet they are difficult to image at high resolution, because they are photosensitive and highly scattering samples. Biofilms are often investigated using confocal microscopy [52,53], and image analysis techniques for cytometry inside biofilms have greatly improved recently [54–56]. Two-objective LSFM techniques have been used recently to successfully image bacterial biofilms [56,57], and single-objective LSFM promises to provide the high resolution and low phototoxicity needed for tracking dynamic events in biofilms at cellular resolution in the widely-used flow chamber sample geometries for biofilm cultivation.
Image detection and remote image rotation is achieved in DC-APOM using a pair of identical objectives to ensure minimal aberrations. To achieve single-cell resolution of bacterial biofilms, the smallest resolvable distance must be below the bacterial cell diameter of ∼1 µm. We therefore evaluated the DC-APOM performance in terms of point spread function (PSF) and in terms of image quality of V. cholerae bacterial biofilms for three different 100x oil immersion objectives with high numerical aperture: a NA = 1.49 oil objective, a NA = 1.45 oil objective, and a NA = 1.35 silicon oil objective (Fig. 2(A)). While the NA 1.49 and NA 1.35 objectives have a correction collar, which we found to be important during the alignment (Supplement 1 (3.8MB, pdf) , Section S1), the NA 1.45 objective does not have such a correction collar, resulting in comparatively larger PSF sizes. For the two objectives that are designed for regular immersion oil (refractive index 1.518), DC-APOM yields strong aberrations for z-positions that are a few µm above the cover slip. These aberrations manifest as axial smearing and loss of signal further away from the coverslip. In comparison, images obtained with the identical objectives using spinning disk confocal microscopy showed much less axial smearing, yet a loss of signal intensity with increasing z is also observed. In the case of the objective lens with silicon oil immersion (refractive index 1.406), there is almost no axial smearing observed for the DC-APOM and spinning disk confocal, and also the loss of signal intensity along z is also much smaller (Fig. 2(A)). The aberrations that we observed for the regular oil-immersion objectives can therefore be attributed to a refractive index-mismatch between the immersion oil and the sample. These results for the different objective lenses show that the silicon oil objective is best-suited for the imaging of bacterial biofilm communities, and likely other biological samples, due to the matching of the refractive index between the silicon oil and the cellular cytoplasm and/or extracellular matrix.
Fig. 2.

Optimization of illumination and detection conditions for DC-APOM. (A) Comparison of the performance for three different objectives with high numerical aperture, using spinning disk confocal and DC-APOM in xz-images of fixed V. cholerae biofilms. For DC-APOM, a pair of two identical objectives was used in each configuration. The insets show the point spread function measured directly on the coverslip using 100 nm fluorescent beads embedded either in medium matching the refractive index of the immersion oil (marked here as oil) or in water. (B) Comparison of the effect of different illumination beam profiles on image contrast (left axis, red) and on signal-to-background ratio (right axis, blue). The confocal slit width is varied for light sheets based on Gaussian beams of a given width: FWHMxy = 0.6 µm (circles), FWHMxy = 1.3 µm (diamonds), FWHMxy = 2.2 µm (squares). Similarly, the confocal slit width is varied for Bessel beams of given width: FWHMxy = 0.4 µm (pyramids), FWHMxy = 0.6 µm (circles), FWHMxy = 1.3 µm (squares). Measurements connected by a line are acquired for the same biofilm. Image contrast is quantified using the variance of Laplacian (LAPV) score. The LAPV and SBR values for an infinitely large confocal slit width correspond to measurements for static light sheets and all LAPV scores are normalized to this point.
The point spread function size, measured on the coverslip surface with 100 nm fluorescent beads, is similar between DC-APOM and spinning disk confocal microscopy for all three objectives (Fig. 2(A)). As expected, the PSF size increases with decreasing NA of the objective. Given the strongly improved image quality of DC-APOM with the silicon oil objective above the coverslip, we therefore recommend using a silicon oil immersion objective for DC-APOM with a NA that is high enough for resolving the biological samples of interest. The highest NA of a commercially available silicon oil-immersion objective is currently 1.35, resulting in a PSF with a full width at half maximum (FWHM) of 269 ± 23 nm in the x-direction and 657 ± 25 nm in the z-direction for fluorescence emission in the range 500-550 nm in DC-APOM (Fig. 2(A)). For all further DC-APOM optimizations and measurements, we therefore used the 100x silicon oil immersion objective lens.
To optimize the DC-APOM excitation illumination conditions for 3D bacterial biofilms, we created different beam types, shapes, and sizes, ranging from Gaussian to Bessel beams using a beam amplitude mask (Supplement 1 (3.8MB, pdf) , Fig. S1). For each beam, we measured the resulting light sheet sizes (Supplement 1 (3.8MB, pdf) , Table S1), which are consistent with predictions from scalar diffraction theory, using either the full theory or its paraxial approximation (Supplement 1 (3.8MB, pdf) , Fig. S2). Using fixed V. cholerae biofilms, we then compared the imaging performance for the different Gaussian and Bessel light sheets for different digital confocal slit sizes. To quantitatively compare the quality of the resulting 3D biofilm images, we calculated the local image contrast using the variance of Laplacian (LAPV) score and the signal-to-background ratio (SBR), as a function of the confocal slit width, normalized by the FWHM of the light sheet (Fig. 2(B)). For both Gaussian and Bessel beams the highest contrast is achieved for smallest slit sizes, as expected. The smallest possible confocal slit width is 1 px on the camera, which correspond to 58.5 nm in the sample in our system. However, smaller confocal slits also result in the rejection of more light, so that a lower overall signal is detected. The SBR curves for both beam types show a peak at ratios of the slit width/FWHM between 1 to 4 and the SBR drops significantly for slit width/FWHM < 1. Interestingly, the highest LAPV scores correspond to the lowest SBR, yet low SBR values can also correspond to low LAPV scores. The comparison between the two beam shapes also shows that for a similar beam size, the Gaussian beams give a higher SBR (Fig. 2(B)). However, Gaussian and Bessel beams give similar local contrast (Fig. 2(B)) and inspection of biofilm images show a lower background level for images obtained with Bessel beams (Supplement 1 (3.8MB, pdf) , Fig. S3). For Bessel beams, a considerable amount of energy is in the side lobes, which do not contribute to the imaged fluorescence signal in DC-APOM if the confocal slit is appropriately small, because the slit rejects the out-of-focus fluorescence generated by the sidelobes [58,59]. However, the energy in the side lobes of the Bessel beam means that the samples need to be exposed to more excitation light to achieve the same SBR, compared with Gaussian beams. Overall, Fig. 2(B) shows that a good trade-off between high local contrast and high SBR levels is achieved when the confocal slit size equals the FWHM of the beam, which was chosen for all subsequent experiments.
A key advantage of using light sheet microscopy for biological samples is the strongly reduced phototoxicity compared with confocal microscopy. As quantitative characterizations of phototoxicity are difficult, we instead investigated photobleaching as a proxy to indirectly estimate phototoxicity. To quantitatively compare the photobleaching rate of DC-APOM and spinning disk confocal microscopy (Fig. 3), we acquired volumetric imaging data of V. cholerae biofilms that were stained with a nucleic acid dye followed by formaldehyde-fixation. For spinning disk confocal microscopy, a drastic reduction in the fluorescence signal is observed after just one acquired volume. For DC-APOM light sheet imaging, the reduction of fluorescence intensity is significantly slower - after 16 imaging cycles the fluorescence signal intensity is still close to 50% of the first imaging cycle for the Gaussian beam and around 40% for the Bessel beam. Bessel beam light sheet imaging causes stronger photobleaching, because the side lobes of the beam bleach the sample without contributing to the detected fluorescence signal. As a control condition, we applied the same illumination energy to a given plane inside biofilms with all three imaging modalities and observed the same bleaching dynamics for all conditions (Supplement 1 (3.8MB, pdf) , Fig. S4). The design of the DC-APOM emission detection beam path leads to a loss of 50% of the emission light at the polarizing beam splitter, and further loss of 52.8% of the signal due to clipping of the NA of the remote objective, resulting in a 23.6% total collection efficiency (see Appendix A for a detailed discussion of the DC-APOM collection efficiency). Despite the relatively low collection efficiency of the DC-APOM system, the photobleaching curves reveal that DC-APOM allows 3D imaging with significantly less photobleaching than traditional confocal microscopes (Fig. 3).
Fig. 3.
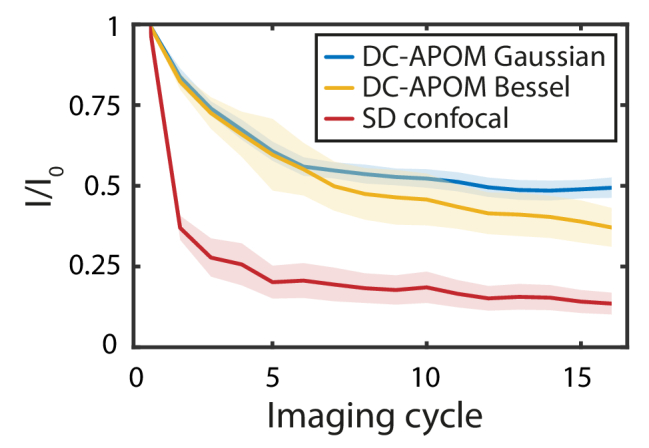
Photobleaching performance. Photobleaching is quantified using fluorescence intensity values I normalized to the first imaging cycle I0, for DC-APOM based on Bessel or Gaussian beams, and for spinning disk confocal. To compare the photobleaching, 3D image volumes of the same resolution were acquired using all imaging modalities, with the same signal-level in the images. Lines denote the mean of n = 10 biofilm volumes and the shaded area indicates the standard deviation.
To demonstrate the capabilities of DC-APOM for living samples, we imaged dynamic 3D processes that require high-NA oil objectives for both prokaryotic and eukaryotic cells (Fig. 4). First, we used DC-APOM to image the dispersal of V. cholerae biofilms over time (Fig. 4(A)). Previously, we discovered that V. cholerae biofilm dispersal can be triggered by carbon source removal for biofilms that are primed by high levels of self-produced quorum sensing autoinducers [60]. DC-APOM imaging revealed that after carbon source removal, biofilm dispersal occurs by the detachment of individual cells from the outer regions of the biofilm, while cells deeper inside the biofilm can only depart once the outer cells have left (Fig. 4(A)). The imaging of dynamic events in the xz-plane that DC-APOM enables is particularly useful for processes that occur so quickly that they are difficult to image with confocal microscopy (which acquires consecutive z-slices), such as the biofilm response to an osmotic shock (Fig. 4(B)). DC-APOM imaging revealed that only 1.6 s into the osmotic shock, the biofilm structure became less dense and several cells already detached from the biofilm, and within 6.4 s the complete biofilm was completely disrupted. Lastly, to test DC-APOM for other cell types, we imaged phagocytosis of Escherichia coli cells by murine macrophages (Fig. 4(C)). Using DC-APOM with the 100x silicon oil immersion objective clearly resolves individual cells encapsulated within phagosomes.
Fig. 4.

Imaging of dynamic three-dimensional processes at high resolution with DC-APOM. (A) Time-lapse of the dispersal process of a V. cholerae biofilm colony (cells express sfGFP), induced by removing glucose from the growth medium. Different cells in the outer periphery of the biofilm depart from the biofilm colony over the course of several minutes. The bottom of the biofilm remains firmly attached to the glass coverslip. (B) Time-lapse at high time resolution of a V. cholerae biofilm (cells express sfGFP) exposed to an osmotic shock, caused by replacing the growth medium with distilled water, showing a massive disruption of the biofilm structure over the course of several seconds. (C) Image overlay showing the interaction of murine macrophages stained with CellTracker (magenta) and E. coli cells expressing sfGFP (green). Individual E. coli cells that have been phagocytosed by the macrophages are visible inside phagosomes. The magnified inset shows the two fluorescence channels separately. The dotted gray lines in the images denote the position of the glass coverslip.
4. Discussion
We implemented a digital-scanning confocal light sheet imaging technique that is based on APOM, which combines the benefits of selective plane illumination, confocal detection, high-NA objectives, and standard sample geometries based on coverslip mounting. While the optical design of DC-APOM is based on the APOM technique [44], the optimized silicon oil objectives, the improved imaging contrast via confocal detection and custom beam shapes, and the improved the fluorescence detection efficiency now make this technique suitable for 3D live-cell imaging. We demonstrated that the DC-APOM system can be used for a range of challenging eukaryotic and prokaryotic samples that require high optical resolution. Unlike in standard two-objective light sheet microscopy, where elaborate sample chambers and agarose-embedding of samples are usually required, and unlike in reflected light sheet microscopy [23,25], where particular sample geometries that include micromirrors are required, DC-APOM only requires a coverslip for sample mounting. Therefore, the DC-APOM technique is compatible with 96-well plates, microfluidic chambers, and standard sample geometries that can also be used on standard confocal microscopes.
The photobleaching comparison between DC-APOM and spinning disk confocal microscopy showed that this single-objective light sheet imaging technique causes much less photobleaching while obtaining comparable signal level. This indicates that for a given signal level, a substantially reduced amount of excitation light energy is needed, enabling a more gentle imaging of the dynamics of biological samples. We have also shown that DC-APOM achieves comparable spatial resolution as spinning disk confocal microscopy, albeit at a higher time resolution when volumes with a large z-component are required, as demonstrated for biofilm dispersal and the biofilm response to osmotic shocks (Fig. 4). DC-APOM is therefore particularly well-suited for imaging biological process that occur perpendicular to the coverslip.
The present implementation of the DC-APOM system has several limitations that need further attention in future developments. The current alignment procedure is relatively complex and requires some time to master (see Supplement 1 (3.8MB, pdf) , Section S1). In addition, due to the image rotation achieved by the 45° mirror (M5) and the remote objective (O2), the system is very sensitive to refractive index changes in the sample medium. The optimal image quality is achieved when the sine and Herschel conditions are fulfilled and the refractive index of all parts of the space above the primary objective are matched to the refractive index of all parts of the space above the remote objective – but small differences can be compensated using the correction collars of the objectives. Furthermore, DC-APOM requires high NA objective lenses in order to minimize the loss of signal due to aperture clipping during image rotation. Based on the geometric considerations of the aperture clipping by Kim et al. [45], we calculated an effective NA along the tilt direction of NAy = 0.677 and along NAx = 1.345 (Appendix A). Finally, current commercial objective lenses are optimized for imaging with minimal aberrations in the focal plane and display aberrations further away from the focal plane. These effects can become noticeable when imaging the xz-plane in an APOM configuration, and specialized objectives could further improve the APOM image quality.
Overall, DC-APOM combines the significant benefits that are inherent to selective plane illumination and the spatial resolution of high-NA confocal microscopy on a commercial inverted microscope body. As the illumination and detection beam paths can be assembled as separate units, this light sheet implementation can be added as a module to standard commercial research microscopes, via the epi-illumination and emission port, respectively. We therefore hope that DC-APOM will make light sheet microscopy more widely used in biological research laboratories.
Acknowledgements
We are grateful to Kerstin Volz and Wolfgang Stolz for providing access to the focused ion beam facility of the Materials Science Centre, Philipps-Universität Marburg.
Appendix A: Beam path and principle of operation
A schematic drawing of the experimental setup is shown in Fig. 1(B). It consists of three main beam paths: excitation (Laser-CL-TL3), illumination-detection (O1-TL1), and detection (TL2-O2-TL4). In the excitation beam path, light from a 488 nm and a 552 nm laser (OBIS LX 488-50 mW and LS 552-60 mW, Coherent) is coupled into to a single-mode fiber SMF (S405-XP, 0.12 NA, 400-680 nm, Thorlabs). After passing throughout the fiber, the light is outcoupled by a collimator CL (achromat, focal length f = 25 mm, G052007000, Qioptiq) to a collimated beam of ca. 6 mm in diameter (measured on 1/e2 intensity level). After collimation, the light passes through a custom-made chromium mask GBM (JD Photodata, UK), which performs amplitude modulation of the beam to shape a Gaussian or a Bessel beam, which results in a light sheet with a selectable width and length (for more details see Supplement 1 (3.8MB, pdf) , Section S3 and Supplement 1 (3.8MB, pdf) , Section S5).
In order to produce a Bessel light sheet, the mask must be positioned in the focus of the lens L1 so that the image of the mask is conjugated to the back focal plane (BFP) of the objective O1 (which then acts as a Fourier lens resulting in a Bessel beam in the focal plane (FP). The lenses L1 (achromat, f = 100 mm, AC254-100-A-ML, Thorlabs) and L2 (achromat, f = 25 mm, AC127-025-A-ML, Thorlabs) form a telescope, which demagnifies the beam 4x (from ≈ 6 mm to ≈ 1.5 mm).
The beam then falls on a pair of mirrors, M1 and the galvo mirror GX (GVS011/M, Thorlabs) as shown in Fig. 1(B): The GX galvo (x-axis control, see coordinate system in Fig. 1(A)) creates the digital (scanned) light sheet while the orientation of the mirror M1 determines the location of the light sheet along y-axis in the O1 space. The lenses L3 and L4 (identical achromats, f = 50 mm, AC254-050-A-ML, Thorlabs) form a relay system, which images the mirror M1 onto the GX galvo. The mirror M1 and galvo GX are in the focuses of the respective back or front lenses for the relay system to work properly. This is done to avoid any potential vignetting effect. Note that the focus of L2 coincides with the focus of L3, and that the same is valid for the other lenses along the excitation beam path. After the L3-L4 relay system, the light passes through a second relay system L5-L6 (identical achromats, f = 50 mm, AC254-050-A-ML, Thorlabs), and then falls on the scan lens SL (achromat, f = 125 mm, AC254-125-A-ML, Thorlabs) and the tube lens TL3 (achromat, f = 160 mm, 67-334, Edmund Optics). This pair of lenses (SL and TL3) forms a telescope (1.28x beam magnification), which finally images the beam on the BFP of the illumination objective O1 (100x/1.35NA Silicone Oil, Olympus). This results in a light sheet, in the sample space of O1, propagating along the z-axis (i.e. vertically, see coordinate system in Fig. 1(A)) of the optical system. Note that the excitation beam is coupled to the objective O1 through the back port of a microscope (Nikon Eclipse Ti-E Inverted Microscope) using a dichroic mirror DM (reflection 488/543/635 nm, Di01-R488/543/635-25 × 36, Semrock). A motorized xy-stage (MS-2000 XY, Applied Scientific Instrumentation) was used for the positioning of the sample and for scanning the sample through the light sheet (for more details on the hardware wiring and control see Supplement 1 (3.8MB, pdf) , Section S4). As the resulting light sheet is vertical (xz-plane) a 3D stack is obtained by scanning with the stage along y-axis: After each step of the stage, a xz-image is obtained and from a series of such images a 3D stack of the object can be reconstructed.
Since in DC-APOM the illumination and detection are performed by the same objective, O1 is used simultaneously for the illumination of the sample (see the blue vertical light sheet in Fig. 1(B)) and detection of the fluorescent signal. Thus, the signal originating from the sample is collected back by O1 passes through the dichroic. Subsequently, the emission signal is transmitted towards the microscope’s tube lens TL1 (achromat, f = 200 mm, G063205000, Qioptiq) and deflected to the side port of the microscope by a right-angle prism mirror (M1). Next the image formed (by the O1-TL1 system) in the focal plane of TL1 is subsequently re-imaged and relayed by another tube lens (TL2, achromat, f = 200 mm, G063205000, Qioptiq) to a polarizing beam splitter PBS (420-680 nm, PBS201, Thorlabs), quarter waveplate QWP (zero-order, 532 nm, WPQ10ME-532, Thorlabs) and a second objective O2 (100x/1.35NA Silicone Oil, Olympus), with identical specifications to the first one, O1. Thus, in the objective space of O2, along the z-axis, an intermediate image is formed: a 3D representation of the sample with the same orientation and magnification as in the O1 objective xyz-space. As the illumination of the sample is due to a light sheet, only a single vertical xz-plane (a cross-section of the sample) is present in the intermediate image formed by objective O2. In order to detect this vertical plane, a tilted mirror M5 (D-shaped mirror, PFD05-03-P01, Thorlabs) at 45° degrees to the optical axis is employed in the space of O2. In order to obtain a high alignment accuracy, we employed a combination of a two axis translational positioner (CXY1, Thorlabs), a rotational positioner (LRM1, Thorlabs) and goniometric stage (GN1/M, Thorlabs). It can be shown by ray tracing (see Supplement 1 (3.8MB, pdf) , Fig. S5) that such a mirror, after reflection, rotates the vertical plane into a horizontal one (Li, et al., 2014). If the refractive index of the immersion oil differs from the refractive index of glass, it is necessary to place a coverslip between O2 and M5. The resulting xy-plane is then detected by the O2 objective and transmitted back towards the QWP, PBS, and mirror M6. Here, the emission filters EF (for GFP, ET525/50, Chroma; for mRuby ET600/50, Chroma) are mounted in a motorized Filter Wheel (FW102C, Thorlabs). Then, another tube lens TL4 (achromat, f = 200 mm, G063148000, Qioptiq) focuses the image on a sCMOS camera (Zyla 4.2 PLUS, Andor). This camera has a rolling shutter readout mode, whose speed and active pixel rows (slit width) can be controlled, creating a virtual confocal slit moving throughout the sCMOS chip. This virtual slit is synchronized with the scanned light sheet by the GX galvo to produce a confocal detection along x-axis (for more details see Supplement 1 (3.8MB, pdf) , Section S4).
In the beam path, it is in principle not mandatory that the distances between O1-TL1, TL1-TL2, and O2-TL2 are in a 4f configuration, but this depends on the choice of initial distance O1-TL1 (in our case the O1-TL1 distance is defined by the microscope manufacturer). Hence, the distances TL1-TL2 and O2-TL2 must be adjusted such that the aberrations and distortions are minimized during the alignment procedure (see Supplement 1 (3.8MB, pdf) , Section S1 for more details).
Due to the geometry that is used in the mechanism of the rotation of the vertical plane in the O2 space, the objective’s aperture is clipped. This aperture clipping can be quantified by following the geometric considerations described by Kim et al [45]. First, only the signal originating from half the objective’s aperture can be detected. In addition, light that originates close to the optical axis cannot be collected, which is indicated by the gray area in Supplement 1 (3.8MB, pdf) , Fig. S5B, which is defined by the collection half angle of the lens. In order to calculate the collection efficiency (CE) of an objective, the surface area of a spherical segment enclosed by the collection half angle of the objective must be divided by the total surface of the sphere ,
| (1) |
The spherical segment enclosed by the objective’s half angle can be calculated using
| (2) |
| (3) |
The total surface of the sphere is given by
| (4) |
With this information, we can first calculate the collection efficiency of the primary objective lens,
| (5) |
Therefore, the primary objective collects 36% of all fluorescence signal that is emitted by the excited fluorophores. To calculate the collection efficiency for the remote objective, we have to consider the aperture clipping described above. As we can only use half of the aperture of our secondary objective, we have to divide by 2. In addition, we lose a small cone of light near the optical axis, as indicated in Supplement 1 (3.8MB, pdf) , Fig. S5B by the gray area [45]. Considering this, the overall surface area becomes
| (6) |
This leads to an overall collection efficiency for NA = 1.35 and n = 1.406 (silicon oil) of
| (7) |
This means that 47.2% of the fluorescence light captured by our primary objective is recollected by our remote objective. This collection efficiency is equivalent to that of a silicon immersion objective with NA = 1.06.
Furthermore, taking into account that the fluorescent signal is non-polarized, we note that at the PBS only one of the two polarizations (horizontal or vertical polarization) is reflected to O2. This leads to another 50% loss of signal, which results in an overall detection efficiency of 23.6%, without accounting for further losses due to other optical components. However, compared with the original APOM design [44], the double pass through the quarter wave plate QWP in the DC-APOM system rotates the polarization by 90° and therefore preserves it so that in the DC-APOM beam path no loss of signal occurs with respect to the chosen polarization. Without such an improvement, the overall theoretical detection efficiency would drop to 11.8%. Based on the geometric and numerical considerations by Kim et al. [45], we also calculated the effective numerical aperture along the x- and y-direction. Along the y-direction, the effective full aperture angle is 57.55°, which results in an effective NAy of
| (8) |
On the other hand, the aperture clipping in the xz direction is almost negligible. Here, the aperture half angle is 73.075° which results in an effective NA of
| (9) |
We therefore have no significant reduction in the aperture along xz or resolving power. A more rigorous discussion of the aperture clipping in APOM can be found in the literature by Kim et al. [45].
The principle of remote focusing is central to APOM. The remote focusing concept was introduced in order to optically refocus (without aberrations) for high NA objectives in z-planes different from the focal one [35,36]. This strategy allows fast z-scanning, without mechanically interfering with the specimen. For remote focusing it is important to note that microscope objectives are not designed to have optimal performance away from the focal plane. The further away from the focal plane, the higher the spherical aberrations of the obtained microscopy image. However, it was shown theoretically and experimentally [34,35,37] that when two objectives are paired to form an optical system with a uniform magnification M = n1/n2 (n1 and n2 are the refractive indexes of the immersion media of objective O1 and O2) in xy and in z, the intermediate image is largely aberration-free within a significant z-range around the focal plane [37]. The reason for this effect is that such a pair of objectives cancels out their mutual aberrations to a great extent [37]. According the remote focusing principle, to achieve a uniform magnification simultaneously along xy- and z-planes, the sine and Herschel conditions for perfect (stigmatic) imaging in xy and along z, respectively, must be fulfilled [35]. In the present case, uniform magnification can be achieved when the pair of objectives and their respective tube lenses (see O1, TL1, TL2 and O2 in Fig. 1(B)) satisfy the following relationship [35]:
| (10) |
Here, M is the magnification of the combined optical system (O1-TL1-TL2-O2), while M1 is the magnification of the first (O1-TL1) and M2 is the magnification of the second system (O2-TL2), respectively. fTL1 and fTL2 are the focal lengths of the tube lenses TL1 and TL2, respectively. fO1 and fO2 are the focal lengths of the objective lenses O1 and O2, respectively. Equation (10) implies that when the overall magnification M (lateral or axial) follows this relationship, the system images a 3D object with a uniform magnification factor n1/n2 in all three directions xyz, in consistence with the conditions for perfect (stigmatic) imaging.
When identical objectives O1 and O2 are employed, the refractive indexes of the immersion media are the same (i.e. n1 = n2), hence the magnification factor M = 1, as in the present case. Thus, the role of the M5 mirror (see Supplement 1 (3.8MB, pdf) , Fig. S5) is to transform a 2D section from the intermediate image of the 3D object into a horizontal plane that coincides with the focal plane FP of O2. Hence, the points away from FP are imaged optimally, i.e. the vertical plane V can be imaged with minimum aberrations by rotating it 90° and thus converting it into a FP, where the aberrations are compensated the best. Note that in practice, the z-range within which this condition (for stigmatic imaging) holds is defined by , i.e. for z displacements from FP much smaller than the focal length f of a given objective. Previously, using the Strehl ratio, a quantitative measure for the quality of imaging along z has been derived [35]. For a typical high NA objective (NA = 1.4) and = 633 nm, the z-range within which the Strehl ratio S is greater than 0.8 is ±70 µm around the FP. This can be seen as the maximum z-range within which the system still operates acceptably well. However, for best performance, the Strehl ratio must be S1 [35] which reduces the z-range of the exact same objective down to ±35 µm. For the objectives that are used in the present DC-APOM implementation, we observed best performance within a range of ±30 µm, which is similar to the theoretical value mentioned above.
Appendix B: Sample preparation and image analysis
Fluorescent bead preparation
For measuring the point spread function and the performance of different microscopy modalities (spinning disk confocal or DC-APOM), we used fluorescent polystyrene beads with 100 nm diameter (FluoSpheres Carboxy, yellow-green 505/515, Thermo Fisher Scientific Inc.). The stock beads solution was diluted 100-1000x and deposited on a microscopy coverslip by drop-casting and drying at room temperature. Subsequently, we employed this bead-covered coverslip for the measurement of the PSF for different objectives in different media. The following media were used: water (n = 1.33), silicone oil (n = 1.406), PVA (polyvinyl alcohol, n = 1.51). The latter two media match the refractive indexes of the respective immersion liquids for the Olympus silicon oil and Nikon oil objectives, and thus give optimal conditions for PSF measurements.
Bacteria-macrophage interaction
E. coli AR3110 expressing gfp under the constitutive Ptac promoter (strain KDE1469) was grown in liquid lysogeny broth (LB) medium overnight at 37 °C under shaking conditions. The following day, LB medium was removed by centrifugation (13,000 rpm, 1 min) and the cells were resuspended in an equal volume of DMEM medium containing 10% fetal calf serum (FCS). For interaction studies of macrophages and E. coli, 150 µl of DMEM medium containing 40,000 murine macrophages (Raw267.4), previously grown in DMEM (with 10% FCS), were stained with 3 µM CellTracker Red CMTPX for 15 min at 37 °C and 5% CO2. This suspension of macrophages was then mixed with 30 µl of the bacterial suspension. After co-incubation for 30 min at 37 °C and 5% CO2, the cells were imaged with the DC-APOM technique in 9-mm diameter wells made out of polydimethylsiloxane (PDMS) with a coverslip bottom surface.
Biofilm growth
In order to grow biofilms, V. cholerae strains were grown overnight at 28 °C in liquid LB medium under shaking conditions. For biofilm architecture characterizations, we used V. cholerae strains derived from the N16961 wild type strain: KDV148 (carrying no fluorescent protein expression system) or KDV615 (carrying a plasmid for constitutive sfGFP expression). For biofilm dispersal experiments, a quorum sensing capable wild type strain KDV296 was created by conjugating a plasmid for constitutive sfGFP expression into the V. cholerae C6706 wild type strain background. The overnight culture was diluted into fresh LB medium and regrown under the same conditions until the culture reached an OD600 between 0.4 and 0.6. This culture was then diluted 1:200 and inoculated into microfluidic chambers, which were made from PDMS and coverslips and internal dimensions of 500 µm width, 90 µm height, and 7000 µm length. After inoculation, the channels were incubated without flow to give the cells time to attach to the glass surface for 1 h. The channels were then flushed for 45 s with a flow rate of 100 µl/min with M9 minimal medium (containing 0.5% glucose and supplemented with 2 mM MgSO4, 100 µM CaCl2, MEM vitamins, and 15 mM triethanolamine (pH 7.1)) to wash away the remaining LB medium and unattached cells. Afterwards, the flow rate was set to 0.1 µl/min until either the osmotic shock experiment or the dispersal experiment started, or until the fixation protocol was applied. Flow rates were controlled with a syringe pump (Harvard Apparatus).
Microfluidic channel fabrication
Microfluidic devices were designed and prepared following standard soft lithography techniques. Briefly, the device design was drawn using CAD Software and printed onto a film photomask (JD Photodata, UK). The design was then transferred to a SU-8 coated silicon wafer by exposure to 365 nm collimated UV light through the photomask, and processed to remove the unexposed photoresist. The resulting silicon master mould was vapour-silanized with chlorotrimethylsilane (Sigma-Aldrich) to facilitate PDMS removal from the mold. PDMS was prepared by mixing the elastomer with the curing agent in a 10:1 (w/w) ratio prior to pouring onto the silicon master mould, degassing, and curing at 65 °C for at least 8 hr. Afterwards the cured PDMS devices are cut and bonded to glass coverslips using an oxygen plasma (Diener Electronic).
Fixation of biofilm samples
For the preparation of fixed samples, biofilms were grown as described above. When the biofilms reached the desired size (at around 16-24 h after inoculation), the flow of M9 medium was stopped and the channels were slowly infused with 0.5 ml of M9 medium containing 0.2% of the nucleic acid stain SYTO 9 (Life Technologies). After 30 min of staining, the stain was flushed out with 0.5 ml of a fixation solution (4% formaldehyde in M9). After 30 min of fixation, the channel was flushed with 1 ml of phosphate-buffered saline (PBS). The fixed samples were either used immediately or stored at 4 °C until further use.
Osmotic shock experiment
Biofilms for the osmotic shock experiment were grown for 15 h after inoculation of the microfluidic channels, using the protocol described above. After identifying a biofilm colony within the channel, the syringe that was connected to the channel for the continuous infusion of M9 medium was exchanged for a syringe containing only distilled water. Images were taken every 800 ms and the flow of distilled water with a flowrate of 10 µl/min was started after 5 images were acquired. Imaging was stopped after 1 min.
Dispersal experiment
Biofilms for the dispersal experiment were grown for 15 h after inoculation, as described above. After identifying a biofilm colony in the channel for imaging, the syringe containing M9 medium that was connected to the growth channel was exchanged for a syringe containing M9 without glucose, which is a condition that is known to induce biofilm dispersal for sufficiently large biofilms [60]. The flow rate of new medium without the carbon source (glucose) was set to 1 µl/min and images were acquired every 3 min for 1 h.
Bleaching experiments and analysis
To compare the photobleaching of the spinning disk confocal microscope with the DC-APOM technique (using Bessel and Gaussian light sheets) for stained and fixed biofilms (Fig. 3), 3D image volumes of the same resolution and the same signal level were acquired using all imaging modalities. For this calculation, we define the signal level as the signal intensity value of the 99.9% percentile of the pixel intensity distribution. Confocal spinning disk image data was acquired with a Yokogawa CSU device (50 µm pinhole size), mounted on a Nikon Ti-E inverted microscope using a 100x/1.35 silicon oil objective (Olympus). Images were acquired using an Andor iXon 888 Ultra EMCCD camera with an exposure time of 90 ms. The EM-gain option of this camera was not used, to enable a comparison of the pixel intensity with the Andor Zyla 4.2 PLUS sCMOS camera used in the DC-APOM. To keep the signal level (defined above) constant across the different imaging modalities, different laser power levels were needed for the excitation. The laser power at the BFP was measured as µW for the spinning disk confocal. The image stacks consisting of 430 slices were acquired at an xy-resolution of 63.2 nm, and a z-spacing of 58.5 nm with the spinning disk confocal. Data for photobleaching of the DC-APOM technique was acquired for scanned light sheets based on a Bessel beam (dimensions: FHWMxy = 1.15 µm; FHWMz = 31.4 µm; µW) and a Gaussian beam (dimensions: FHWMxy = 1.27 µm; FHWMz = 25.7 µm; µW), with a virtual confocal slit exposure time of 0.48 ms and a slit width of 20 px (corresponding to a width of 130 µm on the camera sensor and 1.17 µm at the sample). The laser power at the BFP was measured as 13.1 µW for the Bessel beam and 18 µW for the Gaussian beam for the DC-APOM. The scanning step size along the y-direction was 63 nm and 430 slices were imaged.
To determine the photobleaching rate for both imaging techniques, we first subtracted the background signal for the image volume, followed by splitting the image volume into 10 sub-volumes of equal size. For these sub-volumes, the individual pixel values were summed up and normalized by the first acquired volume.
To validate the method of comparing photobleaching for axially and horizontally acquired images, the following control experiment was performed: Instead of imaging volumes of biofilms with the same volumetric resolution, only a single plane with a fixed field of view was imaged, for which the same light energy was deposited for all three imaging modalities, instead of keeping the signal-level constant. In order to measure the exact laser power per area for the spinning disk confocal microscope, the excitation region was limited by a square aperture which constrained the field of view to 49.2 µm x 49.2 µm. For all imaging modalities we kept the laser power in the back focal plane constant ( µW) and deposited the same energy into the sample by adjusting the exposure time. For the DC-APOM imaging, the deposited energy per image can be calculated by the following formula: . The number of pixel rows was set to 842 px along the light sheet scanning direction and to 780 px perpendicular to the scanning, which results in a field of view of 47.97 µm x 45.63 µm. With a slit exposure time of ms, the deposited energy per plane is µJ. In order to deposit the same energy per plane into the sample for the spinning disk confocal, the exposure time was set to ms. This results in a deposited energy of . The results of the control experiment (Supplement 1 (3.8MB, pdf) Fig. S4) show that under these conditions, the photobleaching is very similar across the three imaging modalities.
3D biofilm image reconstruction
Three-dimensional images of biofilms (Fig. 1(C) were reconstructed from image stacks using Matlab (Mathworks) and BiofilmQ [54] as described previously [52]. First, noise was removed by convolution of the raw data with an averaging kernel, followed by convolution with a 3D Laplacian-of-Gaussian (LoG) kernel of 0.76 µm cube length (corresponding to 13 px) for 3D edge detection. The zero values in the LoG representation represent those locations, where the cells’ fluorescence intensities drop approximately by 50% in comparison to the local intensity maxima and were used as cellular outlines. Second, after subsequent filling of these outlines, a 3D watershed algorithm was applied for cell de-clumping followed by 3D median filtering. Third, the obtained binary objects were further processed to correct segmentation errors as described previously [52]. Segmented biofilms were exported using the mVTK library [61] implemented in the BiofilmQ software tool [54] and rendered in ParaView 5.1.2 (Kitware) using OSPRay rendering with shadows.
Biofilm contrast analysis
The biofilm signal-to-background ratio (SBR) was calculated by sorting the intensity values of the image stack and dividing the thousandth-brightest value (99.9% percentile) by the background intensity (40% percentile). The variance of the Laplacian (LAPV score) is a second-derivative-based measure of the local image contrast and was calculated according to [62] using a local window size of 0.4 µm (7 px). LAPV values were normalized by the LAPV score obtained for the static light-sheet. A static light-sheet was achieved by operating the camera with the normal rolling shutter and a galvo scanning frequency set to ≥2x of the inverse of the camera exposure time.
Funding
Deutsche Forschungsgemeinschaft10.13039/501100001659 (DR 982/5-1, SFB987); Minna-James-Heineman-Stiftung; European Research Council10.13039/501100000781 (716734); Bundesministerium für Bildung und Forschung10.13039/501100002347 (TARGET-Biofilms); Human Frontier Science Program10.13039/501100000854 (CDA00084/2015-C); Max-Planck-Gesellschaft10.13039/501100004189.
Disclosures
The authors declare no conflicts of interests.
Supplemental document
See Supplement 1 (3.8MB, pdf) for supporting content.
References
- 1.Huisken J., Swoger J., Del Bene F., Wittbrodt J., Stelzer E. H. K., “Optical sectioning deep inside live embryos by selective plane illumination microscopy,” Science 305(5686), 1007–1009 (2004). 10.1126/science.1100035 [DOI] [PubMed] [Google Scholar]
- 2.Power R. M., Huisken J., “A guide to light-sheet fluorescence microscopy for multiscale imaging,” Nat. Methods 14(4), 360–373 (2017). 10.1038/nmeth.4224 [DOI] [PubMed] [Google Scholar]
- 3.Mcgorty R., Xie D., Huang B., “High-NA open-top selective-plane illumination microscopy for biological imaging,” Opt. Express 25(15), 17798 (2017). 10.1364/OE.25.017798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu Y., Shroff H., “Faster, sharper, and deeper: structured illumination microscopy for biological imaging,” Nat. Methods 15(12), 1011–1019 (2018). 10.1038/s41592-018-0211-z [DOI] [PubMed] [Google Scholar]
- 5.Royer L. A., Lemon W. C., Chhetri R. K., Keller P. J., “A practical guide to adaptive light-sheet microscopy,” Nat. Protoc. 13(11), 2462–2500 (2018). 10.1038/s41596-018-0043-4 [DOI] [PubMed] [Google Scholar]
- 6.Greger K., Swoger J., Stelzer E. H. K., “Basic building units and properties of a fluorescence single plane illumination microscope,” Rev. Sci. Instrum. 78(2), 023705 (2007). 10.1063/1.2428277 [DOI] [PubMed] [Google Scholar]
- 7.Reynaud E. G., Kržič U., Greger K., Stelzer E. H. K., “Light sheet-based fluorescence microscopy: more dimensions, more photons, and less photodamage,” HFSP J. 2(5), 266–275 (2008). 10.2976/1.2974980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao L., Shao L., Higgins C. D., Poulton J. S., Peifer M., Davidson M. W., Wu X., Goldstein B., Betzig E., “Noninvasive imaging beyond the diffraction limit of 3D dynamics in thickly fluorescent specimens,” Cell 151(6), 1370–1385 (2012). 10.1016/j.cell.2012.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim J., Lee H. K., Yu W., Ahmed S., “Light sheet fluorescence microscopy (LSFM): past, present and future,” Analyst 139(19), 4758–4768 (2014). 10.1039/C4AN00624K [DOI] [PubMed] [Google Scholar]
- 10.Strobl F., Schmitz A., Stelzer E. H. K., “Improving your four-dimensional image: traveling through a decade of light-sheet-based fluorescence microscopy research,” Nat. Protoc. 12(6), 1103–1109 (2017). 10.1038/nprot.2017.028 [DOI] [PubMed] [Google Scholar]
- 11.Olarte O. E., Andilla J., Gualda E. J., Loza-Alvarez P., “Light-sheet microscopy: a tutorial,” Adv. Opt. Photonics 10(1), 111 (2018). 10.1364/AOP.10.000111 [DOI] [Google Scholar]
- 12.McDole K., Guignard L., Amat F., Berger A., Malandain G., Royer L. A., Turaga S. C., Branson K., Keller P. J., “In toto imaging and reconstruction of post-implantation mouse development at the single-cell level,” Cell 175(3), 859–876.e33 (2018). 10.1016/j.cell.2018.09.031 [DOI] [PubMed] [Google Scholar]
- 13.Wu Y., Wawrzusin P., Senseney J., Fischer R. S., Christensen R., Santella A., York A. G., Winter P. W., Waterman C. M., Bao Z., Colón-Ramos D. A., McAuliffe M., Shroff H., “Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy,” Nat. Biotechnol. 31(11), 1032–1038 (2013). 10.1038/nbt.2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bishop K. W., Glaser A. K., Liu J. T. C., “Performance tradeoffs for single- and dual-objective open-top light-sheet microscope designs: a simulation-based analysis,” Biomed. Opt. Express 11(8), 4627 (2020). 10.1364/BOE.397052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barner L. A., Glaser A. K., Huang H., True L. D., Liu J. T. C., “Multi-resolution open-top light-sheet microscopy to enable efficient 3D pathology workflows,” Biomed. Opt. Express 11(11), 6605 (2020). 10.1364/BOE.408684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sala F., Castriotta M., Paiè P., Farina A., D’Annunzio S., Zippo A., Osellame R., Bragheri F., Bassi A., “High-throughput 3D imaging of single cells with light-sheet fluorescence microscopy on chip,” Biomed. Opt. Express 11(8), 4397 (2020). 10.1364/BOE.393892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye F., Avants B. W., Veeraraghavan A., Robinson J. T., “Integrated light-sheet illumination using metallic slit microlenses,” Opt. Express 26(21), 27326 (2018). 10.1364/OE.26.027326 [DOI] [PubMed] [Google Scholar]
- 18.Chen B.-C., Legant W. R., Wang K., Shao L., Milkie D. E., Davidson M. W., Janetopoulos C., Wu X. S., Hammer J. A., Liu Z., English B. P., Mimori-Kiyosue Y., Romero D. P., Ritter A. T., Lippincott-Schwartz J., Fritz-Laylin L., Mullins R. D., Mitchell D. M., Bembenek J. N., Reymann A.-C., Böhme R., Grill S. W., Wang J. T., Seydoux G., Tulu U. S., Kiehart D. P., Betzig E., “Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution,” Science 346(6208), 1257998 (2014). 10.1126/science.1257998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fadero T. C., Gerbich T. M., Rana K., Suzuki A., DiSalvo M., Schaefer K. N., Heppert J. K., Boothby T. C., Goldstein B., Peifer M., Allbritton N. L., Gladfelter A. S., Maddox A. S., Maddox P. S., “LITE microscopy: Tilted light-sheet excitation of model organisms offers high resolution and low photobleaching,” J. Cell Biol. 217(5), 1869–1882 (2018). 10.1083/jcb.201710087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunsby C., “Optically sectioned imaging by oblique plane microscopy,” Opt. Express 16(25), 20306 (2008). 10.1364/OE.16.020306 [DOI] [PubMed] [Google Scholar]
- 21.Bouchard M. B., Voleti V., Mendes C. S., Lacefield C., Grueber W. B., Mann R. S., Bruno R. M., Hillman E. M. C., “Swept confocally-aligned planar excitation (SCAPE) microscopy for high-speed volumetric imaging of behaving organisms-SI,” Nat. Photonics 9(2), 113–119 (2015). 10.1038/nphoton.2014.323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar S., Wilding D., Sikkel M. B., Lyon A. R., MacLeod K. T., Dunsby C., “High-speed 2D and 3D fluorescence microscopy of cardiac myocytes,” Opt. Express 19(15), 13839 (2011). 10.1364/OE.19.013839 [DOI] [PubMed] [Google Scholar]
- 23.Galland R., Grenci G., Aravind A., Viasnoff V., Studer V., Sibarita J.-B., “3D high- and super-resolution imaging using single-objective SPIM,” Nat. Methods 12(7), 641–644 (2015). 10.1038/nmeth.3402 [DOI] [PubMed] [Google Scholar]
- 24.Meddens M. B. M., Liu S., Finnegan P. S., Edwards T. L., James C. D., Lidke K. A., “Single objective light-sheet microscopy for high-speed whole-cell 3D super-resolution,” Biomed. Opt. Express 7(6), 2219 (2016). 10.1364/BOE.7.002219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zagato E., Brans T., Verstuyft S., van Thourhout D., Missinne J., van Steenberge G., Demeester J., De Smedt S., Remaut K., Neyts K., Braeckmans K., “Microfabricated devices for single objective single plane illumination microscopy (SoSPIM),” Opt. Express 25(3), 1732 (2017). 10.1364/OE.25.001732 [DOI] [PubMed] [Google Scholar]
- 26.Miura T., Mikami H., Isozaki A., Ito T., Ozeki Y., Goda K., “On-chip light-sheet fluorescence imaging flow cytometry at a high flow speed of 1 m/s,” Biomed. Opt. Express 9(7), 3424 (2018). 10.1364/BOE.9.003424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mangeol P., Peterman E. J. G., “High-resolution real-time dual-view imaging with multiple point of view microscopy,” Biomed. Opt. Express 7(9), 3631 (2016). 10.1364/BOE.7.003631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engelbrecht C. J., Voigt F., Helmchen F., “Miniaturized selective plane illumination microscopy for high-contrast in vivo fluorescence imaging,” Opt. Lett. 35(9), 1413 (2010). 10.1364/OL.35.001413 [DOI] [PubMed] [Google Scholar]
- 29.Kashekodi A. B., Meinert T., Michiels R., Rohrbach A., “Miniature scanning light-sheet illumination implemented in a conventional microscope,” Biomed. Opt. Express 9(9), 4263 (2018). 10.1364/BOE.9.004263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ponjavic A., Ye Y., Laue E., Lee S. F., Klenerman D., “Sensitive light-sheet microscopy in multiwell plates using an AFM cantilever,” Biomed. Opt. Express 9(12), 5863 (2018). 10.1364/BOE.9.005863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cutrale F., Gratton E., “Inclined selective plane illumination microscopy adaptor for conventional microscopes,” Microsc. Res. Tech. 75(11), 1461–1466 (2012). 10.1002/jemt.22089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J., Wojcik M., Wang Y., Moon S., Zin E. A., Marnani N., Newman Z. L., Flannery J. G., Xu K., Zhang X., “Oblique-plane single-molecule localization microscopy for tissues and small intact animals,” Nat. Methods 16(9), 853–857 (2019). 10.1038/s41592-019-0510-z [DOI] [PubMed] [Google Scholar]
- 33.Hoffmann M., Judkewitz B., “Diffractive oblique plane microscopy,” Optica 6(9), 1166 (2019). 10.1364/OPTICA.6.001166 [DOI] [Google Scholar]
- 34.Botcherby E. J., Juskaitis R., Booth M. J., Wilson T., “Aberration-free optical refocusing in high numerical aperture microscopy,” Opt. Lett. 32(14), 2007 (2007). 10.1364/OL.32.002007 [DOI] [PubMed] [Google Scholar]
- 35.Botcherby E. J., Juškaitis R., Booth M. J., Wilson T., “An optical technique for remote focusing in microscopy,” Opt. Commun. 281(4), 880–887 (2008). 10.1016/j.optcom.2007.10.007 [DOI] [Google Scholar]
- 36.Botcherby E. J., Booth M. J., Juškaitis R., Wilson T., “Real-time slit scanning microscopy in the meridional plane,” Opt. Lett. 34(10), 1504 (2009). 10.1364/OL.34.001504 [DOI] [PubMed] [Google Scholar]
- 37.Anselmi F., Ventalon C., Bègue A., Ogden D., Emiliani V., “Three-dimensional imaging and photostimulation by remote-focusing and holographic light patterning,” Proc. Natl. Acad. Sci. U. S. A. 108(49), 19504–19509 (2011). 10.1073/pnas.1109111108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voleti V., Patel K. B., Li W., Perez Campos C., Bharadwaj S., Yu H., Ford C., Casper M. J., Yan R. W., Liang W., Wen C., Kimura K. D., Targoff K. L., Hillman E. M. C., “Real-time volumetric microscopy of in vivo dynamics and large-scale samples with SCAPE 2.0,” Nat. Methods 16(10), 1054–1062 (2019). 10.1038/s41592-019-0579-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar M., Kozorovitskiy Y., “Tilt (in)variant lateral scan in oblique plane microscopy: a geometrical optics approach,” Biomed. Opt. Express 11(6), 3346–3359 (2020). 10.1364/BOE.389654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar M., Kozorovitskiy Y., “Tilt-invariant scanned oblique plane illumination microscopy for large-scale volumetric imaging,” Opt. Lett. 44(7), 1706–1709 (2019). 10.1364/OL.44.001706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang B., Chen X., Wang Y., Feng S., Pessino V., Stuurman N., Cho N. H., Cheng K. W., Lord S. J., Xu L., Xie D., Mullins R. D., Leonetti M. D., Huang B., “Epi-illumination SPIM for volumetric imaging with high spatial-temporal resolution,” Nat. Methods 16(6), 501–504 (2019). 10.1038/s41592-019-0401-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sapoznik E., Chang B. J., Huh J., Ju R. J., Azarova E. V., Pohlkamp T., Welf E. S., Broadbent D., Carisey A. F., Stehbens S. J., Lee K. M., Marín A., Hanker A. B., Schmidt J. C., Arteaga C. L., Yang B., Kobayashi Y., Tata P. R., Kruithoff R., Doubrovinski K., Shepherd D. P., Millett-Sikking A., York A. G., Dean K. M., Fiolka R. P., “A versatile oblique plane microscope for large-scale and high-resolution imaging of subcellular dynamics,” eLife 9, 1–39 (2020). 10.7554/eLife.57681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang B., Millett-Sikking A., Lange M., Solak A. C., Kobayashi H., York A., Royer L. A., “High-resolution, large field-of-view, and multi-view single objective light-sheet microscopy,” bioRxiv 2020.09.22.309229 (2020).
- 44.Li T., Ota S., Kim J., Wong Z. J., Wang Y., Yin X., Zhang X., “Axial plane optical microscopy,” Sci. Rep. 4(1), 7253 (2015). 10.1038/srep07253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J., Li T., Wang Y., Zhang X., “Vectorial point spread function and optical transfer function in oblique plane imaging,” A Proc. R. Soc. Lond. A Math. Phys. Sci 4, 1354–1360 (2014). [DOI] [PubMed] [Google Scholar]
- 46.An S., Ziegler K. F., Zhang P., Wang Y., Kwok T., Xu F., Bi C., Matosevic S., Yin P., Li T., Huang F., “Axial plane single-molecule super-resolution microscopy of whole cells,” Biomed. Opt. Express 11(1), 461 (2020). 10.1364/BOE.377890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fahrbach F. O., Simon P., Rohrbach A., “Microscopy with self-reconstructing beams,” Nat. Photonics 4(11), 780–785 (2010). 10.1038/nphoton.2010.204 [DOI] [Google Scholar]
- 48.Planchon T. A., Gao L., Milkie D. E., Davidson M. W., Galbraith J. A., Galbraith C. G., Betzig E., “Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination,” Nat. Methods 8(5), 417–423 (2011). 10.1038/nmeth.1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baumgart E., Kubitscheck U., “Scanned light sheet microscopy with confocal slit detection,” Opt. Express 20(19), 21805 (2012). 10.1364/OE.20.021805 [DOI] [PubMed] [Google Scholar]
- 50.Silvestri L., Bria A., Sacconi L., Iannello G., Pavone F. S., “Confocal light sheet microscopy: micron-scale neuroanatomy of the entire mouse brain,” Opt. Express 20(18), 20582 (2012). 10.1364/OE.20.020582 [DOI] [PubMed] [Google Scholar]
- 51.Flemming H.-C., Wuertz S., “Bacteria and archaea on Earth and their abundance in biofilms,” Nat. Rev. Microbiol. 17(4), 247–260 (2019). 10.1038/s41579-019-0158-9 [DOI] [PubMed] [Google Scholar]
- 52.Hartmann R., Singh P. K., Pearce P., Mok R., Song B., Díaz-Pascual F., Dunkel J., Drescher K., “Emergence of three-dimensional order and structure in growing biofilms,” Nat. Phys. 15(3), 251–256 (2019). 10.1038/s41567-018-0356-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Díaz-Pascual F., Hartmann R., Lempp M., Vidakovic L., Song B., Jeckel H., Thormann K. M., Yildiz F. H., Dunkel J., Link H., Nadell C. D., Drescher K., “Breakdown of Vibrio cholerae biofilm architecture induced by antibiotics disrupts community barrier function,” Nat. Microbiol. 4(12), 2136–2145 (2019). 10.1038/s41564-019-0579-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hartmann R., Jeckel H., Jelli E., Singh P. K., Vaidya S., Bayer M., Rode D. K. H., Vidakovic L., Díaz-Pascual F., Fong J. C. N., Dragoš A., Lamprecht O., Thöming J. G., Netter N., Häussler S., Nadell C. D., Sourjik V., Kovács ÁT, Yildiz F. H., Drescher K., “Quantitative image analysis of microbial communities with BiofilmQ,” Nat. Microbiol. 6(2), 151–156 (2021). 10.1038/s41564-020-00817-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeckel H., Drescher K., “Advances and opportunities in image analysis of bacterial cells and communities,” FEMS Microbiol. Rev. (2020). [DOI] [PMC free article] [PubMed]
- 56.Zhang M., Zhang J., Wang Y., Wang J., Achimovich A. M., Acton S. T., Gahlmann A., “Non-invasive single-cell morphometry in living bacterial biofilms,” Nat. Commun. 11(1), 6151 (2020). 10.1038/s41467-020-19866-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin B., Fei C., Bridges A. A., Mashruwala A. A., Stone H. A., Wingreen N. S., Bassler B. L., “Cell position fates and collective fountain flow in bacterial biofilms revealed by light-sheet microscopy,” Science 369(6499), 71–77 (2020). 10.1126/science.abb8501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fahrbach F. O., Gurchenkov V., Alessandri K., Nassoy P., Rohrbach A., “Light-sheet microscopy in thick media using scanned Bessel beams and two-photon fluorescence excitation,” Opt. Express 21(11), 13824 (2013). 10.1364/OE.21.013824 [DOI] [PubMed] [Google Scholar]
- 59.Zhang P., Phipps M. E., Goodwin P. M., Werner J. H., “Confocal line scanning of a Bessel beam for fast 3D Imaging,” Opt. Lett. 39(12), 3682 (2014). 10.1364/OL.39.003682 [DOI] [PubMed] [Google Scholar]
- 60.Singh P. K., Bartalomej S., Hartmann R., Jeckel H., Vidakovic L., Nadell C. D., Drescher K., “Vibrio cholerae combines individual and collective sensing to trigger biofilm dispersal,” Curr. Biol. 27(21), 3359–3366.e7 (2017). 10.1016/j.cub.2017.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flandin G., “mVTK: a VTK library for MATLAB,” https://www.artefact.tk/software/matlab/mvtk/.
- 62.Pech-Pacheco J. L., Cristobal G., Chamorro-Martinez J., Fernandez-Valdivia J., “Diatom autofocusing in brightfield microscopy: a comparative study,” in Proceedings 15th International Conference on Pattern Recognition. ICPR-2000 (IEEE Comput. Soc, 2000), Vol. 3, pp. 314–317. [Google Scholar]