

SUMMARY
Host genetic landscapes can shape microbiome assembly in the animal gut by contributing to the establishment of distinct physiological environments. However, the genetic determinants contributing to the stability and variation of these microbiome types remain largely undefined. Here, we use the free-living nematode Caenorhabditis elegans to identify natural genetic variation among wild strains of C. elegans strains that drives assembly of distinct microbiomes. To achieve this, we first established a diverse model microbiome that represents the strain-level phylogenetic diversity naturally encountered by C. elegans in the wild. Using this community, we show that C. elegans utilizes immune, xenobiotic and metabolic signaling pathways to favor the assembly of different microbiome types. Variations in these pathways were associated with enrichment for specific commensals, including the Alphaproteobacteria Ochrobactrum. Using RNAi and mutant strains, we showed that host selection for Ochrobactrum is mediated specifically by host insulin signaling pathways. Ochrobactrum recruitment is blunted in the absence of DAF-2/IGFR and modulated by the competitive action of insulin signaling transcription factors DAF-16/FOXO and PQM-1/SALL2. Further, the ability of C. elegans to enrich for Ochrobactrum as adults is correlated with faster animal growth rates and larger body size at the end of development. These results highlight a new role for the highly conserved insulin signaling pathways in the regulation of gut microbiome composition in C. elegans.
Keywords: host-microbe interactions, genetics, gnotobiotic models, insulin signaling, model microbiome
INTRODUCTION
Across kingdoms, shifts in microbiome composition accompany and contribute to host development, health, and physiology 1–3. Along with diet and lifestyle, host genetics can regulate the size and composition of the microbiome 4–7. This is apparent in human diseases with altered microbiome composition such as inflammatory bowel disease and obesity 6. While predicted host polymorphic loci for the development of these diseases have been identified 8, the directionality of impact or molecular mediators remain ill-defined for most cases. Thus, there is a great need to identify causal genetic host determinants that contribute to the stability and variation of microbiome types in order to effectively develop microbiome interventions as potential therapies.
To address this problem, we used wild strains of the nematode Caenorhabditis elegans and established a new diverse 63-member model microbiome, termed ‘BIGbiome’, that better represents the phylogenetic and functional diversity of the C. elegans wild microbiome. This system proves several key advantages. C. elegans itself has a transparent body plan and robust genetic toolbox, and its short lifespan and amenability to high-throughput methods increase experimental throughput 9,10. C. elegans also shares many conserved pathways with higher organisms that could regulate microbiome recruitment, including metabolic, stress and innate immune pathways 11–13. Yet, it has been difficult to determine which of these pathways may contribute to microbiome community outcomes because much of our current understanding comes from decades of studies of the C. elegans lab strain N2-Bristol in association with Escherichia coli or human pathogens 14. By contrast, wild C. elegans strains encounter a large variety of microbes and selectively recruit only some of these from the environment to form its gut microbiome 15–17. Thus, probing host genetics with a representative natural microbial community may more completely reveal causal host determinants that contribute to microbiome outcomes.
We make progress towards this goal here. We present the most comprehensive examination to date of the causal influence of C. elegans natural genetic variation on the establishment of its gut microbiome. To achieve this, we utilized our newly developed model microbiome to test natural variation in its acquisition by a panel of nearly 40 ‘germ-free’ wild strains of C. elegans 18. We found that these strains selected for and acquired one of only three distinct types of gut microbiomes: (i) one dominated by Ochrobactrum, (ii) another dominated by Bacteroidetes, and (iii) one similar in composition to the bacterial lawn. Selection of these microbes was robust and consistent within the host strain, suggestive of a deterministic process driven by host genetic variation. To probe this variation, we conducted phenotypic, genetic and transcriptional profiling of wild strains representative of each microbiome type. This analysis revealed fundamental differences in host immune, stress and metabolic responses specific for the acquisition of each microbiome. Genetic loss of function studies further reveal a key role for insulin signaling pathways in microbiome regulation. In particular, we identify a previously uncharacterized role for insulin signaling in wild strains of C. elegans in the promotion of selective acquisition and maintenance of the gut microbiome via a DAF-2/PQM-1 pathway. Finally, we find that the ability of particular host genetic backgrounds to acquire a given microbiome directly influences host health fate. Higher levels of insulin signaling and broad activation of immune pathways promoted intestinal acquisition of otherwise rare Ochrobactrum from the bacterial lawns, and this was associated with faster growth rates. In contrast, low levels of insulin signaling activated non-selective stress responses and resulted in gut microbiomes that resemble the lawn and were associated with lower rates of host growth. Together, these studies both establish wild C. elegans and their natural microbes as a robust microbiome system and identify novel roles for host insulin signaling in regulation of gut microbiome composition.
RESULTS
Establishment of a diverse and representative gut microbiome of C. elegans.
Effective identification of host genes that drive assembly of distinct microbiomes requires a diverse model microbial community that closely resembles the variation a host may encounter in the wild. We reasoned that such a community should be: (i) reflective of the major microbial taxa found in natural microbiomes of wild C. elegans; (ii) highly functionally redundant; and (iii) easy to use and create in the lab. To achieve this, we expanded on previous analyses of the core microbiome of wild C. elegans populations 19 and selected bacterial strains from our collections (>500 strains) that matched the 14 core families of the C. elegans microbiome— Enterobacteriaceae, Pseudomonadaceae, Xanthomonadaceae, Sphingomonadaceae, Sphingobacteriaceae, Flavobacteriaceae, Weeksellaceae, Acetobacteraceae, Moraxellaceae, Oxalobacteraceae, Comamonadaceae, Rhodobacteraceae, Microbacteriaceae, and Actinomycetales. The resulting community, termed BIGbiome001 (referred to as ‘BIGbiome’ hereafter), comprises 63 strains from 23 genera (10 of 14 core families). Together, it represents 50–80% of the biomass in natural microbiomes of wild C. elegans [Table 1; see strain origins in Data S1AA; Figure S1A–B]. We also sought to model the functional redundancy observed in natural communities by including several taxonomically related bacterial strains from distinct wild C. elegans strains or habitats (range of 2–22 strains). BIGbiome complements simplified microbiomes like the recently developed 12-member CeMbio community 20 as it reflects the extensive strain level microbial diversity found in C. elegans natural microbiomes while still remaining experimentally tractable.
Table 1.
Summary of microbial strains in the BIGb iome model microbiome
| Division | Family | Genera | Strains |
|---|---|---|---|
| Proteobacteria | Brucellaceae | Ochrobactrum | BH3 |
| Acetobacteraceae | Gluconobacter | BIGb0611 | |
| Rhizobiaceae | Rhizobium | JUb45 | |
| Comamonadaceae | Delftia | JUb8 | |
| Limnohabitans | JUb58, BIGb0172 | ||
| Ramlibacter | BIGb0124 | ||
| Moraxellaceae | Acinetobacter | JUb89, BIGb0102, BIGb0196 | |
| Pseudomonadaceae | Pseudomonas | BIGb0272, BIGb0273, BIGb0404, BIGb0408, BIGb0470, BIGb0473, BIGb0477, BIGb0525, JUb28, JUb52, JUb85, JUb96 | |
| Xanthomonadaceae | Stenotrophomonas | JUb19, JUb23, BIGb0145, BIGb0219 | |
| Enterobacteriaceae | Raoultella | JUb54, BIGb0138. BIGb0399 | |
| Erwinia | BIGb0193, BIGb0393, BIGb0435 | ||
| Enterobacter | JUb30, JUb66, JUb101, BIGb0359, BIGb0383 | ||
| Citrobacter | BIGb0149, BIGb0188, BIGb0211, BIGb0267 | ||
| Buttiauxella | BIGb0552 | ||
| Yersinia | JUb53, BIGb0156, BIGb0236 | ||
| Providencia | JUb39, JUb102, BIGb0506 | ||
| Bacteroidetes | Flavobacteriaceae | Chryseobacterium | JUb44, BIGb0186, BIGb0215 |
| Myroides | BIGb0243 | ||
| Sphingobacteriaceae | Sphingobacterium | JUb20, JUb56, JUb78 | |
| Actinobacteria | Microbacteriaceae | Curtobacterium | JUb34, JUb65 |
| Leucobacter | JUb18, BIGb0106, BIGb0117 | ||
| Micrococcaceae | Arthrobacter | JUb115 | |
| Nocardiaceae | Rhodococcus | JUb83 | |
| Firmicutes | Streptococcaceae | Lactococcus | BIGb0210 |
Development of distinct gut microbiome types in adult C. elegans
We next tested the robustness of the BIGbiome community for microbiome studies by profiling how it is acquired in the lab strain (N2) and a wild C. elegans strain (JU1218). To achieve this, we established a phenotyping pipeline for microbiome-based measures that include gut colonization density and composition [Figure 1A]. These methods allow for high-throughput determination of both the levels of overall bacterial colonization and proportions of bacteria that colonize the C. elegans gut. In this approach, strains are first made ‘germ-free’ by bleaching eggs followed by synchronization at the L1 stage. L1 animals are then exposed to the BIGbiome community (proportional mixture of each strain) on agar plates and monitored over their development and into adulthood. Using this approach, we found that appreciable C. elegans gut colonization could not be observed until day 1 of adulthood (48 hours post-L1). Differences between N2 and JU1218 host strains were observed by day 3 of adulthood and appeared to stabilize at that time [Figure S1C–D]. These results are consistent with previous studies of bacterial colonization of the C. elegans N2 intestine by E. coli 21. For these reasons, we chose days 1 and 3 of adulthood for further studies.
Figure 1. Natural genetic variation in C. elegans drives distinct gut microbiome types.
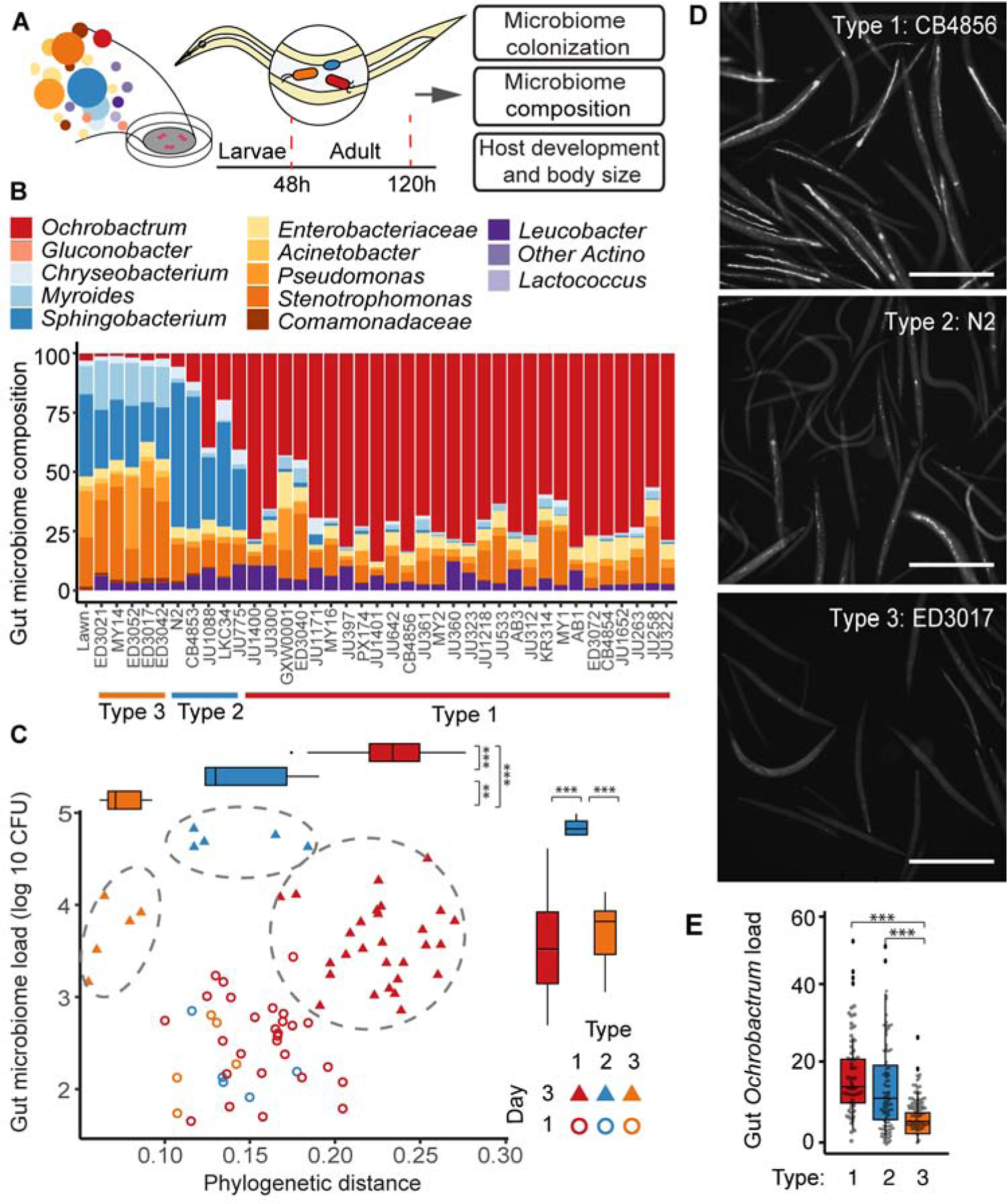
Schematic diagram illustrating the pipeline to measure gut microbiome and host phenotypes of 38 C. elegans strains grown on microbiome mixture. Worm samples were collected at 48 h (day 1 adults) and 120 h (day 3 adults) after exposing synchronized L1 populations to BIGbiome. B. Gut microbiome composition of the 38 C. elegans strains in day 3 adulthood. Relative microbiome abundance was presented here as the mean of biological duplicates for each strain. C. The 38 strains were clustered into three distinct microbiome types based on their gut microbiome load per animal (y-axis) and phylogenetic distances to BIGbiome lawn (x-axis). Solid symbols showed samples collected in day 3 adulthood and open symbols showed samples in day 1 adulthood. Inset: Box-whisker plot of microbiome load per animal in three microbiome types. Type 2 strains (n=10) carried significantly higher gut microbiome load than Type 1 (n=56) and Type 3 strains (n=10). Box-whisker plot of phylogenetic distances to BIGbiome for the three microbiome types. Phylogenetic distances between each strain and BIGbiome lawn were calculated by weighted UniFrac. Type 1 strains (n=56) showed further distance to the BIGbiome lawn than Type 2 (n=10) and Type 3 strains (n=10). n represents the number of independent worm populations. See also Figure S3 and Data S1A. D. Representative images of C. elegans strains in day 3 adulthood from each of the three microbiome types grown on BIGbiome with an isogenic GFP expressing Ochrobactrum strain. Bar = 500 µm. E. Box-whisker plot of GFP intensity quantified from fluorescent images of C. elegans strains grown on BIGbiome (GFP-Ochrobactrum) showed higher GFP-Ochrobactrum colonization in Type 1 (n=55) and 2 (n=94) strains than Type 3 strains (n= 73). n, individual animals quantified by microscopic images; P-values were generated from one-way ANOVA, followed by Tukey posthoc test with 95% confidence level and adjusted for multiple comparisons (***p<0.001, **p<0.01). See also Data S1B. See also Table S1, plus Figures S2 and S3.
To assess the impact of host genetic variation on microbiome selection, we next used a genetically tractable but diverse host community. We selected 38 well characterized, fully genome sequenced and genetically distinct C. elegans strains 18 [Table S1]. Together with the lab strain (N2), these wild strains were first made ‘germ-free’ by bleaching eggs, and synchronized L1 animals were exposed to the BIGbiome community on agar plates. Animals from each strain were collected in bulk as adults at early (day 1) and later (day 3) stages of microbiome establishment and then assayed for differences in microbiome composition and gut colonization density. All of the worm strains exhibited both low levels of colonization and a comparable, lawn-like composition of their gut microbiomes at day 1 [Figure S2]. By day 3 of adulthood, however, the gut microbiomes became largely distinct from the surrounding bacterial lawn, and hosts exhibited up to a 30-fold range in levels of colonization [Figure 1, Figure S2].
We next asked whether particular microbiome representations were favored more than others. We performed weighted UniFrac-based clustering of the animals by microbiome types on day 3 of adulthood and found that gut microbiome composition robustly separated into three microbiome types. Clustering was driven by dominant microbial taxa, and we termed the host clusters as Type 1, 2, or 3 [Figure 1A–B, Figure S4A–B]. The Type 1 hosts contained the largest group of C. elegans strains, harboring 28 strains. Notably, these strains were dominated by Ochrobactrum pituitosum BH3 [>40% relative abundance; Figure 1B], a microbe previously identified as a common beneficial member of the C. elegans microbiome in the wild 22–24. The microbiomes of Type 2 strains (lab strain N2 and four wild strains) were dominated by several Bacteroidetes taxa (e.g., Myroides, Chryseobacterium and Sphingobacterium). These animals displayed reduced levels of Ochrobactrum in the gut [10–40% relative abundance; Figure 1B] and higher levels of gut colonization overall [2 to 30-fold higher than Type 1 or Type 3 strains; Figure 1C, Figure S2C]. Type 3 animals (five wild worm strains) were nearly devoid of gut Ochrobactrum and instead were dominated by high levels of Bacteroidetes, Pseudomonas and Stenotrophomonas [Figure 1B]. Overall, the microbiome of Type 3 strains resembled that of the bacterial lawn [Figure 1C, Figure S4]. Analyses of overall gut microbiome alpha-diversity within samples indicates that the dominance of Ochrobactrum in the Type 1 strains had a tempering impact on microbiome diversity [Faith’s phylogenetic diversity of 6.3±0.9 in Type 1 vs. 8.9±0.7 in Type 3; Figure S4C]. Enrichment of otherwise rare microbes like Ochrobactrum from the lawn in the gut microbiome also increased beta-diversity between samples. The highest enrichment differential between the host microbiome composition relative to the lawn was observed for Type 1 animals, with more moderate enrichment observed in Types 2 and 3 [Figure 1C, Figure S4D].
We next capitalized on the inherent transparency of C. elegans to assess microbial enrichment on a single animal basis in order to examine individual variation within a given host strain. To accomplish this, we created BIGbiome mixtures where Ochrobactrum BH3 was replaced by an isogenic GFP-expressing strain. Both microscopy- and large particle flow cytometry (Biosorter)-based analyses supported our finding that Ochrobactrum enrichment was greater in individuals from Type 1 strains (CB4856), particularly when compared to Type 3 animals (ED3017) that have limited Ochrobactrum colonization [P<0.0001; Figure 1DE]. Type 2 animals (N2 or LKC34) exhibited a broader distribution of Ochrobactrum levels on a per animal basis [Figure 1E]. This may be due to an inherent stochasticity in microbial levels and composition during the colonization process, as has been shown for the lab strain of C. elegans (N2) under certain conditions 25. Together, our results highlight three robust modes of microbiome regulation by host strains that vary in their selectivity for the microbes that colonize and their relative levels within the gut.
C. elegans natural genetic variation is associated with adult microbiome composition
Through our analysis of these microbiome communities, we found that the majority of the strains within the BIGbiome community colonized the guts of at least two independent worm strains [91.6%, 55 strains]. For example, Enterobacteriaceae exhibit significant genomic plasticity and are common in wild C. elegans microbiomes 19,26. Though resolution of this family in our samples is limited due to high identity of small subunit (SSU) rRNA genes, Enterobacteriaceae were consistent colonizers as a group (5–10% relative abundance). Other consistent colonizers included Pseudomonas, Stenotrophomonas, and Comamonas; the rarer Leucobacter was also enriched 30–60 fold in the worm gut relative to the bacterial lawn [Data S1AB]. Meanwhile, we also observed those that exhibit more strain-specific or stochastic colonization of the C. elegans gut [Figure 2A–B; Data S1AB]. The assemblages and proportions of microbes observed in each worm strain were unique.
Figure 2. Microbial taxa are associated with natural genetic variation in C. elegans gut microbiome types.

A. Box-whisker plot of enrichment factors for microbial taxa for 38 C. elegans strains on day 3 adulthood colored by microbiome types. Enrichment factors for each microbial taxa were generated by log 2 transformation of fold changes of relative abundance in worm samples to BIGbiome lawn. B. Bar plot of commonality for each microbial taxa is calculated as the percentage of worm strains that was colonized by the corresponding microbial taxon at a minimum threshold of 0.01% in relative abundance. See also Data S1AB. C. GWAS analyses identify genetic loci that are associated with gut microbiome abundance. GWAS plot for traits of absolute abundance of specific microbiome members. Points represent significance and genome region and are colored by microbe. Dashed lines indicate genomic region enriched for microbe-specific trait and are similarly colored by microbe. D. GWAS plot for traits of relative abundance of specific microbiome members. Points represent significance and genome region and are colored by microbe. Dashed lines indicate genomic region enriched for microbe-specific trait and are similarly colored by microbe. See also Table S2 for a full list of genomic positions and associated microbial taxa.
To explore the potential for specific natural variation in host genetics in driving the selection of particular microbiome communities, we used the extensive genomics resources available through the Million Mutation Project for our wild C. elegans panel [>3.8M single nucleotide variants, ~65,000 missense mutations versus N2 reference genome 18. We performed GWAS analyses (see Methods) to identify regions of the genome associated with taxa abundance, colonization level, alpha diversity and beta diversity as trait values per strain. We identified several regions that were associated with taxa abundance of Chryseobacterium, Enterobacteriaceae, Gluconobacter, Acinetobacter, Curtobacterium and Leucobacter across host strains [17–56% variance explained per taxa (relative and/or absolute abundance); 1308 total genes in 9 loci; Figure 2C–D; see full list in Table S2]. As a whole, nine loci were enriched for genes with previously unknown functions [419 genes; Q=0.0039; WormCat tool 27], suggesting that microbiome studies may help ascribe phenotypes for these genes and the 40% of the genome that remains without ascribed functions no doubt due to limited exposure its microbes 22. Notably, the most significant overlap between the loci was observed for genes that are upregulated in insulin receptor (daf-2/IGFR) mutants [316 genes; Q=1.3e-32 to dataset 28; WormExp tool 29]. Together, these analyses indicate that natural genetic variation may drive microbiome compositional differences.
C. elegans growth rates and body size correlate with adult microbiome composition.
We next examined representative C. elegans strains from each microbiome type for changes in growth rates or body sizes after development. Each of the strains were grown on agar plates containing either BIGbiome or E. coli OP50 lawns from L1 until adulthood (46–58hrs). Notably, all strains tested exhibited faster growth rates on the BIGbiome community compared to those grown on E. coli OP50 alone [Figure 3A–B]. The extent of the growth promotion did differ by microbiome type, however. Type 1 strains (JU1400) exhibited 75% faster growth versus 65% and 40% for Type 2 and 3 strains, respectively [Figure 3A–B]. Type 3 (ED3017) animals were also significantly smaller than the other microbiome types after 48hrs of development, although these differences did normalize by day 3 of adulthood [Figure 3C, Figure S4A]. Both faster developmental growth rates and/or larger body sizes at the L4 stage correlated with higher gut colonization of Ochrobactrum [Pearson of 0.74 and 0.49, respectively, P<0.002; Figure 3D–E] and lower Enterobacter [~3% relative abundance; Pearson of 0.45 with body size only, P<0.005] and Leucobacter colonization [~5% relative abundance; Pearson = 0.49 with growth rate only, P<0.005; Table S3]. Conversely, slower growth rates and smaller body size were associated with more permissive colonization by nine other genera: Bacteroidetes (Chryseobacterium and Myroides), Betaproteobacteria (Limnohabitans, Ramlibacter, and Delftia), Gammaproteobacteria (Acinetobacter and Stenotrophomonas), and Actinobacteria (Arthrobacter and Curtobacterium) [P<0.05; Table S3]. No significant correlations were observed between the overall gut microbiome load and either growth rates or body size [Figure S4B–C]. These results indicate that the microbiome can influence host growth and development and may drive acquisition of a selected microbial community in adulthood.
Figure 3. C. elegans developmental growth rates and body size during development correlate with adult microbiome.

A. Developmental growth rates of representative strains from each microbiome type [Type 1 (JU1400 and CB4856), Type 2 (N2 and LKC34) and Type 3 (ED3017)] grown on BIGbiome and E. coli OP50. Percentage of adults are represented as mean ± SD with 4 replicates for each condition; representative of 3 independent experiments. See also Data S1F. B. Box-whisker plots of percent adults at 52 h post L1 stage (from A). Number of individual animals: BIGbiome (JU1400: n=145, CB4856: n=141, N2: n=132, LKC34: n=176, ED3017: n=157); E. coli OP50 (JU1400: n=173, CB4856: n=166, N2: n=142, LKC34: n=142, ED3017: n=153). C. Box-whisker plot of C. elegans body size by microbiome types at 48 h and 120 h post L1 stage. Type 1 strains (n=1076) had longer body size than Type 2 (n=168) and Type 3 strains (n=158) at 48 h. No significant difference among Type 1 (n=501), 2 (n=68), and 3 (n=56) at 120 h. P-values (for B and C) were generated from: one-way ANOVA, followed by and post hoc Tukey Honest Significant Difference test with 95% confidence level and adjusted for multiple comparisons (*** p<0.001, ** p<0.01,* p<0.05, n.s not significant). See also Data S1G for body size at strain level. D. Pearson correlations of microbial taxa abundance (day 3 adults) with host developmental rates (52 h post L1) and body size (48 h post L1). The test statistic is based on Pearson’s product moment correlation coefficient and follows a t distribution with length(x)-2 degrees of freedom at the level of 95% confidence interval. Ochrobactrum (colored in red) is the only microbial taxa with positive correlation with both host phenotypes (p<0.05). 9 microbial taxa (colored in blue) show negative correlations with both host phenotypes (p<0.05). See also Table S3 and Figure S4.
Type 1 animals express a broad repertoire of microbial response pathways to create selectivity.
To more specifically identify the host signaling networks regulating selection of the gut microbiome, we transcriptionally profiled the host responses to colonization of a panel of representative C. elegans strains from each of the three microbiome types [Type 1, JU1400 and ED3040; Type 2, N2, LKC34, and CB4853; and Type 3, ED3017, MY14, and ED3042] [Figure 4A, Data S1AC]. At day 3 of adulthood, we observed a large set of differentially expressed genes between Types 1 and 3 [1507 higher in Type 1 (‘Type 1 Up’), 1706 higher in Type 3 (‘Type 3 Up’); Figure 4B, Data S1AC], consistent with the differences in microbiome composition between these strains. We first tested for correlations between transcript and taxa abundance across all of the C. elegans strains. We identified 2844 genes that were differentially expressed by microbiome type and significantly correlated with taxa abundance of one or more microbes [Figure 4C]. Interestingly, Ochrobactrum-correlated genes dominated the taxa-specific signatures, and genes that were positively correlated with Ochrobactrum were negatively correlated with Bacteroidetes Myroides (259 genes) and vice versa (857 genes). Smaller subsets of genes were correlated with the abundance of 16 other taxa, and these genes sets were largely distinct from those of Ochrobactrum and Myroides [Figure 4C]. These data could indicate that similar transcriptional networks coordinate the enrichment Ochrobactrum and the exclusion of Myroides. To begin to identify the function of these and other host genes that were upregulated in association with particular microbial communities, we used the WormExp tool 29. We observed broad increases in expression in genes involved in three main pathways: microbial and immune response, general stress response, and insulin signaling [Figure 4D–G].
Figure 4. Transcriptional changes in insulin signaling, microbial and stress response genes define microbiome types.
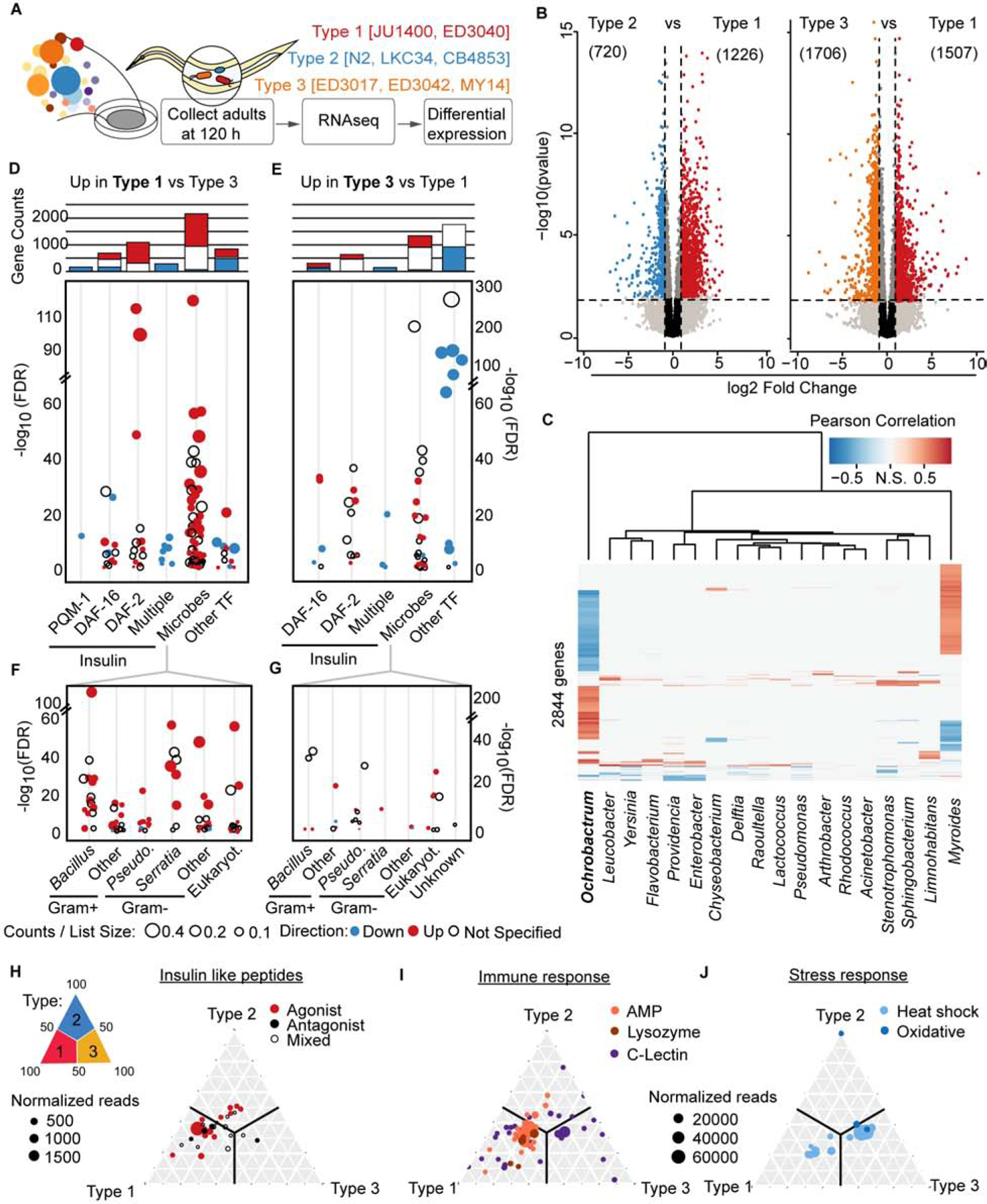
Representative strains from each of the microbiome types grown on BIGbiome to Day 3 adulthood were collected for RNAseq. B. Volcano plots displaying genes differentially expressed between Types 1 and 2 and Types 1 and 3. Type 1 vs Type 2; Significantly differentially expressed genes (Benjaminii-Hochberg adjusted p-value < 0.05) are colored red if they are upregulated in Type 1, or log2FC > 1, or colored blue if upregulated in Type 2, or log2FC < −1. Type 1 vs Type 3; Significantly differentially expressed genes (Benjamini-Hochberg adjusted p-value < 0.05) are colored red if they are upregulated in Type 1, or log2FC > 1, or colored orange if upregulated in Type 3, or log2FC < −1. See also Data S1AC for a full gene list. C-F. Significant (FDR < 0.05) WormExp enrichments from Type 1 Up gene set (C) and Type 3 Up gene set (D). Barplots represent counts of unique genes for each category. ‘Multiple’ category includes daf-16;daf-2 double mutants. ‘Microbes’ category subset separated into specific terms in the Type 1 Up set (E) and Type 3 Up set (F). G. Heatmap depicting genes that are significantly differentially expressed between microbiome types and significantly correlated (Pearson correlation, Benjamini-Hochberg adjusted p-value < 0.05) with the absolute abundance of at least one BIGbiome member. H-J. Ternary plots illustrating the microbiome type enrichment patterns of genes belong to insulin-like peptides (H), immune (I) and stress responses (J). Each dot is an individual gene and dot sizes are proportional to normalized read counts in the transcriptional dataset. Due to a large number in the immune and stress response gene, only genes with significant changes (p< 0.05) in expression between the microbiome types are shown. Only one gene (ctl-1) is expressed almost exclusively in Type 2 (in J). See also Figure S6.
Microbial response pathway genes varied significantly between the strain groups. We found that genes more highly expressed in Type 1 animals were broadly enriched in genes altered in response to a wide array of microbes [60.4% (81/134 datasets) for ‘Type 1 Up’ versus 42.9% (27/63 datasets) for ‘Type 3 Up’; Figure 4E–F]. Interestingly, ‘Type 1 Up’ genes overlap with those upregulated upon exposure to pathogens [Figure 4E] (e.g., B. thuringiensis, S. marcescens, E. faecalis, P. aeruginosa and others 30–32) while ‘Type 3 Up’ genes overlap more with those downregulated upon pathogen exposure [Figure 4F]. Though no pathogens are included in the BIGbiome, Type 1 animals seem to be using similar responses to related microbes to exclude most everything but Ochrobactrum from the gut. Consistent with this idea, ‘Type 1 Up’ genes overlap with 13 datasets of upregulated genes in response to the pathogen P. aeruginosa PA14 [162 genes in total; e.g., 39 genes from 33, Q = 1.8e-8]. Under these conditions, the twelve Pseudomonas strains in the BIGbiome are excluded from the guts of Type 1 animals [1.2% relative abundance compared to 6.4% and 7.4% for Type 3 and lawns, respectively]. Further, several canonical C. elegans immune effectors from multiple pathways 34 were expressed more highly in Type 1 animals [Figure 4I, Data S1AC]— e.g., irg-5 [2.81-fold and Pearson=0.75 to Ochrobactrum; p38/MAPK and FSHR-1], lys-5 [2.80-fold; Wnt/β-catenin and HLH-30/TFEB], and irg-2 [2.9-fold; ZIP-2]. These specific responses are likely to promote Ochrobactrum colonization in the process. In contrast, more limited immune pathway expression was observed in Type 3 animals which instead more highly express general stress-related pathways [Figure 4I–J, Data S1AC]— e.g., gcn-1 [3.2-fold; SKN-1/Nrf2, oxidative stress] and hsp-6 [2.4-fold; ATFS-1, unfolded protein stress]. Type 3 animals did express a subset of c-type lectins more significantly than the other microbiome types [Figure 4I]. Thus, Type 1 animals appear to employ a suite of immune pathways in parallel to create the highly selective environment within the gut for Ochrobactrum colonization, which are largely absent in Type 3 animals.
Transcriptional variation in insulin signaling networks distinguish microbiome types
Among the pathways enhanced in the microbiome types, we observed a particular enrichment for insulin signaling. Both ‘Type 1 Up’ and ‘Type 3 Up’ gene sets were highly enriched for DAF-2- and/or DAF-16-dependent genes, though overlap was more extensive in Type 1 animals [47 datasets for ‘Type 1 Up’ and 20 datasets for ‘Type 3 Up’; both high- and low-insulin conditions observed; Figure 4C–D]. In addition, the vast majority of the microbially responsive genes identified above are also associated with changes in insulin signaling pathways in the lab strain N2 [1066 genes (80%) in Type 1 versus 464 genes (46%) overlap with insulin signaling datasets; Data S1AC, Figure 4C–D]. Interestingly, the enrichment observed for ‘Type 1 Up’ genes have been associated with both low- and high-insulin signaling conditions in the lab strain N2, which may reflect plasticity in gene expression driven by natural genetic variation in these wild strains.
To more clearly gauge the insulin signaling balance in these animals, we examined the expression of the nearly 40 insulin-like peptides (ILPs) that compete for binding of DAF-2/IGFR. The mixture of ILPs serves to activate (agonists) or repress (antagonists) downstream insulin signaling pathways to provide phenotypic specificity and coordination of responses across tissues 35,36. There was a notable shift in ILP expression between Type 1 and Type 3 animals: 9 of the 40 ILPs were expressed significantly higher in Type 1 strains compared to minimal ILP expression in Type 3 strains [Figure 4H]. Type 2 animals expressed intermediate levels and a mix of agonist and antagonist ILPs, consistent with the intermediate expression of insulin pathway genes [Figure S6A]. Nearly all of the genes in the canonical insulin signaling pathway, including daf-2/IGFR, age-1/PI3K, akt-1/AKT, daf-18/PTEN and daf-16/FOXO were expressed higher in Type 3 than Type 1 animals [Figure S6A].
Insulin signaling pathways drive microbiome composition and its impact on host physiology.
We next sought to test directly whether host insulin signaling mediates microbiome selection and its resulting effects on host physiology. To do this, we used RNAi to knock down daf-2/IGFR and daf-16/FOXO gene expression in representative strains for each microbiome type: Type 1, JU1400; Type 2, N2; and Type 3, ED3017. If high levels of insulin signaling positively select for Type 1 microbial communities, then reducing the activation of these pathways may result in these hosts adopting communities and host physiological attributes that more closely resemble those in Type 3 strains. Indeed, this is what we observed. Knockdowns of daf-2 resulted in slower development [Figure S5AB] and reduced body size in JU1400 [Figure S5C] when grown on BIGbiome lawns versus vector controls. Conversely, knockdowns of the transcription factor daf-16/FOXO generally accelerated development [Figure S7A,B] and increased animal body size [Figure S5C]. In Type 2 animals (N2), we observed lower Ochrobactrum colonization in daf-2 RNAi (P<0.001) and higher in daf-16 RNAi, as shown via microbiome sequencing and fluorescence quantification of GFP-Ochrobactrum [P<0.001, Figure 5A–C]. The impact of these knockdowns was most dramatic in Type 1 and 3 strains: daf-2 RNAi reduced the recruitment of Ochrobactrum in Type 1 (JU1400) animals by 30–50% compared to vector controls [P<0.001, Figure 5A–C], while daf-16 RNAi increased Ochrobactrum colonization by more than 20-fold in non-selective Type 3 animals (ED3017) [P<0.001, Figure 5A–C].
Figure 5. Insulin signaling pathways mediate recruitment of Ochrobactrum.
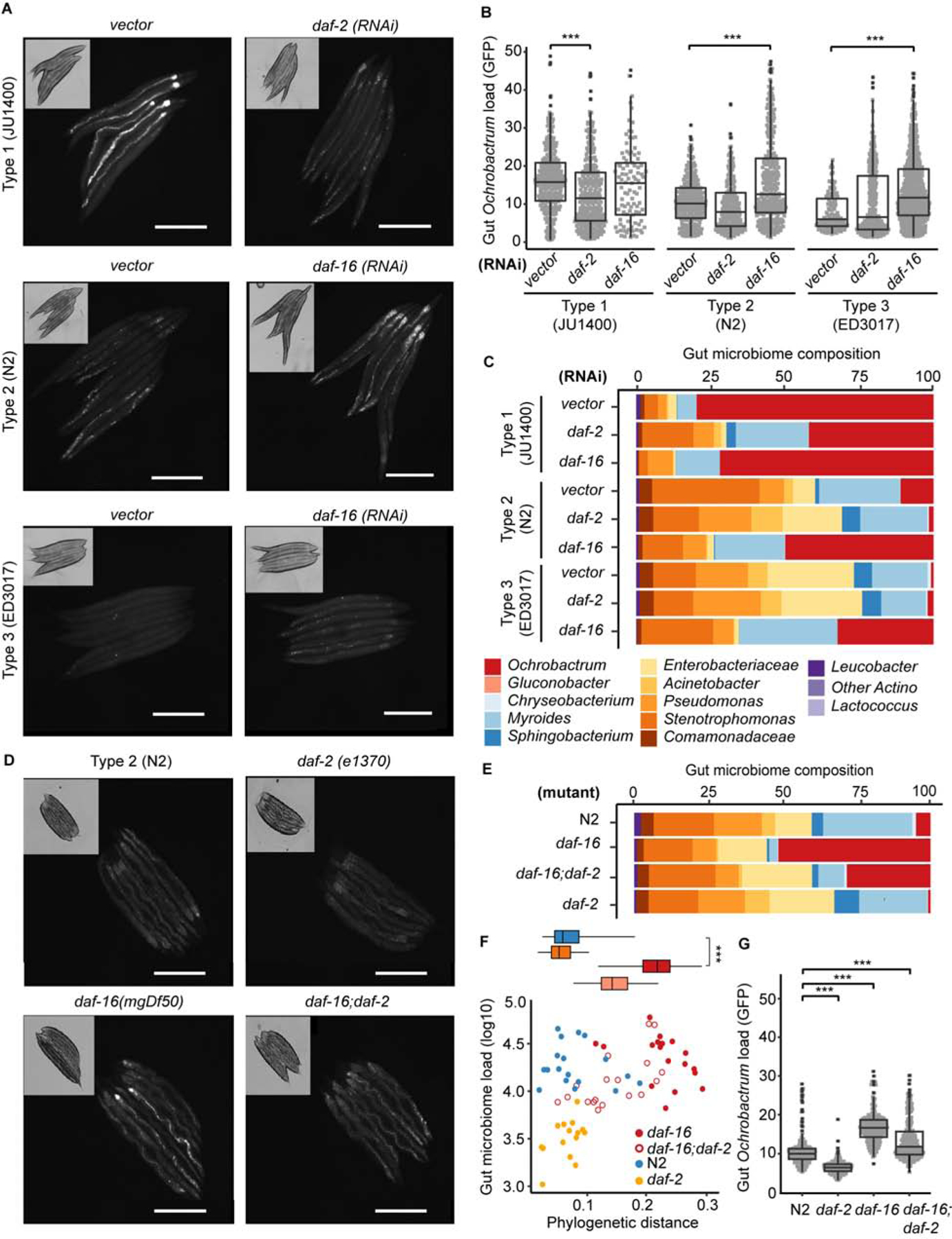
A-C. Ochrobactrum colonization in JU1400(Type 1) decreased with daf-2(RNAi) and increased in N2(Type 2) and ED3017(Type 3) with daf-16(RNAi). Similar trends are shown in representative images of Day 3 adults grown on BIGbiome with GFP-Ochrobactrum (A, Bar = 500 µm), GFP signal per individual animal (B, JU1400(vector): n=290, JU1400(daf-2): n=274, JU1400(daf-16): n=97, N2(vector): n=233, N2(daf-2): n=202, N2(daf-16): n=79, ED3017(vector): n=103, ED3017(daf-2): n=247, ED3017(daf-16): n=292, see also Data S1K), and bulk gut microbiome sequence of the corresponding population (C, see also Data S1J). D,G. Ochrobactrum colonization decreased in daf-2(e1370), increased in daf-16(mgDf50) and by a lesser extent in daf-16(mgDf50);daf-2(e1370);daf-16(mgDf50) mutants. Similar trends are shown in representative images of Day 3 adults grown on BIGbiome with GFP-Ochrobactrum (D, Bar = 500 µm), bulk gut microbiome sequence of the corresponding population (E, see also Data S1J), and GFP signal per individual animal (G, N2: n=216, daf-2: n=198, daf-16: n=184, daf-16;daf-2;daf-16: n=207, see also Data S1M). F. N2 and insulin signaling mutants daf-2, daf-16, daf-16;daf-2 host distinct microbiome types based on gut microbiome load per animal (y-axis) and phylogenetic distances to BIGbiome lawn (x-axis). Inset: Box-whisker plot of phylogenetic distances to BIGbiome for the three microbiome types. (B,G) n=individual animals; P-values were generated from one-way ANOVA, followed by and post hoc Tukey test with 95% confidence level and adjusted for multiple comparisons (***p<0.001, **p<0.01, *p<0.05). See also Figure S5.
Since Type 2 animals are intermediate in their selectivity for Ochrobactrum, we next sought to test whether Type 2 insulin signaling mutants exhibit altered phenotypic responses reflective of other microbiome types. Loss-of-function mutants of the insulin peptide receptor daf-2(e1370) in the lab strain (N2) mimic low agonist insulin levels. On BIGbiome lawns, daf-2/IGFR mutants exhibited Type 3-like developmental delays and smaller body size compared to wild type animals [P<0.001; Figure S5D–E]. The daf-2/IGFR mutants had lower Ochrobactrum colonization at the population level by microbiome sequencing [P<0.001; Figure 5E] and at individual level by fluorescence quantification of GFP-Ochrobactrum [P<0.001; Figure 5G]. Conversely, loss-of-function mutants of the downstream transcription factor daf-16(mgDf50) developed much faster, had larger body sizes in early adulthood [P<0.001; Figure S5D–E], and had greater Ochrobactrum colonization [P<0.001, Figure 5D–G]. Finally, double-mutants of daf-16;daf-2 increased Ochrobactrum colonization by one-third compared to daf-16 mutants [P<0.001; Figure 5D–G], suggesting other potential regulators may be acting in the low insulin signaling conditions to suppress Ochrobactrum colonization. Together, these data indicate that under low insulin signaling, DAF-16 regulated processes either limit Ochrobactrum colonization or fail to effectively exclude other microbiome members.
To test the generalizability of these responses, we then expanded our RNAi analyses to both additional representative strains [ED3042 (Type 3), CB4856 (Type 1) and N2 (Type 2)] and additional genes in the canonical insulin signaling pathway. We observed that RNAi-mediated knockdown genes that activate the insulin signaling like daf-2/IGFR, age-1/PI3K and akt-1/AKT all reduced Ochrobactrum colonization levels in Type 1 and 2 animals [Figure S6C]. Conversely, knockdowns of akt-2/AKT and pathway suppressor daf-18/PTEN increased Ochrobactrum colonization [Figure S6C] in Type 2 and 3 animals, which is a rare phenotypic separation of the largely redundant AKT orthologs. Further, through RNAi of each of the 20 insulin-like peptides in the intestine of Type 2 (N2) animals, we identified increased Ochrobactrum colonization for 6 of the 9 antagonist ILPs [ins-1, −11, −18, −21, −24, −31; P<0.001, Figure S6B]; many of these are upregulated in daf-2 mutant background 37. Together, these data indicate that subsets of canonical insulin signaling pathways regulate microbiome composition and, in turn, impact host physiology and growth.
Interplay of downstream insulin signaling transcription factors drives microbiome regulation.
We next sought to determine what genes in the insulin signaling regulons influence microbiome composition. To achieve this, we examined two mutually exclusive transcription factors known to orchestrate insulin signaling in C. elegans, DAF-16/FOXO and PQM-1/SALL2. PQM-1 has also previously been associated with regulation of both development and immunity into adulthood 38,39. To directly test its role in regulation of the microbiome we knocked down pqm-1 by RNAi in representative strains of each microbial community type: JU1400 (Type 1), N2 (Type 2) and ED3017 (Type 3). We observed significantly delayed development and reduced body sizes on BIGbiome in Type 1 animals [P = 0.02 and 0.004, respectively; Figure 6A–B]. Consistent with the effect on development, we observed a decrease of Ochrobactrum colonization after pqm-1 knockdown by RNAi [P<0.001; Figure 6E–F]. Knockdowns of pqm-1 in Type 2 (N2) and Type 3 (ED3017) animals showed similar but non-significant decreases in developmental rates [Figure S7A–B], and the already low levels of Ochrobactrum colonization were decreased to the limit of detection for both strains [Figure S7C].
Figure 6. PQM-1 regulates microbiome impact on host physiology and recruitment of Ochrobactrum to the gut microbiome.

A. Box-whisker plot of adult percentage of vector (n=4) and pqm-1 (n=4) RNAi knockdown mutants in Type 1 JU1400 background at 54 h post L1 stage. B. Box-whisker plot of body size of vector (n=66) and pqm-1 (n=85) RNAi knockdown mutants in Type 1 JU1400 background at 48 h post L1 stage. C. Box-whisker plot of adult percentage of vector (n=4) and pqm-1 (n=4) RNAi knockdown mutants in daf-16(mgDf50) background at 54 h post L1 stage. D. Box-whisker plot of body size of vector (n=136) and pqm-1 RNAi knockdown mutants (n=179) in daf-16(mgDf50) background at 48 h post L1 stage. (B,D) n represents the number of independent worm populations. (C,E) n represents the number of individual animals quantified by microscopic images. See also Figure S7A,B,D,E. E-G. Ochrobactrum colonization in JU1400(Type 1) and daf-16(−) decreased with pqm-1(RNAi). Similar trends are shown in representative images of day 3 adults grown on BIGbiome with GFP-Ochrobactrum (E, Bar = 500 µm) and GFP signal per individual animal (F,G). (G,H) n represents the number of individual animals quantified by Biosorter. P-values were generated from student’s t-test (***p<0.001, **p<0.01,*p<0.05). See also Figure S7C,F,G. H. Sunburst plot illustrating significantly enriched (WormCat-reported padj < 0.05) WormCat subcategories from Class II targets upregulated in Type 1 strains and Class I targets upregulated in Type 3 strains. I. Representative images show nuclear localization of PQM-1 GFP in day 3 adults grown on BIGbiome, compared to no nuclear localization on E. coli OP50. No nuclear localization of DAF-16 GFP in day 3 adults grown on BIGbiome and E. coli OP50 (Bar = 100 µm). J. Day 3 adults grown on BIGbiome express higher PQM-1::GFP and DAF-16::GFP than on E. coli OP50, quantified by GFP signal per individual animal. K. Schematic diagram of insulin signaling targets drives Ochrobactrum colonization (Type 1, red arrows; Type 3, blue arrows). See also Figure S7.
To further dissect the genetic interaction of pqm-1 with daf-2 and daf-16, we knocked down pqm-1 in daf-2(e1370) mutants by RNAi and observed significantly slower developmental rates [40% less, P<0.001; Figure S7D] and reduced body sizes [13%, P<0.001; Figure S7E], but Ochrobactrum colonization remained similar to the empty vector at a low level. RNAi knockdown of pqm-1 in daf-16(mgDf50) mutants also delayed development by 10% [P<0.001; Figure 6C] and significantly lowered Ochrobactrum colonization compared to the empty vector [Figure 6E,G]. These data suggest that pqm-1 promotes Ochrobactrum colonization independent of daf-16.
Finally, we examined the transcriptional networks themselves based on promoter binding elements for each of these transcription factors. DAF-16 activates genes that contain a DAF-16 binding element (DBE; Class I) under stressful or low insulin conditions, while PQM-1 activates genes under favorable or high insulin conditions containing the DAF-16 associated element (DAE; Class II) 38. Analysis of the transcriptional datasets identified 170 (10.2%) Class I and 219 (12.6%) Class II genes that were differentially regulated between Types 1 and 3. WormCat analyses of these genes highlighted two very different responses in Type 1 and Type 3 animals. The Class II genes from the ‘Type 1 Up’ set [Figure 6H] are enriched for multiple detoxification and immune responses against pathogens, including cytochrome P450 genes (cyp-13A3, cyp-32A1, cyp-25A1), which can metabolize toxic compounds, and c-type lectins (clec-57, clec-49, clec-204), which are involved in antimicrobial immunity 40 . In contrast, the Class I genes from the ‘Type 3 Up’ set are enriched for general oxidative and heat stress responses rather than pathogen specific responses. Metabolism categories also differed between Types 1 and 3, with Type 3 enrichment for glycolysis, lipid (fatty acid and phospholipid), short chain dehydrogenase, and carbohydrates. Favorable insulin signaling may therefore promote the selection of more specialized microbial communities via regulation of immune and xenobiotic response genes that may help establish a selective environment for Ochrobactrum to colonize the gut. We further compared PQM-1::GFP and DAF-16::GFP reporter strains grown on BIGbiome and E. coli OP50. We observed both greater PQM-1::GFP expression and nuclear localization [Figure 6IJ] for adults grown on BIGbiome than on E. coli OP50, while DAF-16::GFP expression also increased but no differences in nuclear localization were observed. Together, our results suggest higher insulin signaling activates PQM-1 to promote microbial specific immune response in microbiome selection from the environment, while lower insulin signaling levels drive DAF-16 mediated broad stress responses that suppress microbiome selection.
DISCUSSION
Insulin signaling shapes the microbiome landscape in C. elegans.
C. elegans flourish in natural habitats of rotten fruit and plant matter, an environment with abundant and diverse microbes. They rapidly respond to environmental fluctuation and adjust growth, defense and reproduction strategies to ensure their success in the wild. To learn more about the genetic circuits in microbiome response from this widely used model organism, we reunite wild C. elegans with microbial consortia isolated from their natural habitats. Although grown on the same microbiome mixture BIGbiome, 38 C. elegans strains established distinct gut microbiome types in adulthood. Wild C. elegans with faster growth and development showed stronger recruitment of Ochrobactrum, a commensal member of their core microbiome in nature. Transcriptomic analysis suggested host insulin signaling was driving establishment of the Ochrobactrum dominant gut microbiome. We used RNAi knockdowns and mutants to confirm that IIS modulates the Ochrobactrum-driven microbiome type variation through downstream transcription factors of DAF-16/FOXO and PQM-1/SALL2.
Insulin signaling mediated selection of Ochrobactrum dominates among wild C. elegans strains.
The influences of highly conserved insulin signaling pathways are found in nearly all aspects of animal physiology, including development, fertility, stress resistance and longevity 41–43. Our findings underscore a distinct role for insulin signaling in establishing a selective environment for microbiome enrichment among Type 1 strains. Insulin-like peptides (ILPs) represent the most upstream components of insulin signaling and we show that they act in the intestine to mediate the microbiome composition. ILPs are important in regulating the balance energy expenditure in growth, reproduction, and defense as a function of animal age 35, suggesting their critical roles in modulating insulin signaling during adulthood and coordinating across tissues activities. INS-7 is an agonist of DAF-2/IGFR and is regulated by DAF-2/IGFR and DAF-16/FOXO in the intestine to provide positive feedback regulation in coordination of animal physiology across tissues 35,37. Thus, we hypothesize that the observed high expression of ins-7 and other ILPs in Type 1 keeps DAF-16 activity low to prevent overstimulation of general stress responses, or indiscriminate microbial response leading to commensal exclusion. On the other hand, antagonistic ILPs like ins-11 could suppress host insulin signaling to reduce host selection of commensals, forming the microbiome Types 2 and 3 we observed. While the differences in ILPs landscape are likely driven by natural variation among wild worms, it is also possible that the BIGbiome community may shape ILP production, as some studies have found pathogen infections induced antagonist ins-11 expression 44.
Downstream of DAF-2/IGFR there two orthologs of the AKT are observed in C. elegans, akt-1 and akt-2, which act redundantly to regulate most physiologic processes in part by preventing DAF-16 nuclear localization 45. Surprisingly, we observed different responses in Ochrobactrum colonization when akt-1 and akt-2 were knocked down. Although both AKT-1 and AKT-2 are activated by insulin signaling, they may compensate each other since dauer-c phenotype requires knockdown of akt-1 and akt-2 simultaneously 46. Thus, it is possible that knocking down akt-2 promotes akt-1 expression in Type 2 and Type 3 animals, thus promoting Ochrobactrum colonization in Type 2 and 3 animals. Some studies have indicated individual roles for akt-1 and akt-2 in regulation of lifespan and reproduction in C. elegans 47,48. Therefore, the roles of akt-1 and akt-2 in differentially regulating microbiome deserve further investigation.
Insulin signaling-regulated PQM-1/SALL2 activates downstream targets by binding DAE promoter elements 49, likely contributing to gut microbiome selection. In the Ochrobactrum-dominant microbiome Type 1, up-regulated PQM-1 targets are enriched in host immune response genes, including C-type lectins and antimicrobial peptides 50. C-type lectins are known to recognize microbial molecular patterns, implying their roles in bacterial specific immunity 40. Antimicrobial peptides like the saposin genes spp-2 and −5 have been shown to be induced by Ochrobactrum MYb71 colonization 23. In addition, the Ochrobactrum-dominant microbiome type is associated with elevated xenobiotic response gene families like CYP, GST, and UGT. These enzymes can detoxify microbial products and act as a sink of reactive oxygen species (ROS), thus reducing oxidative stress for cellular protection and maintenance. Taken together, elevated insulin signaling in Type 1 strains may establish a suitable gut environment for increased colonization of commensal microbes, establishing an Ochrobactrum-dominant microbiome type in adulthood.
Trade-offs in microbiome regulation in wild C. elegans with reduced insulin signaling.
Our studies indicate that Type 3 strains with reduced insulin signaling upregulated broad-spectrum stress responses that limit microbiome colonization, but also abolished the ability to select commensals from the environment. Without selection, Type 3 gut microbiomes mirror the lawn in composition. The non-selective microbiome Type 3 also mimics the long-lived daf-2 mutant in higher expression of catalase genes, as well as mitochondria and ER stress markers like hsp-4 and hsp-6 43,51. Other signatures of daf-2 mutants include shift of lipid metabolism and reduced brood size. Similarly, Type 3 strains increased the expression of mitochondrial β-oxidation genes like acdh-2 and glycogen synthesis genes like gsy-1, indicating a switch from lipid metabolism to carbohydrate storage. In addition, transcription factors (lin-11, lin-13b, mep-1) that negatively regulated reproduction were highly expressed in non-selective Type 3 strains and, suggesting a reduced investment in reproduction. As the worm ages, reduced insulin signaling during adulthood activates DAF-16 dependent immune response to defend against microbes, compensating for the immune-senescence in other protective pathways like the MAP kinase 52. Although reduced insulin signaling provides benefit to the host in pathogen resistance and lifespan extension, the trade-off in adulthood might be loss of commensal colonization and reduced reproduction. Interestingly, increased fertility was observed in C. elegans colonized by Ochrobactrum, driven by genes with enriched GATA motifs 23. Since GATA-like sequences are overrepresented in PQM-1 activated DAE genes, therefore it is possible that PQM-1 activates these Ochrobactrum-responsive genes in adulthood, boosting commensal recruitment for the benefit of enhanced host reproduction or because of concomitant impacts on reproduction. The evolutionary benefits of microbiome selection in long term phenotypes like lifespan and healthspan remain to be explored.
Potential for microbiome modulation of insulin signaling networks in C. elegans
The gradient of Ochrobactrum colonization among wild strains reflects their various degrees of insulin signaling activation. There are a large number of SNPs that contributed to intrinsic natural genetic variation in the insulin signaling regulatory network among wild worm strains 53, many were found in our microbiome GWAS analysis and are differentially expressed in insulin receptor (daf-2/IGFR) mutants. The fact that prior to colonization as adults C. elegans grew and developed faster on the BIGbiome community versus E. coli OP50 suggests that it may stimulate host insulin signaling that accelerated their growth and development. PQM-1/SALL2 has been shown to influence developmental growth rates 38, and may therefore be responding to these BIGbiome cues both in development and in adulthood. Gut microbes have been shown to engage insulin signaling pathways in several other animals, including hydra, Drosophila, zebrafish, mice, and humans 1,54–56.
Further, individual natural microbes from this community have been shown to have a dramatic impact on the physiology and development of C. elegans as well 13,16. For example, many Alphaproteobacteria and Enterobacteriaceae strains generally promote growth, while most Bacteroidetes and Stenotophomonas strains delay the growth of N2 worms 16,17. This could indicate either that Type 3 animals that have slower growth rates and higher levels of Bacteroidetes colonization due to impaired responses to these bacteria, or that Type 1 strains are more resistant and are therefore able to restrict their colonization. Taken together, microbial engagement of insulin signaling could potentially form a feed forward loop during development that could influence distinct microbiome composition in adulthood.
Broader signaling networks in regulation of the microbiome
Acting in the same direction of insulin signaling, TGF-β signaling was also up-regulated in the Ochrobactrum dominant microbiome Type 1 animals, likely the result of extensive crosstalk between the two pathways 57. TGF-β signaling from neurons and epidermis can activate ILPs secretion that feed into insulin signaling to modulate DAF-16 activities in the intestine 58,59. C. elegans TGF-β mutants dbl-1 were highly colonized by Enterobacter with enhanced pathogenicity when grown on a synthetic natural microbiome, and though these genes remain unchanged in our studies of this pathway in microbiome regulation 60.
Similar to DAF-16, transcription factor SKN-1 was also enriched in the intestine and acts downstream of insulin signaling as an AKT-1 phosphorylation target 61. Under reduced insulin signaling in the microbiome Type 3 strains, SKN-1 likely synergized with DAF-16 to induce oxidative and heat shock stress that suppress microbiome selection and colonization, which explained why daf-16;daf-2 double mutants only partially reduced Ochrobactrum colonization compared to daf-2 mutants. Interestingly, the longevity effect of SKN-1 was dependent on the type of E. coli strains, suggesting the pathway is under the influence of microbial content 62. SKN-1 may be also responsible for higher expression of collagen genes in microbiome Type 3, as these extracellular matrix (ECM) genes were known to up-regulated by SKN-1 and play critical roles in pathogen defense as weakened cuticles were associated with increased susceptibility to Microbacterium nematophilum infection 63,64.
Prospectus
Animals have partnered with microbes throughout evolution to extend their genetic repertoire and metabolic capacity 65. This partnership is now deeply imprinted in animal physiology and the disruption of this commensal relationship can compromise animal health. Here, we presented a genetically tractable platform that integrates a natural microbiome with the rich molecular tools in C. elegans. Our results demonstrated that natural variation in insulin signaling drives microbiome selection, suggesting that the regulation of DAF-16/FOXO and PQM-1/SALL2 play major roles in the formation of C. elegans microbiome types. From the microbial side, increasing genomic information from natural microbiomes 20 will undoubtedly aid in discovery of microbial factors that engage host pathways like insulin signaling. Exposure to natural microbes and their products may help to ascribe phenotypes and functions to the over 40% of microbiome type enriched genes in the C. elegans genome, many of which have orthologs in other animals as well. In addition, external factors like nutrients, temperature, pH, and liquid growth can modulate the metabolic state of C. elegans hosts and associated microbiome, contributing to shifting response in host signaling pathways like insulin signaling and altered outcomes of gut microbiome colonization 20,66,67. Ultimately, we believe that this system will allow for greater understanding of the interplay of host, microbial and environmental factors that regulate microbiome impact on broad aspects of host physiology.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Buck Samuel (buck.samuel@bcm.edu).
Materials Availability
All microbial strains and other materials used in these studies are available upon request.
Data and Code Availability
All datasets have been included as raw data [Data S1]. Sequencing based datasets have been deposited at NCBI Sequence Read Archive database (Bioproject PRJNA540192) with the following sample accession numbers for RNAseq reads (SAMN13050735–13050742) and microbiome sequencing reads (SAMN13068200–13068238, 13071563–13071602, 16597785–16597833, 16611296–16611371, 17054579–17054627). All code used in the analysis of datasets is available through the Bitbucket link, including those for overall microbiome compositional analyses (‘Microbiome_analysis_scripts.txt’), processing and analysis of RNAseq datasets (‘Kallisto_bbmap_bbduk_Script.txt’ and ‘DifferentialExpression_Script.txt’), correlation of microbial taxa with gene expression profiles (‘MicrobialAbundance_vs_GeneExpression_Correlation_Script.txt’) and figure generation in R environment (‘R_plot_figures.txt). (https://bitbucket.org/the-samuel-lab/natural-variation/src/master/).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Maintenance of Caenorhabditis elegans strains
Caenorhabditis elegans strains utilized in this study can be obtained from the Caenorhabditis Genetics Center (CGC), including N2-Bristol, CB1370 [daf-2(e1370)], GR1307 [daf-16(mgDf53)], HT1890 [daf-2(e1370);daf-16(mgDf53)], OP201[unc-119(ed3);wgIs201(pqm-1::TY1 EGFP FLAG C;unc-119)], HT1889[daf-16(mgDf50);unc-119(ed3);lpIs14(daf-16f::GFP;unc-119)] and several natural isolates: AB1, AB3, CB4853, CB4854, CB4856, ED3017, ED3021, ED3040, ED3042, ED3052, ED3072, GXW0001, JU1088, JU1171, JU1218, JU1400, JU1401, JU1652, JU258, JU263, JU300, JU312, JU322, JU323, JU360, JU361, JU397, JU533, JU642, JU775, KR314, LKC34, MY1, MY14, MY16, MY2, and PX174 [Table S1]. The intestinal RNAi strain JM45 (rde-1(ne219); Is[Pges-1::RDE-1::unc54 3′UTR; Pmyo2::RFP3]) was a gift from Dr. Meng Wang. All C. elegans strains were grown and maintained on nematode growth media (NGM; Research Products International) seeded with Escherichia coli strain OP50 at 20°C. E. coli OP50 and HT115 RNAi strains can be requested from the CGC.
Preparation of C. elegans populations
Prior to each experiment, worm populations were rendered ‘germ-free’ and synchronized to L1 stage79 by treating gravid hermaphrodites with bleach solution (mixture of Clorox bleach and 5M NaOH in 2:1 volume ratio), followed by multiple washes with M9 buffer79 to remove bleach solution. Germ-free L1s were then allowed to hatch and synchronize in sterile M9 buffer 15–18 hours rotating at 20°C.
Preparation of microbiome mixtures
All microbial strains used were originally isolated from C. elegans natural isolates or habitats [Data S1AA] and stored at −80°C as glycerol stocks 16. Ochrobactrum pituitosum BH3 and an isogenic strain expressing GFP [Tn7 insertion of GFP on the chromosome 68] were generous gifts from Dr. Emily Troemel. JUb strains were originally isolated by Dr. Marie-Anne Félix.
To begin all experiments, we stamped out fresh cultures from glycerol stocks onto a rectangular LB plate, then incubated overnight at 28°C. The colonies on the plate were then used to inoculate a 1 ml 96 deep well plate (Axygen) filled with 300 µl lysogeny broth (10g Tryptone, 5g yeast extract, 10g NaCl in 1L distilled water adjust to pH=7.5) in each well. After overnight growth (14–16 h) at 28°C and 250 rpm shaking, bacterial cells were pelleted down by centrifuge at 4000 x g for 10 min. Supernatants were discarded and replaced with 200 µL sterile M9 buffer in each well. Pellets were then fully resuspended by pipetting then transferred to a clear bottom 96 well plate (Costar, Corning). Growth of each microbe was assessed by measurement of optical density (OD) readings at 600nm using a Multiskan FC Microplate Photometer (Thermo Scientific). Bacterial density in each well in the parent plate was then normalized individually to an OD600 of 1.0 using sterile filtered M9 buffer. BIGbiome001 master mixes (referred to as ‘BIGbiome’ throughout) were created by combining equal volumes of each bacterial strain, which was then used to seed (30 µL) Nematode Growth Medium (NGM) agar in 12 well plates (Costar, Corning). Seeded plates were grown overnight at 20°C (80% humidity) before use.
METHOD DETAILS
Measurement of gut microbiome colonization in C. elegans
Existing methods that use surface sterilization with antibiotics, pestle-based disruption of animals and enumeration of bacterial colonies on agar plates21, though robust, were optimized for determination of bacterial densities of an individual strain or small set bacteria of interest rather than communities. Discrimination of bacteria by colony morphologies is similarly intractable within complex communities. We addressed these challenges by: (i) replacing antibiotic treatment, which is ineffective in a large community that contains variable antibiotic resistance profiles, with a more consistent dilute bleach treatment to kill surface associated microbes; and (ii) replacing the mortar-and-pestle with bead-based, multi-well format disruption of C. elegans to release gut microbes into solution. Further, to quantify live bacteria in the gut, we also adapted a liquid-based CFU quantification method to remove the need for laborious colony counting on plates.
Creation of standard curves for CFU estimations:
Overnight grown BIGbiome lawn was sampled and resuspended in M9 buffer. The mixture was subjected to a serial dilution from 10−1 to 10−6. The number of live bacteria from the dilution series were determined by counting CFU from 10 µL of each dilution onto a LB plate. The same dilution was inoculated into a 96 well flat bottom plate containing 100 µL LB medium in each well. The plate was incubated at 28 °C and bacterial growth curve in each dilution was recorded by measuring OD600 every 15 min for 18 h. Within the range of linear portion of growth, OD600 equal to 0.2 was used as a threshold to interpolate the corresponding growth time, designated as CGT 80. Exponential regression between CFU number and CGT (R² = 0.99) was used to infer the CFU number from sample CGT at OD600 threshold of 0.2. Regression derived trendline equation was applied: total bacterial cells = (8E+11)*e(−1.114 * CGT).
Collection, surface sterilization and lysis of animals:
Around 100 L1 animals were seeded in duplicate on the BIGbiome lawn at 20°C with 80% humidity. Worm populations were assayed at 48 h and 120 h post seeding. On sampling day, worms were washed from a bacterial lawn with 600 µL of M9 buffer (0.01% triton X-100) to a sterilized 2 ml 96-well deep plate (Axygen). The deep well plate was centrifuged at 300 g for 1 minute to pellet down worms, bacteria in the liquid were removed by an aspirating manifold (VP1171A, V&P scientific). These washing steps were repeated 5 times with M9 buffer (0.01% triton X). 100 µL of 10 mM levamisole in M9 buffer (0.01% triton X) was then added to paralyze the worms for 5 min. Then 200µl of 4% bleach solution (diluted from of Clorox bleach and 5M NaOH in 2:1 mixture) in M9 treatment for 2 min, further eliminate residual bacteria in liquid and on worm cuticle. 2 more washing with M9 buffer (0.01% triton X) was done to remove bleach and levamisole solution. After the last wash, an aliquot of liquid volume from each well was transferred to a new flat bottom 96 well plate (Costar 3370, Corning) for bright field imaging under a Nikon TiE Inverted Microscope. Generated images were used to estimate the number and size of adult animals in each well. An aliquot of supernatant from the imaging plate was taken as a negative control to assess background residual live bacteria before host lysis. The remaining worms were then lysed by adding 1.0 mm sterilized garnet beads (Biospect) in a Mixer Mill (Restch) at 25 Hz for 5 min to release live bacteria into solution.
Quantification of bacterial densities using growth curve estimations:
Worm lysates were diluted 10-fold with M9 buffer to reduce debris, and 20 µL of the lysate dilution was inoculated into a 96 well flat bottom plate with 100 µL LB medium. The plate was incubated at 28°C for 18 h. OD600 values were recorded every 15 min to generate bacterial growth curves for each well. Threshold growth time (CGT) at OD600 equal to 0.2 was derived from the corresponding growth curve. Total bacterial cells in each well were calculated based on the BIGbiome equation with corresponding CGT number. Colonization level per animal was then calculated using the following formula:
Measurement of gut microbiome composition in C. elegans
Collection and lysis of animals:
Worm lysate from the previous step by centrifuged at 4000 x g for 10 min. extraction, a freeze-thaw process in −80°C freezer overnight was first applied, then 0.1 mm sterile zirconia/silica beads (BioSpec products) were added (enough to cover well bottom), bead-beating in Mixer Mill (Restch) at 25 Hz for 5 min to disrupt bacterial cells. Immediately followed by enzymatic treatment of 1 mg/mL proteinase K (NEB) at 60°C for 60 min, then 95°C for 15 min to deactivate the proteinase K. After the treatment, samples were centrifuged at 4000 g for 10 min to pellet down cellular fractions.
Amplicon library construction and sequencing:
Supernatant from lysate was transferred to a clean 96 well PCR plate as DNA template. 16S rRNA gene primer set (515F/806R) targeting variable region 4 in bacteria 81. Barcode information was added to the reverse primer 806r. Amplicons for each library were normalized based on the PCR product quantified by image processing package in Fiji, then pooled into a single tube for Illumina MiSeq. A detailed protocol for high throughput colonization assay can be found on protocols.io (DOI: dx.doi.org/10.17504/protocols.io.rtzd6p6).
Analysis of gut microbiome composition:
Fastq files for each library were split by barcode and quality trimmed in the QIIME software package (v1.9.0) 76 with an average quality score of 30. Chimeras were removed by usearch61 and Greengenes 13.8 database. Resulting fasta files were imported to Deblur 75 with default parameters with all sequences trimmed to 250 bp and positive filter based on 16S rRNA sequences of the 63 strains in the core microbiome. A phylogenetic tree with all Amplicon Sequence Variant (ASV) detected was generated using maximum likelihood method in Mega7 with default parameters. Diversity indices were computed in QIIME using core_diversity_analyses.py with default parameters and rarefied to 3,000 sequences. Alpha diversity was determined using Faith’s phylogenetic diversity and Beta-diversity (between samples) distance matrices were computed within QIIME using default parameters. Phylogenetic-based weighted UniFrac metric was used to compare compositional overlap between worm microbiomes and BIGbiome lawns; the weighted UniFrac metric refers to the degree of overlap in two communities as a function of taxa abundance and shared branches on a combined phylogenetic tree 82. Large ‘distances’ indicate less overlap and distinct community compositions. A detailed working pipeline can be found in Bitbucket [See Key Resource Table for link].
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial Strains | ||
| Escherichia coli OP50 | Caenorhabditis Genetics Center | OP50 |
| Ochrobactrum pituitosum BH3 | Buck Samuel 68 | BH3 |
| Acinetobacter sp. BIGb0102 | Buck Samuel 16 | BIGb0102 |
| Leucobacter sp. BIGb0106 | Buck Samuel 16 | BIGb0106 |
| Leucobacter sp. BIGb0117 | Buck Samuel 16 | BIGb0117 |
| Ramlibacter sp. BIGb0124 | Buck Samuel 16 | BIGb0124 |
| Raoultella sp. BIGb0138 | Buck Samuel 16 | BIGb0138 |
| Stenotrophomonas sp. BIGb0145 | Buck Samuel 16 | BIGb0145 |
| Citrobacter sp. BIGb0149 | Buck Samuel 16 | BIGb0149 |
| Yersinia sp. BIGb0156 | Buck Samuel 16 | BIGb0156 |
| Limnohabitans sp. BIGb0172 | Buck Samuel 16 | BIGb0172 |
| Chryseobacterium sp. BIGb0186 | Buck Samuel 16 | BIGb0186 |
| Citrobacter sp. BIGb0188 | Buck Samuel 16 | BIGb0188 |
| Erwinia sp. BIGb0193 | Buck Samuel 16 | BIGb0193 |
| Acinetobacter sp. BIGb0196 | Buck Samuel 16 | BIGb0196 |
| Citrobacter sp. BIGb0211 | Buck Samuel 16 | BIGb0211 |
| Chryseobacterium sp. BIGb0215 | Buck Samuel 16 | BIGb0215 |
| Stenotrophomonas sp. BIGb0219 | Buck Samuel 16 | BIGb0219 |
| Lactococcus sp. BIGb0220 | Buck Samuel 16 | BIGb0220 |
| Yersinia sp. BIGb0236 | Buck Samuel 16 | BIGb0236 |
| Myroides sp. BIGb0244 | Buck Samuel 16 | BIGb0244 |
| Citrobacter sp. BIGb0267 | Buck Samuel 16 | BIGb0267 |
| Pseudomonas sp. BIGb0272 | Buck Samuel 16 | BIGb0272 |
| Pseudomonas sp. BIGb0273 | Buck Samuel 16 | BIGb0273 |
| Enterobacter sp. BIGb0359 | Buck Samuel 16 | BIGb0359 |
| Enterobacter sp. BIGb0383 | Buck Samuel 16 | BIGb0383 |
| Erwinia sp. BIGb0393 | Buck Samuel 16 | BIGb0393 |
| Raoultella sp. BIGb0399 | Buck Samuel 16 | BIGb0399 |
| Pseudomonas sp. BIGb0404 | Buck Samuel 16 | BIGb0404 |
| Pseudomonas sp. BIGb0408 | Buck Samuel 16 | BIGb0408 |
| Erwinia sp. BIGb0435 | Buck Samuel 16 | BIGb0435 |
| Pseudomonas sp. BIGb0470 | Buck Samuel 16 | BIGb0470 |
| Pseudomonas sp. BIGb0473 | Buck Samuel 16 | BIGb0473 |
| Pseudomonas sp. BIGb0477 | Buck Samuel 16 | BIGb0477 |
| Providencia sp. BIGb0506 | Buck Samuel 16 | BIGb0506 |
| Pseudomonas sp. BIGb0525 | Buck Samuel 16 | BIGb0525 |
| Buttiauxella sp. BIGb0552 | Buck Samuel 16 | BIGb0552 |
| Gluconobacter sp. BIGb0611 | Buck Samuel 16 | BIGb0611 |
| Enterobacter sp. JUb101 | Marie-Anne Félix 16 | JUb101 |
| Providencia sp. JUb102 | Marie-Anne Félix 16 | JUb102 |
| Arthrobacter sp. JUb115 | Marie-Anne Félix 16 | JUb115 |
| Leucobacter sp. JUb18 | Marie-Anne Félix 16 | JUb18 |
| Stenotrophomonas sp. JUb19 | Marie-Anne Félix 16 | JUb19 |
| Sphingobacterium sp. JUb20 | Marie-Anne Félix 16 | JUb20 |
| Stenotrophomonas sp. JUb23 | Marie-Anne Félix 16 | JUb23 |
| Pseudomonas sp. JUb28 | Marie-Anne Félix 16 | JUb28 |
| Enterobacter sp. JUb30 | Marie-Anne Félix 16 | JUb30 |
| Curtobacterium sp. JUb34 | Marie-Anne Félix 16 | JUb34 |
| Providencia sp. JUb39 | Marie-Anne Félix 16 | JUb39 |
| Chryseobacterium sp. JUb44 | Marie-Anne Félix 16 | JUb44 |
| Neorhizobium sp. JUb45 | Marie-Anne Félix 16 | JUb45 |
| Pseudomonas sp. JUb52 | Marie-Anne Félix 16 | JUb52 |
| Yersinia sp. JUb53 | Marie-Anne Félix 16 | JUb53 |
| Raoultella sp. JUb54 | Marie-Anne Félix 16 | JUb54 |
| Sphingobacterium sp. JUb56 | Marie-Anne Félix 16 | JUb56 |
| Limnohabitans sp. JUb58 | Marie-Anne Félix 16 | JUb58 |
| Curtobacterium sp. JUb65 | Marie-Anne Félix 16 | JUb65 |
| Enterobacter sp. JUb66 | Marie-Anne Félix 16 | JUb66 |
| Sphingobacterium sp. JUb78 | Marie-Anne Félix 16 | JUb78 |
| Delftia sp. JUb8 | Marie-Anne Félix 16 | JUb8 |
| Rhodococcus sp. JUb83 | Marie-Anne Félix 16 | JUb83 |
| Pseudomonas sp. JUb85 | Marie-Anne Félix 16 | JUb85 |
| Acinetobacter sp. JUb89 | Marie-Anne Félix 16 | JUb89 |
| Pseudomonas sp. JUb96 | Marie-Anne Félix 16 | JUb96 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: Natural isolate: AB1 | Caenorhabditis Genetics Center 18 | AB1 |
| C. elegans: Natural isolate: AB3 | Caenorhabditis Genetics Center 18 | AB3 |
| C. elegans: Natural isolate: CB4853 | Caenorhabditis Genetics Center 18 | CB4853 |
| C. elegans: Natural isolate: CB4854 | Caenorhabditis Genetics Center 18 | CB4854 |
| C. elegans: Natural isolate: CB4856 | Caenorhabditis Genetics Center 18 | CB4856 |
| C. elegans: Natural isolate: ED3017 | Caenorhabditis Genetics Center 18 | ED3017 |
| C. elegans: Natural isolate: ED3021 | Caenorhabditis Genetics Center 18 | ED3021 |
| C. elegans: Natural isolate: ED3040 | Caenorhabditis Genetics Center 18 | ED3040 |
| C. elegans: Natural isolate: ED3042 | Caenorhabditis Genetics Center 18 | ED3042 |
| C. elegans: Natural isolate: ED3052 | Caenorhabditis Genetics Center 18 | ED3052 |
| C. elegans: Natural isolate: ED3072 | Caenorhabditis Genetics Center 18 | ED3072 |
| C. elegans: Natural isolate: GXW0001 | Caenorhabditis Genetics Center 18 | GXW0001 |
| C. elegans: Natural isolate: JU1088 | Caenorhabditis Genetics Center 18 | JU1088 |
| C. elegans: Natural isolate: JU1171 | Caenorhabditis Genetics Center 18 | JU1171 |
| C. elegans: Natural isolate: JU1218 | Caenorhabditis Genetics Center 18 | JU1218 |
| C. elegans: Natural isolate: JU1400 | Caenorhabditis Genetics Center 18 | JU1400 |
| C. elegans: Natural isolate: JU1401 | Caenorhabditis Genetics Center 18 | JU1401 |
| C. elegans: Natural isolate: JU1652 | Caenorhabditis Genetics Center 18 | JU1652 |
| C. elegans: Natural isolate: JU258 | Caenorhabditis Genetics Center 18 | JU258 |
| C. elegans: Natural isolate: JU263 | Caenorhabditis Genetics Center 18 | JU263 |
| C. elegans: Natural isolate: JU300 | Caenorhabditis Genetics Center 18 | JU300 |
| C. elegans: Natural isolate: JU312 | Caenorhabditis Genetics Center 18 | JU312 |
| C. elegans: Natural isolate: JU322 | Caenorhabditis Genetics Center 18 | JU322 |
| C. elegans: Natural isolate: JU323 | Caenorhabditis Genetics Center 18 | JU323 |
| C. elegans: Natural isolate: JU360 | Caenorhabditis Genetics Center 18 | JU360 |
| C. elegans: Natural isolate: JU361 | Caenorhabditis Genetics Center 18 | JU361 |
| C. elegans: Natural isolate: JU397 | Caenorhabditis Genetics Center 18 | JU397 |
| C. elegans: Natural isolate: JU533 | Caenorhabditis Genetics Center 18 | JU533 |
| C. elegans: Natural isolate: JU642 | Caenorhabditis Genetics Center 18 | JU642 |
| C. elegans: Natural isolate: JU775 | Caenorhabditis Genetics Center 18 | JU775 |
| C. elegans: Natural isolate: KR314 | Caenorhabditis Genetics Center 18 | KR314 |
| C. elegans: Natural isolate: LKC34 | Caenorhabditis Genetics Center 18 | LKC34 |
| C. elegans: Natural isolate: MY1 | Caenorhabditis Genetics Center 18 | MY1 |
| C. elegans: Natural isolate: MY14 | Caenorhabditis Genetics Center 18 | MY14 |
| C. elegans: Natural isolate: MY16 | Caenorhabditis Genetics Center 18 | MY16 |
| C. elegans: Natural isolate: MY2 | Caenorhabditis Genetics Center 18 | MY2 |
| C. elegans: Natural isolate: PX174 | Caenorhabditis Genetics Center 18 | PX174 |
| C. elegans: Wild-type: N2 | Caenorhabditis Genetics Center | N2 |
| daf-2(e1370) | Caenorhabditis Genetics Center | CB1370 43 |
| daf-16(mgDf50) | Caenorhabditis Genetics Center | GR1307 69 |
| daf-16(mgDf50);daf-2(e1370) | Caenorhabditis Genetics Center | HT1890 47 |
| unc-119(ed3);wgIs201 | Caenorhabditis Genetics Center | OP201 70 |
| daf-16(mgDf50);unc-119(ed3);lpIs14 | Caenorhabditis Genetics Center | HT1889 47 |
| Chemicals and Commercial Assays | ||
| Triton X-100 | Sigma-Aldrich | Cat#: T8787 |
| Trizol | Thermo-Fisher | Cat#: 15596026 |
| Garnet beads (1.0 mm) | Biospect | Cat#: 11079110gar |
| Silica beads (0.1 mm) | Biospect | Cat#: 11079101Z |
| Proteinase K | New England Biolabs | Cat#: P8107S |
| Nematode Growth Medium | RPI | Cat#: N81800–1000.0 |
| Levamisole | Fisher | Cat#: AC187870100 |
| Carbenicillin | Sigma-Aldrich | Cat#: C1389 |
| IPTG | Sigma-Aldrich | Cat#: I6758 |
| MirVANA total RNA kit | Thermo-Fisher | Cat#: A27828 |
| Miseq (paired-end 250bp) | Illumina | N.A. |
| Hiseq4000 (paired-end 150bp) | Illumina | N.A. |
| Deposited Data | ||
| Code for data analysis | Bitbucket | https://bitbucket.org/the-samuel-lab/natural-variation/src/master/ |
| Original 16S rRNA amplicon of gut microbiome sequences | NCBI | Bioproject PRJNA540192 (SAMN13068200–13068238, 13071563–13071602, 16597785–16597833, 16611296–16611371, 17054579–17054627) |
| Original RNAseq data from wild worms | NCBI | Bioproject PRJNA540192 (SAMN13050735–13050742) |
| Software and Algorithms | ||
| RStudio | GNU | Version 1.3.1093 |
| ggplot: Various R Programming Tools for Plotting Data. | R package | Version 3.3.2 |
| ggbeeswarm | R package | Version 0.6.1 |
| ggtern | R package | Version 3.1.0 |
| FASTQC | 71 | Version 0.11.9 |
| bbmap | JGI-DOE | N.A. |
| bbduk | JGI-DOE | N.A. |
| kallisto | 72 | Version 0.45.0 |
| DESeq2 | 73 | Version 1.30.0 |
| WormExp | 29 | Version 1.0 |
| WormCat | 27 | N.A. |
| Worm machine | 74 | N.A. |
| CeNDR | 53 | Version 1.2.9 |
| ImageJ | NIH | Version 2.0.0 |
| Deblur | 75 | Version 1.0.2 |
| QIIME | 76 | Version 1.8.0 |
| Oligonucleotides | ||
| primer set (515F/806R) for 16S rRNA | 77,78 | N.A. |
GWAS analyses of genetic associations with gut microbiome abundance
The Caenorhabditis elegans Natural Diversity Resource (CeNDR) was used to perform GWAS 53 using the EMMA algorithm via the rrBLUP package 83,84. The EMMA algorithm used within CeNDR takes into account prevalent linkage disequilibrium observed in C. elegans 85. The gut microbiome taxa abundance values and C. elegans strain names were used as input for GWAS. The CeNDR version used was 1.2.9, with data release 20180527 and cegwas version 1.01. Version WS263 of the worm genome was used in this data release. Representative strains for isotypes with more than one strain tested were randomly selected prior GWAS analyses in CeNDR.
RNAi knockdown of C. elegans genes
L1 animals were grown on NGM plates with 25 µg/ml carbenicillin and 1 mM IPTG and seeded with 30 µL (OD=1) of E. coli HT115 expressing dsRNA to C. elegans target genes. To separate exposures to E. coli and BIGbiome communities, RNAi treated gravid adults were treated with bleach solution to generate synchronized L1 progeny. Around 100 L1 animals (RNAi F1s) were transferred to NGM plates with BIGbiome lawn to assess gut microbiome colonization and composition after 120 hrs. Previous studies have shown that progeny typically maintain the RNAi-mediated silencing for at least one generation 86. Natural variation in RNAi effectiveness in wild strains of C. elegans was also assessed by measuring adult body size following dpy-13 RNAi knockdowns, and no significant differences were observed [JU1400(vector): 1360±154 µm n=16, JU1400(dpy-13): 632±156 µm n=15, N2(vector): 1373±199 µm n=30, N2(dpy-13): 655±185 µm n=25, ED3017(vector): 1396±236 µm n=25, ED3017(dpy-13): 687±173 µm n=22, Data S1N].
Transcriptional profiling of C. elegans animals
RNA isolation, library preparation and sequencing:
C. elegans strains [CB4853, ED3017, ED3040, ED3042, JU258, JU775, JU300, JU1400, LKC34, MY14, and N2] for RNAseq were grown on BIGbiome in triplicate for 120 hrs at 20ºC. Animals were then washed off plates using M9 buffer (plus 0.01% triton X-100), and progeny were removed by filtering through a sterile 40 µm Nylon mesh (Fisher Scientific). Approximately 500 adult worms were aliquoted in 1.5 mL Eppendorf tubes and placed on ice for 1 min to settle animals, then combined with 200µL of Trizol and 10–20 1.0mm garnet beads. Animals were lysed using a Mixer-mill (Restch) at 25 Hz for 5 min and then incubated at 4ºC for 5 min. 200 µL of chloroform was then added to each tube and vortexed for 30 s to mix, and allowed to incubate at room temperature for 3 minutes. Debris was removed by centrifugation at 13,800 x g for 15 minutes at 4ºC, and supernatants (~200 µL) were transferred into RNase-free Eppendorf tubes and stored at −80 ºC until extraction. Frozen supernatants were thawed at room temperature and loaded to a KingFisher flex purification system (Thermo Scientific) for automatic RNA processing using MirVANA total RNA kit (Thermo Scientific) following the manufacturer’s protocol. Purified RNAs were stored at −20ºC in elution buffer until use. Aliquots of RNA (0.5–2 µg) were used for creation of RNA sequencing libraries and sequenced by Illumina HiSeq4000 (paired end 150bp reads; QuickBiology).
RNAseq processing and analysis:
An average of 19,638,470 reads were obtained from each dataset, and samples with less than 2 replicates were not utilized in analyses. RNAseq result quality was examined using FASTQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/; 71 ), and reads were filtered and trimmed using bbmap (https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbmap-guide/) and bbduk, respectively (https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk-guide/; https://sourceforge.net/projects/bbmap/). Reads that did not map to the C. elegans genome (build WBcel235) with high quality were removed from the analysis [3.8–5.6% of reads for each dataset]. This was based on an internal evolutionary probability model score ‘minid’, which was set to 0.92, and described in more detail in the bb tools user guide referenced above. Acceptable reads were trimmed using bbduk with the following parameters: ktrim=r, k-23, mink=11, and hdist=1. These and other parameters are described in detail in the bb tools user guide referenced above. Filtered and mapped reads (average 19,586,505 per dataset) were pseudoaligned to the WBcel235 genome assembly using kallisto [https://pachterlab.github.io/kallisto/; 72] with default settings [89.0–93.3% aligned for each dataset]. DESeq2 was used to estimate differential expression in an R workspace [https://bioconductor.org/packages/release/bioc/html/DESeq2.html; 73]. Briefly, DESeq2 models raw counts, normalizes to library depth, estimates and shrinks gene-wise dispersions, and fits a negative binomial model to estimate differential expression based on a Likelihood Ratio Test. Genes with an adjusted p-value of 0.05 or smaller and expressional change greater than two-fold were considered differentially expressed and used in further analyses.
Gene set enrichment analyses:
WormExp [https://wormexp.zoologie.uni-kiel.de/wormexp/; 29] was used for gene set enrichment analyses compared to a comprehensive database of over 1700 curated gene expression datasets in C. elegans. Significance for enrichment scores are calculated using the method developed for the program EASE 87 and reported as uncorrected p-value, Bonferroni-corrected p-value, and False Discovery Rate. Terms were considered significant if the WormExp-reported FDR score was less than 0.05. WormCat [http://wormcat.com; 27] is a similar nematode-specific enrichment analysis and visualization tool that allows for easy categorization and interpretation of datasets based on gene ontology (GO) terms. WormCat is designed for identification of gene sets that are coexpressed or cofunctioning, allowing for drilled-down analysis of specific pathways. Significance scores are reported as Fisher’s exact test p-values. Terms were considered significant if the WormCat-reported P-Value score was less than 0.05.
Quantification of animal body size and Ochrobactrum gut colonization
Microscopy-based quantification:
GFP expressing Ochrobactrum were used to visualize the colonization of this bacterium. Brightfield and fluorescent images taken by a Nikon TiE Inverted Microscope were imported to MATLAB based WorMachine74. A mask was generated for individual worms with default parameters from brightfield images. Worm length and GFP intensity for each mask were measured and compared using one-way ANOVA and Tukey HSD post hoc test in R packages.
Biosorter-based quantification:
Animals on Day 3 adulthood were collected, washed, paralyzed, and surface bleached as described in the gut microbiome colonization steps, then transferred with 150 µl M9 buffer to a flat bottom 96 well plate (Costar 3370, Corning). Individual body size (time of flight, TOF) and level of GFP intensity were measured by a COPAS Biosorter (Union Biometrica) with a 250 micron flow cell and Sapphire488 laser at 310 volt and 1.0 pmt gain settings. Individual events were gated by a combination of TOF and extinction coefficient to filter adult animals from the population. GFP values normalized by TOF from each host strains and RNAi knockdown conditions were compared using one-way ANOVA and Tukey HSD post hoc test in R packages.
Developmental timing assays
Approximately 40 synchronized L1 worms were added to the plates containing BIGbiome mixtures or OP50. Animals were scored every 2 hrs for the number of adult animals on plate from 44 to 60 h at 20°C. Four replicates were scored for each condition, n > 100 animals were scored per strain/condition. Percentages of adults between the three microbiome types from the same time points were compared using one-way ANOVA and Tukey HSD post hoc test R packages.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNAseq analyses
See Method Details for explanation of software and programs used. In brief, reads were checked for quality with FASTQC, filtered for quality with bbmap, trimmed with bbduk, aligned with kallisto, and analyzed for differential expression with DESEQ2 73. Genes were considered differentially expressed between microbiome types if the Benjamini-Hochberg adjusted p-value was less than 0.05. The work was completed locally using a Late 2013 Mac Pro (3.5 GHz 6-Core Intel Xeon E5) and software including Mac Terminal (fastqc, bbmap, bbduk, kallisto) and Rstudio (DESeq2).
Gene set enrichment analyses
The WormExp tool uses a statistical approach designed for gene list interpretation, EASE 87 to determine statistical significance. Default parameters were used and produced adjusted p-values based on False Discovery Rate (FDR) estimations. Adjusted P-values less than 0.05 were considered significant.
Rationale for statistical tools used within the WormCat tool are described in detail in Holdorf, et al., 27. Briefly, WormCat produces Fisher’s exact test p-values. The method was chosen after providing few false positives without being too stringent in a randomized test of 100, 500, 1000, or 1500 genes. In our analyses, results were considered significant if the P-value score output from the WormCat online tool was less than 0.05.
Correlation analyses of microbial taxa abundance and gene expression
Pearson correlation was calculated between absolute abundance of each microbial taxa and expression of each gene for every strain of C. elegans using the ‘stats’ package within RStudio using default parameters. Correlations were considered significant if the Benjamini-Hochberg adjusted p-value was less than 0.05.
Supplementary Material
This table contains raw data to support all of the Figures 1–6, plus supplemental figures and tables in the manuscript. A summary tab describes data types and corresponding figure panels.
INCLUSION AND DIVERSITY.
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
ACKNOWLEDGEMENTS
This work was supported by NIH grants DP2DK116645 (to B.S.S). This project was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the CPRIT Core Facility Support Award (CPRIT-RP180672), the NIH (S10 OD025251, CA125123, and RR024574) and the assistance of Joel M. Sederstrom, plus an instrumentation grant for the Biosorter NIH grant (S10 OD025251). Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). Emily Troemel generously provided Ochrobactrum BH3 and isogenic GFP expressing strains. We also thank Gretchen Diehl Lab, Joseph Hyser Lab, Rachel Arey Lab, Meng Wang Lab and Houston Area Worm Group and Center for Metagenomics and Microbiome Research labs for helpful advice at various stages of this project, plus Diehl and Estes labs for sharing key equipment and the CMMR core facility for microbiome sequencing and Rachel Arey for helpful comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Shin SC, Kim S-H, You H, Kim B, Kim AC, Lee K-A, Yoon J-H, Ryu J-H, and Lee W-J (2011). Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 334, 670–674. [DOI] [PubMed] [Google Scholar]
- 2.Chaparro JM, Badri DV, and Vivanco JM (2014). Rhizosphere microbiome assemblage is affected by plant development. ISME J 8, 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dominguez-Bello MG, Godoy-Vitorino F, Knight R, and Blaser MJ (2019). Role of the microbiome in human development. Gut 68, 1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansen J, Gulati A, and Sartor RB (2010). The role of mucosal immunity and host genetics in defining intestinal commensal bacteria. Curr. Opin. Gastroenterol 26, 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snijders AM, Langley SA, Kim Y-M, Brislawn CJ, Noecker C, Zink EM, Fansler SJ, Casey CP, Miller DR, Huang Y, et al. (2016). Influence of early life exposure, host genetics and diet on the mouse gut microbiome and metabolome. Nat. Microbiol 2, 16221. [DOI] [PubMed] [Google Scholar]
- 6.Hall AB, Tolonen AC, and Xavier RJ (2017). Human genetic variation and the gut microbiome in disease. Nat. Rev. Genet 18, 690–699. [DOI] [PubMed] [Google Scholar]
- 7.Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, Deelen P, Vatanen T, Schirmer M, Smeekens SP, et al. (2016). The effect of host genetics on the gut microbiome. Nat. Genet 48, 1407–1412. [DOI] [PubMed] [Google Scholar]
- 8.Goodrich JK, Davenport ER, Clark AG, and Ley RE (2017). The Relationship Between the Human Genome and Microbiome Comes into View. Annu. Rev. Genet 51, 413–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frézal L, and Félix M-A C. elegans outside the Petri dish. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Girard LR, Fiedler TJ, Harris TW, Carvalho F, Antoshechkin I, Han M, Sternberg PW, Stein LD, and Chalfie M (2007). WormBook: the online review of Caenorhabditis elegans biology. Nucleic Acids Res 35, D472–D475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulenburg H, Kurz CL, and Ewbank JJ (2004). Evolution of the innate immune system: the worm perspective. Immunol. Rev 198, 36–58. [DOI] [PubMed] [Google Scholar]
- 12.Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan M-W, et al. (2002). A Conserved p38 MAP Kinase Pathway in Caenorhabditis elegans Innate Immunity. Science 297, 623–626. [DOI] [PubMed] [Google Scholar]
- 13.MacNeil L, Watson E, Arda HE, Zhu LJ, and Walhout AJM (2013). Diet-Induced Developmental Acceleration Independent of TOR and Insulin in C. elegans. Cell 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sterken MG, Snoek LB, Kammenga JE, and Andersen EC (2015). The laboratory domestication of Caenorhabditis elegans. Trends Genet. TIG 31, 224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berg M, Stenuit B, Ho J, Wang A, Parke C, Knight M, Alvarez-Cohen L, and Shapira M (2016). Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. ISME J 10, 1998–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samuel BS, Rowedder H, Braendle C, Félix M-A, and Ruvkun G (2016). Caenorhabditis elegans responses to bacteria from its natural habitats. Proc. Natl. Acad. Sci 113, E3941–E3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dirksen P, Marsh SA, Braker I, Heitland N, Wagner S, Nakad R, Mader S, Petersen C, Kowallik V, Rosenstiel P, et al. (2016). The native microbiome of the nematode Caenorhabditis elegans: gateway to a new host-microbiome model. BMC Biol 14, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson O, Edgley M, Strasbourger P, Flibotte S, Ewing B, Adair R, Au V, Chaudhry I, Fernando L, Hutter H, et al. (2013). The million mutation project: A new approach to genetics in Caenorhabditis elegans. Genome Res 23, 1749–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang F, Berg M, Dierking K, Félix M-A, Shapira M, Samuel BS, and Schulenburg H (2017). Caenorhabditis elegans as a Model for Microbiome Research. Front. Microbiol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dirksen P, Assié A, Zimmermann J, Zhang F, Tietje A-M, Marsh SA, Félix M-A, Shapira M, Kaleta C, Schulenburg H, et al. (2020). CeMbio - The Caenorhabditis elegans Microbiome Resource. G3 Bethesda Md 10, 3025–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Portal-Celhay C, Bradley ER, and Blaser MJ (2012). Control of intestinal bacterial proliferation in regulation of lifespan in Caenorhabditis elegans. BMC Microbiol 12, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulenburg H, and Félix M-A (2017). The Natural Biotic Environment of Caenorhabditis elegans. Genetics 206, 55–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang W, Petersen C, Pees B, Zimmermann J, Waschina S, Dirksen P, Rosenstiel P, Tholey A, Leippe M, Dierking K, et al. (2019). The Inducible Response of the Nematode Caenorhabditis elegans to Members of Its Natural Microbiota Across Development and Adult Life. Front. Microbiol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimmermann J, Obeng N, Yang W, Pees B, Petersen C, Waschina S, Kissoyan KA, Aidley J, Hoeppner MP, Bunk B, et al. (2020). The functional repertoire contained within the native microbiota of the model nematode Caenorhabditis elegans. ISME J 14, 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vega NM, and Gore J (2017). Stochastic assembly produces heterogeneous communities in the Caenorhabditis elegans intestine. PLOS Biol 15, e2000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brunder W, and Karch H (2000). Genome plasticity in Enterobacteriaceae. Int. J. Med. Microbiol 290, 153–165. [DOI] [PubMed] [Google Scholar]
- 27.Holdorf AD, Higgins DP, Hart AC, Boag PR, Pazour GJ, Walhout AJM, and Walker AK (2020). WormCat: An Online Tool for Annotation and Visualization of Caenorhabditis elegans Genome-Scale Data. Genetics 214, 279–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR, and Ristow M (2012). Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab 15, 451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang W, Dierking K, and Schulenburg H (2016). WormExp: a web-based application for a Caenorhabditis elegans-specific gene expression enrichment analysis. Bioinforma. Oxf. Engl 32, 943–945. [DOI] [PubMed] [Google Scholar]
- 30.Sinha A, Rae R, Iatsenko I, and Sommer RJ (2012). System Wide Analysis of the Evolution of Innate Immunity in the Nematode Model Species Caenorhabditis elegans and Pristionchus pacificus. PLOS ONE 7, e44255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pujol N, Zugasti O, Wong D, Couillault C, Kurz CL, Schulenburg H, and Ewbank JJ (2008). Anti-fungal innate immunity in C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog 4, e1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engelmann I, Griffon A, Tichit L, Montañana-Sanchis F, Wang G, Reinke V, Waterston RH, Hillier LW, and Ewbank JJ (2011). A Comprehensive Analysis of Gene Expression Changes Provoked by Bacterial and Fungal Infection in C. elegans. PLOS ONE 6, e19055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, and Kim DH (2006). p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet 2, e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engelmann I, and Pujol N (2010). Innate Immunity in C. elegans. In Invertebrate Immunity Advances in Experimental Medicine and Biology, Söderhäll K, ed. (Springer US; ), pp. 105–121. [DOI] [PubMed] [Google Scholar]
- 35.Murphy CT, Lee S-J, and Kenyon C (2007). Tissue entrainment by feedback regulation of insulin gene expression in the endoderm of Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A 104, 19046–19050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaplan REW, Maxwell CS, Codd NK, and Baugh LR (2019). Pervasive Positive and Negative Feedback Regulation of Insulin-Like Signaling in Caenorhabditis elegans. Genetics 211, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abreu D.A.F. de, Caballero A, Fardel P, Stroustrup N, Chen Z, Lee K, Keyes WD, Nash ZM, López-Moyado IF, Vaggi F, et al. (2014). An Insulin-to-Insulin Regulatory Network Orchestrates Phenotypic Specificity in Development and Physiology. PLOS Genet 10, e1004225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tepper RG, Ashraf J, Kaletsky R, Kleemann G, Murphy CT, and Bussemaker HJ (2013). PQM-1 complements DAF-16 as a key transcriptional regulator of DAF-2-mediated development and longevity. Cell 154, 676–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dowen RH, Breen PC, Tullius T, Conery AL, and Ruvkun G (2016). A microRNA program in the C. elegans hypodermis couples to intestinal mTORC2/PQM-1 signaling to modulate fat transport. Genes Dev 30, 1515–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pees B, Yang W, Zárate-Potes A, Schulenburg H, and Dierking K (2016). High Innate Immune Specificity through Diversified C-Type Lectin-Like Domain Proteins in Invertebrates. J. Innate Immun 8, 129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baugh LR, and Sternberg PW (2006). DAF-16/FOXO regulates transcription of cki-1/Cip/Kip and repression of lin-4 during C. elegans L1 arrest. Curr. Biol. CB 16, 780–785. [DOI] [PubMed] [Google Scholar]
- 42.Kimura KD, Tissenbaum HA, Liu Y, and Ruvkun G (1997). daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942–946. [DOI] [PubMed] [Google Scholar]
- 43.Kenyon C, Chang J, Gensch E, Rudner A, and Tabtiang R (1993). A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464. [DOI] [PubMed] [Google Scholar]
- 44.Lee K, and Mylonakis E (2017). An Intestine-Derived Neuropeptide Controls Avoidance Behavior in Caenorhabditis elegans. Cell Rep 20, 2501–2512. [DOI] [PubMed] [Google Scholar]
- 45.Paradis S, and Ruvkun G (1998). Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev 12, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans EA, Chen WC, and Tan M-W (2008). The DAF-2 insulin-like signaling pathway independently regulates aging and immunity in C. elegans. Aging Cell 7, 879–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kwon E-S, Narasimhan SD, Yen K, and Tissenbaum HA (2010). A new DAF-16 isoform regulates longevity. Nature 466, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gami MS, Iser WB, Hanselman KB, and Wolkow CA (2006). Activated AKT/PKB signaling in C. elegans uncouples temporally distinct outputs of DAF-2/insulin-like signaling. BMC Dev. Biol 6, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang P, Judy M, Lee S-J, and Kenyon C (2013). Direct and Indirect Gene Regulation by a Life-Extending FOXO Protein in C. elegans: Roles for GATA Factors and Lipid Gene Regulators. Cell Metab 17, 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miltsch SM, Seeberger PH, and Lepenies B (2014). The C-type lectin-like domain containing proteins Clec-39 and Clec-49 are crucial for Caenorhabditis elegans immunity against Serratia marcescens infection. Dev. Comp. Immunol 45, 67–73. [DOI] [PubMed] [Google Scholar]
- 51.Hsu A-L, Murphy CT, and Kenyon C (2003). Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300, 1142–1145. [DOI] [PubMed] [Google Scholar]
- 52.McHugh DR, Koumis E, Jacob P, Goldfarb J, Schlaubitz-Garcia M, Bennani S, Regan P, Patel P, and Youngman MJ (2020). DAF-16 and SMK-1 Contribute to Innate Immunity During Adulthood in Caenorhabditis elegans. G3 Genes Genomes Genet 10, 1521–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cook DE, Zdraljevic S, Roberts JP, and Andersen EC (2017). CeNDR, the Caenorhabditis elegans natural diversity resource. Nucleic Acids Res 45, D650–D657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hill JH, Franzosa EA, Huttenhower C, and Guillemin K (2016). A conserved bacterial protein induces pancreatic beta cell expansion during zebrafish development. eLife 5, e20145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Im E, Jung J, Pothoulakis C, and Rhee SH (2014). Disruption of Pten speeds onset and increases severity of spontaneous colitis in Il10(−/−) mice. Gastroenterology 147, 667–679.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mortzfeld BM, Taubenheim J, Fraune S, Klimovich AV, and Bosch TCG (2018). Stem Cell Transcription Factor FoxO Controls Microbiome Resilience in Hydra. Front. Microbiol 9, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu T, Zimmerman KK, and Patterson GI (2004). Regulation of signaling genes by TGFβ during entry into dauer diapause in C. elegans. BMC Dev. Biol 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Narasimhan SD, Yen K, Bansal A, Kwon E-S, Padmanabhan S, and Tissenbaum HA (2011). PDP-1 Links the TGF-β and IIS Pathways to Regulate Longevity, Development, and Metabolism. PLOS Genet 7, e1001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shaw WM, Luo S, Landis J, Ashraf J, and Murphy CT (2007). The C. elegans TGF-beta Dauer pathway regulates longevity via insulin signaling. Curr. Biol. CB 17, 1635–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berg M, Monnin D, Cho J, Nelson L, Crits-Christoph A, and Shapira M (2019). TGFβ/BMP immune signaling affects abundance and function of C. elegans gut commensals. Nat. Commun 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blackwell TK, Steinbaugh MJ, Hourihan JM, Ewald CY, and Isik M (2015). SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic. Biol. Med 88, 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mizunuma M, Neumann-Haefelin E, Moroz N, Li Y, and Blackwell TK (2014). mTORC2-SGK-1 acts in two environmentally responsive pathways with opposing effects on longevity. Aging Cell 13, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gravato-Nobre MJ, Nicholas HR, Nijland R, O’Rourke D, Whittington DE, Yook KJ, and Hodgkin J (2005). Multiple genes affect sensitivity of Caenorhabditis elegans to the bacterial pathogen Microbacterium nematophilum. Genetics 171, 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ewald CY, Landis JN, Porter Abate J, Murphy CT, and Blackwell TK (2015). Dauerin-dependent insulin/IGF-1-signalling implicates collagen remodelling in longevity. Nature 519, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McFall-Ngai M, Hadfield MG, Bosch TCG, Carey HV, Domazet-Lošo T, Douglas AE, Dubilier N, Eberl G, Fukami T, Gilbert SF, et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U. S. A 110, 3229–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnke J, Dirksen P, and Schulenburg H (2020). Community assembly of the native C. elegans microbiome is influenced by time, substrate and individual bacterial taxa. Environ. Microbiol 22, 1265–1279. [DOI] [PubMed] [Google Scholar]
- 67.Taylor M, and Vega NM (2020). Host immunity alters successional ecology and stability of the microbiome in a C. elegans model. bioRxiv, 2020.06.26.174706. [DOI] [PMC free article] [PubMed]
- 68.Troemel ER, Félix M-A, Whiteman NK, Barrière A, and Ausubel FM (2008). Microsporidia Are Natural Intracellular Parasites of the Nematode Caenorhabditis elegans. PLOS Biol 6, e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, and Ruvkun G (1997). The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389, 994–999. [DOI] [PubMed] [Google Scholar]
- 70.Zhong M, Niu W, Lu ZJ, Sarov M, Murray JI, Janette J, Raha D, Sheaffer KL, Lam HYK, Preston E, et al. (2010). Genome-Wide Identification of Binding Sites Defines Distinct Functions for Caenorhabditis elegans PHA-4/FOXA in Development and Environmental Response. PLOS Genet 6, e1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Andrews S (2010). FastQC: a quality control tool for high throughput sequence data
- 72.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- 73.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hakim A, Mor Y, Toker IA, Levine A, Neuhof M, Markovitz Y, and Rechavi O (2018). WorMachine: machine learning-based phenotypic analysis tool for worms. BMC Biol 16, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Xu ZZ, Kightley EP, Thompson LR, Hyde ER, Gonzalez A, et al. (2017). Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parada AE, Needham DM, and Fuhrman JA (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol 18, 1403–1414. [DOI] [PubMed] [Google Scholar]
- 78.Apprill A, McNally S, Parsons R, and Weber L (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol 75, 129–137. [Google Scholar]
- 79.Stiernagle T (2006). Maintenance of C. elegans (WormBook) [DOI] [PMC free article] [PubMed]
- 80.Hazan R, Que Y-A, Maura D, and Rahme LG (2012). A method for high throughput determination of viable bacteria cell counts in 96-well plates. BMC Microbiol 12, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lozupone C, and Knight R (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol 71, 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Endelman JB (2011). Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 4, 250–255. [Google Scholar]
- 84.Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, and Eskin E (2008). Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Andersen EC, Gerke JP, Shapiro JA, Crissman JR, Ghosh R, Bloom JS, Félix M-A, and Kruglyak L (2012). Chromosome-scale selective sweeps shape Caenorhabditis elegans genomic diversity. Nat. Genet 44, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Conte D, MacNeil LT, Walhout AJM, and Mello CC (2015). RNA Interference in Caenorhabditis elegans. Curr. Protoc. Mol. Biol 109, 26.3.1–26.3.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hosack DA, Dennis G, Sherman BT, Lane HC, and Lempicki RA (2003). Identifying biological themes within lists of genes with EASE. Genome Biol 4, R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This table contains raw data to support all of the Figures 1–6, plus supplemental figures and tables in the manuscript. A summary tab describes data types and corresponding figure panels.
Data Availability Statement
All datasets have been included as raw data [Data S1]. Sequencing based datasets have been deposited at NCBI Sequence Read Archive database (Bioproject PRJNA540192) with the following sample accession numbers for RNAseq reads (SAMN13050735–13050742) and microbiome sequencing reads (SAMN13068200–13068238, 13071563–13071602, 16597785–16597833, 16611296–16611371, 17054579–17054627). All code used in the analysis of datasets is available through the Bitbucket link, including those for overall microbiome compositional analyses (‘Microbiome_analysis_scripts.txt’), processing and analysis of RNAseq datasets (‘Kallisto_bbmap_bbduk_Script.txt’ and ‘DifferentialExpression_Script.txt’), correlation of microbial taxa with gene expression profiles (‘MicrobialAbundance_vs_GeneExpression_Correlation_Script.txt’) and figure generation in R environment (‘R_plot_figures.txt). (https://bitbucket.org/the-samuel-lab/natural-variation/src/master/).