

Abstract
Background
Hypotrichosis with juvenile macular dystrophy (HJMD) is a rare autosomal recessive inherited disorder caused by biallelic variants in the CDH3 gene encoding P‐cadherin. Here, we report two Japanese sibling patients with HJMD.
Methods
Whole‐exome sequencing (WES) was performed to identify disease‐causing variants. In addition, ophthalmic and dermatological examinations were performed to classify the phenotype of each patient.
Results
The WES analysis revealed novel compound heterozygous CDH3 variants [c.123_129dupAGGCGCG (p.Glu44fsX26) and c.2280+1G>T] in both patients; the unaffected, nonconsanguineous parents each exhibited one of the variants. Both patients showed the same clinical findings. Ophthalmologically, they exhibited progressive loss of visual acuity and chorioretinal macular atrophy, as examined with fundoscopy, fundus autofluorescence imaging, and optical coherence tomography. Full‐field electroretinography, assessing generalized retinal function, revealed nearly normal amplitudes of both rod‐ and cone‐mediated responses. Multifocal electroretinography, reflecting macular function, showed extremely decreased responses in the central area, corresponding to the chorioretinal atrophy. Dermatological examination revealed diffuse thinning of the scalp hair, which was sparse and fragile.
Conclusion
This is the first report of Japanese patients with HJMD and novel compound heterozygous truncating variants in CDH3. Our findings can expand the knowledge and understanding of CDH3‐related HJMD, which could be helpful to ophthalmologists and dermatologists.
Keywords: CDH3, electroretinography, hypotrichosis, Japanese, macular dystrophy, retina
This is the first report of Japanese patients with hypotrichosis with juvenile macular dystrophy (HJMD) and novel compound heterozygous truncating variants in the CDH3 gene. Our findings can expand the knowledge and understanding of CDH3‐related HJMD.

1. INTRODUCTION
Hypotrichosis with juvenile macular dystrophy (HJMD, OMIM #601553) is a rare form of an autosomal recessive multi‐organ disorder, first described in 1935 (Wagner, 1935). The Cadherin 3 gene (CDH3, OMIM *114021), encoding P‐cadherin, was first reported to cause HJMD in 2001 (Sprecher et al., 2001) and was subsequently shown to be linked with its closely associated phenotypes, ectodermal dysplasia, ectrodactyly, and macular dystrophy (EEM syndrome, OMIM # 225280) in 2005 (Kjaer et al., 2005). P‐cadherin plays an important role in cell‐to‐cell adhesion (Angst et al., 2001; Shapiro & Weis, 2009); it is expressed at the cell‐to‐cell border of retinal pigment epithelium (RPE) cells in the human retina (Xu et al., 2002; Yang et al., 2018) and in the hair follicle placode during mouse hair development (Shimomura et al., 2008). Patients with HJMD exhibit hair abnormalities, including short and sparse scalp hair, and progressive macular atrophy (Hull et al., 2016; Indelman et al., 2002; Leibu et al., 2006).
The CDH3 gene is located on the long arm of chromosome 16 (16q22) and has 16 coding exons that span a genomic region of approximately 55 kilobase pairs. To date, 35 pathogenic variants of CDH3 have been reported in the Human Gene Mutation Database (HGMD Professional as of January 2020; http://www.hgmd.cf.ac.uk/ac/index.php). Most cases are found in the Middle‐Eastern population, including in Israeli, Turkish, Arab, and Pakistani populations, while some cases have been found in North American and European populations (Hull et al., 2016; Indelman et al., 2003, 2007; Karti et al., 2017). However, to date, CDH3‐related HJMD/EEM syndrome has not been reported in the Japanese and East Asian populations. Here, we encountered two siblings/patients with HJMD in a single Japanese family. The purpose of this study was to describe the clinical and genetic features of these patients with HJMD and biallelic CDH3 variants.
2. MATERIALS AND METHODS
2.1. Molecular genetic analysis
The Institutional Review Boards of The Jikei University (approval no. 24‐231 6997) and Nippon Medical School Chiba Hokusoh Hospital (approval no. 27‐04) approved the study protocol. The protocol adhered to the tenets of the Declaration of Helsinki, and informed consent was obtained from both the siblings and their unaffected, non‐consanguineous parents. Genomic DNA was extracted from the leucocytes of all the participants, and whole‐exome sequencing (WES) and targeted sequence analyses were performed according to the previously described methods (Katagiri et al., 2013, 2014). All single nucleotide variants (SNVs) and insertions/deletions (INDELs) among the 271 genes registered on the RetNet (https://sph.uth.edu/retnet/), a database of genes causing inherited retinal diseases, were filtered according to allele frequency (less than 0.1%) of the East Asian population in 1000 genome database. CDH3 was the only gene in which two heterozygous variants were identified. The allelic frequency of the variants was estimated in reference to three databases; the Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org), the Human Genetic Variation Database (HGVD) (http://www.hgvd.genome.med.kyoto‐u.ac.jp), and Japanese Multi Omics Reference Panel (jMorp) (https://jmorp.megabank.tohoku.ac.jp/202001/). The CDH3 variants were confirmed by direct sequencing of all family members. The following primer sets for exons 2 and 15 were used: exon 2 forward primer 5′‐ AGGTTTGCTGGCTGCAGTGC ‐3′ and reverse primer 5′‐ GGTCCACACCAAAATGGTCA ‐3′, and exon 15 forward primer 5′‐ CCCATGAGCCAGAGTATCCA ‐3′ and reverse primer 5′‐ ACTCCAGGCCCATGCTTGTT ‐3′. We used the transcript (NM_001793.6) and genomic (NG_009096.1) sequences of CDH3.
3. RESULTS
3.1. Clinical findings
A 22‐year‐old female proband (patient II‐2, JU#1166 in Figure 1a) was referred to The Jikei University Hospital for decreased visual acuity in both eyes (BE). She exhibited a tendency to lose hair easily, and reported congenital absence of scalp hair lasting until early childhood, and decreased visual acuity at 17 years of age. At presentation, her decimal best‐corrected visual acuity (BCVA) was 0.8 (Snellen equivalent, 20/25) with −3.50 diopters (D) in the right eye (RE) and 0.8 with −2.50 D in the left eye (LE). Slit‐lamp examination showed no remarkable findings in anterior segments and media. Horizontal optical coherence tomography (OCT) (Cirrus HD‐OCT; Carl Zeiss Meditec AG, Dublin, CA, USA) showed extensive atrophy of the retina, RPE, and choroid, relatively sparing the fovea, in which the ellipsoid zone (EZ) was preserved (Figure 2a). Outer retinal tubulation secondary to outer retinal damage and focal choroidal excavation were observed in the LE (Figure 2a). Fundoscopy revealed symmetrical chorioretinal atrophy within the vascular arcade in BE (Figure 2b), while fundus autofluorescence imaging (FAI) (Spectralis HRA; Heidelberg Engineering, Heidelberg, Germany) showed loss of autofluorescence, corresponding to the chorioretinal atrophy, and slight hyper‐autofluorescence around the area of autofluorescence loss (Figure 2c). Goldmann perimetry (GP) showed central scotomas (10° to 20°) with the I‐4e stimulus and normal peripheral visual fields in BE (Figure 3a). Full‐field electroretinography (FF‐ERG) to assess generalized retinal function was performed in accordance with the protocols of the International Society for Clinical Electrophysiology of Vision (McCulloch et al., 2015). The procedure and conditions followed have been previously reported (Kutsuma et al., 2019; Takeuchi et al., 2010). FF‐ERG indicated that the amplitudes of dark‐adapted rod ERG (stimulation, 0.01 cd·s·m−2) and combined rod‐cone ERG (stimulation, 3.0 cd·s·m−2) were normal when compared with those of our age‐matched controls (Kutsuma et al., 2019), whereas the amplitudes of light‐adapted cone ERG (stimulation, 3.0 cd·s·m−2; background, 30 cd·m−2) and 30‐Hz flicker ERG (stimulation, 3.0 cd·s·m−2; background, 30 cd·m−2) were in the lower limit compared with those of the age‐matched controls (Figure 1d). Multifocal ERG (VERIS Science; Electro‐Diagnostic Imaging, Inc. Redwood City, CA, USA), reflecting macular function, showed extremely decreased responses in central areas, but normal responses outside the central areas in BE (Figure 1e). Dermatological examination revealed diffuse thinning of the scalp hair, which was sparse and fragile. At 26 years old, her BCVA was 0.2 (Snellen equivalent, 20/100) in RE and 0.7 (Snellen equivalent, 20/29) in LE; ultra‐widefield fundoscopy and FAI (Optos Panoramic 200MA; Optos PLC, Dunfermline, UK) revealed that the chorioretinal atrophy was limited to the posterior pole, and the peripheral retina was preserved (Figure 2d,e). GP showed that central scotomas, with the V‐4e stimulus, progressively worsened in BE (Figure 3b). Moreover, OCT indicated that the foveal thickness was thinner than that at initial examination (Figure 2f).
FIGURE 1.
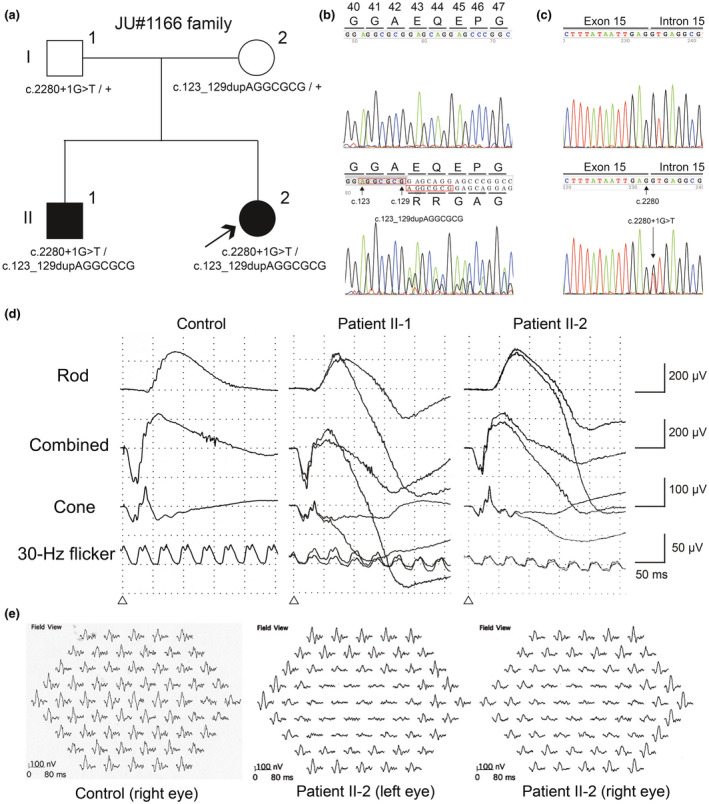
Pedigree chart and genetic and electroretinographic findings. (a) The pedigree of a Japanese family with hypotrichosis with juvenile macular dystrophy. Partial nucleotide sequences of the CDH3 gene showing the c.123_129dupAGGCGCG (b) and c.2280+1G>T (c) variants. The top electropherograms are from a control and the bottom electropherograms are from the patients (II‐1/II‐2) (b,c). (d) Full‐field electroretinography (ERG) of a control, patient II‐1 at 30 years old, and patient II‐2 at 22 years old. (e) Multifocal ERG of a control and patient II‐2 at 22 years old
FIGURE 2.
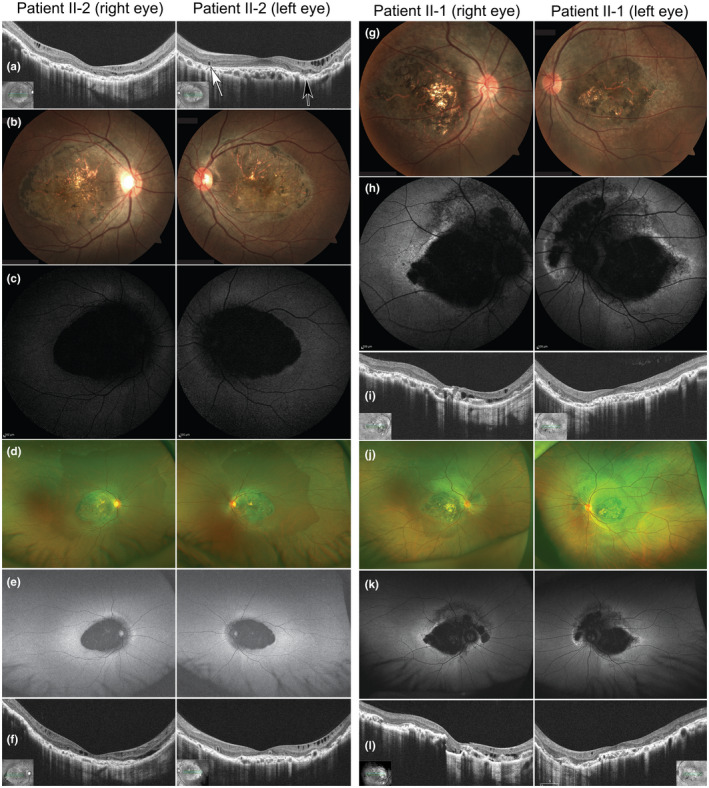
Optical coherence tomography and fundus and fundus autofluorescence images. (a) Optical coherence tomography (OCT) of patient II‐2 at 22 years old. White arrows indicate outer retinal tubulation and the black arrow shows the focal choroidal excavation. Fundus (b) and fundus autofluorescence imaging (FAI) (c) images of patient II‐2 at 24 years old. Ultra‐widefield fundus (d), FAI (e), and OCT (f) images of patient II‐2 at 26 years old. Fundus (g), FAI (h), and OCT (i) images of patient II‐1 at 30 years old. Ultra‐widefield fundus (j) and FAI (k) at 30 years old. (i) OCT of patient II‐1 at 34 years old
FIGURE 3.
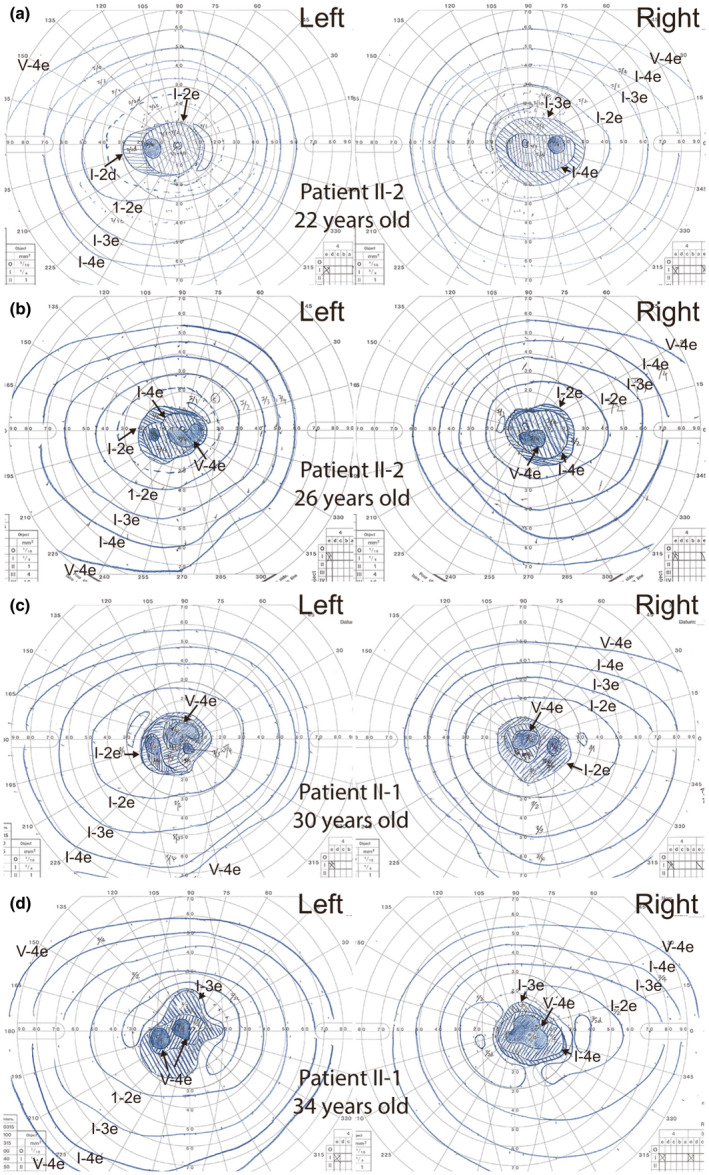
Goldmann visual field testing. (a) Patient II‐2 at 22 years old and (b) 26 years old. (c) Patient II‐1 at 30 years old and (d) 34 years old
A 30‐year‐old elder brother (patient II‐1, JU#1185) was also referred to The Jikei University Hospital. He reported decreased visual acuity at 20 years of age and congenital loss of scalp hair lasting into early childhood. At presentation, his BCVA was 0.1 (Snellen equivalent, 20/200) with −3.75 D in RE and 0.8 with −2.00 D in LE. Slit‐lamp examination showed no remarkable findings. Moreover, the findings of fundoscopy (Figure 2g), FAI (Figure 2h), horizontal OCT images (Figure 2i), ultra‐widefield fundoscopy and FAI (Figure 2j,k), GP (Figure 3c), and FF‐ERG (Figure 1d) were essentially very similar to those of patient II‐2. At 34 years old, his BCVA was 0.07 (Snellen equivalent, 20/286) in RE and 0.5 (Snellen equivalent, 20/40) in LE. Chorioretinal atrophy had not apparently progressed after the initial fundoscopy, GP (Figure 3d), and horizontal OCT (Figure 2l). Further, like his sibling, he also exhibited diffuse thinning of the scalp hair, which were sparse and fragile, as evaluated by dermatological examination. Neither patient exhibited any limb malformation, confirming the diagnosis of HJMD.
3.2. Molecular genetic findings
The WES analyses of patient II‐1 revealed compound heterozygous variants c.123_129dupAGGCGCG [GRCh37/hg19chr16‐68679623‐AGGCGCG (7 bp)] and c.2280+1G>T (GRCh37/hg19chr16‐68729827) in CDH3, and were confirmed by Sanger sequencing (Figure 1b,c). The frameshift variant c.123_129dupAGGCGCG in exon 2 leads to a premature termination codon (p.Glu44fsTer26) (Figure 1b), whereas the splice site variant c.2280+1G>T at the donor site of exon 15 can affect the splicing of exon 15 (Figure 1c). Neither variants were found in the HGMD, HGVD, and jMorp databases. Further, patient II‐2 also exhibited these compound heterozygous variants (Figure 1a). Each variant was found to co‐segregate with the disease in this family (Figure 1a). According to the American College of Medical Genetics standards (Richards et al., 2015), c.123_129dupAGGCGCG (p.Glu44ArgfsTer26) was classified as pathogenic based on the following criteria: PSV [Null variant (frame‐shift) affecting CDH3, which is a known cause of the disease], PM2 (The variant was not found in GnomAD exomes), PM3 (The variant was detected in trans configuration for an autosomal recessive disorder). PP4 (Patient's phenotype was highly specific for CDH3‐related HJMD); and c.2280+1G>T was classified as pathogenic based on the following criteria: PSV [Null variant (within ±2 splice sites) affecting CDH3, which is a known cause of disease], PM2 [The variant was found at extremely low frequency in GnomAD exomes; the allele frequency was 0.00003 (1/31,398 alleles)], PM3, PP3 (Pathogenic computational verdict because there were four pathogenic predictions from DANN, EIGEN, FATHMM‐MKL, and MutationTaster vs. no benign predictions), and PP4. Both pathogenic variants were predicted to cause protein truncation.
4. DISCUSSION
In this report, we described the clinical and genetic findings in two Japanese patients with HJMD. We identified compound heterozygous variants in CDH3 (p.Glu44ArgfsTer26 and c.2280+1G>T), both of which are novel. This report is the first of a Japanese family with CDH3‐related HJMD.
Biallelic CDH3 variants have been reported to cause two different phenotypes: HJMD and EEM syndrome (Basel‐Vanagaite et al., 2010; Hull et al., 2016). However, both phenotypes are considered to be in the same clinical spectrum of the CDH3‐related syndrome. Moreover, no genotype–phenotype correlation has been reported regardless of the variant types and/or positions in CDH3. In fact, some variants, such as c.830delG (p.Gly277AlafsTer20) and c.160+1G>A, are causative for phenotypes of HJMD and EEM syndrome (Hull et al., 2016; Indelman et al., 2003; Kjaer et al., 2005). Therefore, other modifier gene variants and/or environmental factors might modulate the phenotypes.
Abnormal ophthalmic and dermatologic findings of CDH3‐related HJMD and EEM syndrome are common in most cases; for example, progressive chorioretinal atrophy in the posterior pole and preserved peripheral retina and hypotrichosis (Hull et al., 2016; Karti et al., 2017; Leibu et al., 2006; Singh et al., 2016; Vicente et al., 2017), which is consistent with our findings (Figure 2). In terms of disease progression, a 9‐year follow‐up observation of an 18‐year‐old HJMD patient revealed normal FF‐ERG responses, but decreased responses in pattern ERG assessing macular function (Hull et al., 2016). For our patients, the FF‐ERG demonstrated close to normal amplitudes (Figure 1d); however, follow‐up examination showed progressively decreased visual acuity. Furthermore, multifocal ERG showed that the abnormal responses were confined to the central areas (Figure 1e), corresponding to the chorioretinal atrophic lesions. These and other findings demonstrating the normal peripheral visual fields of both patients (Figure 3) indicate that the retinal function outside the chorioretinal macular atrophy is normal. Furthermore, P‐cadherin is expressed in the RPE, and not the photoreceptors; therefore, the decreased central responses (Figure 1e) due to loss of photoreceptors may be secondary to P‐cadherin dysfunction caused by biallelic CDH3 variants. Further follow‐up of the patients will be needed to evaluate if peripheral retinal function and visual fields are preserved.
In conclusion, we report the first two Japanese patients with HJMD and novel compound heterozygous variants in CDH3. Our findings can expand the knowledge and understanding of CDH3‐related genotype–phenotype correlations.
CONFLICT OF INTEREST
All authors declare no competing interests.
AUTHOR CONTRIBUTIONS
T.H., S.K. (Satoshi Katagiri), D.K., K.M., Y.I., A.A., S.K. (Shuhei Kameya), and T.N. contributed to the conception of this work. D.K. and S.K. (Shuhei Kameya) performed molecular genetic analysis. T.H. and S.K. (Satoshi Katagiri) wrote the draft of the manuscript. All authors contributed to manuscript revision and approved the final submitted version.
ACKNOWLEDGMENTS
This work was supported by grants from the Japan Society for the Promotion of Science Grant‐in‐Aid for Scientific Research (17K11434) and Japanese Retinitis Pigmentosa Society (JRPS) Research Grant 2019.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Angst, B. D. , Marcozzi, C. , & Magee, A. I. (2001). The cadherin superfamily: Diversity in form and function. Journal of Cell Science, 114(Pt 4), 629–641. https://www.ncbi.nlm.nih.gov/pubmed/11171368 [DOI] [PubMed] [Google Scholar]
- Basel‐Vanagaite, L. , Pasmanik‐Chor, M. , Lurie, R. , Yeheskel, A. , & Kjaer, K. W. (2010). CDH3‐related syndromes: Report on a new mutation and overview of the genotype‐phenotype correlations. Molecular Syndromology, 1(5), 223–230. 10.1159/000327156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull, S. , Arno, G. , Robson, A. G. , Broadgate, S. , Plagnol, V. , McKibbin, M. , Halford, S. , Michaelides, M. , Holder, G. E. , Moore, A. T. , Khan, K. N. , & Webster, A. R. (2016). Characterization of CDH3‐related congenital hypotrichosis with juvenile macular dystrophy. JAMA Ophthalmology, 134(9), 992–1000. 10.1001/jamaophthalmol.2016.2089 [DOI] [PubMed] [Google Scholar]
- Indelman, M. , Bergman, R. , Petronius, D. , Ciubutaro, D. , Sprecher, E. , Lurie, R. , Richard, G. , Miller, B. , & Leibu, R. (2002). A missense mutation in CDH3, encoding P‐cadherin, causes hypotrichosis with juvenile macular dystrophy. Journal of Investigative Dermatology, 119(5), 1210–1213. 10.1046/j.1523-1747.2002.19528.x [DOI] [PubMed] [Google Scholar]
- Indelman, M. , Bergman, R. , Ramon, M. , Sprecher, E. , Hamel, C. P. , Nischal, K. K. , Thompson, D. , Surget, M.‐O. , Ganthos, H. , Miller, B. , Richard, G. , Lurie, R. , Leibu, R. , & Russell‐Eggitt, I. (2003). Phenotypic diversity and mutation spectrum in hypotrichosis with juvenile macular dystrophy. Journal of Investigative Dermatology, 121(5), 1217–1220. 10.1046/j.1523-1747.2003.12550_1.x [DOI] [PubMed] [Google Scholar]
- Indelman, M. , Eason, J. , Hummel, M. , Loza, O. , Suri, M. , Leys, M. J. , Bayne, M. , Schwartz, F. L. , & Sprecher, E. (2007). Novel CDH3 mutations in hypotrichosis with juvenile macular dystrophy. Clinical and Experimental Dermatology, 32(2), 191–196. 10.1111/j.1365-2230.2006.02335.x [DOI] [PubMed] [Google Scholar]
- Karti, O. , Abali, S. , Ayhan, Z. , Gokmeydan, E. , Nalcaci, S. , Yaman, A. , & Saatci, A. O. (2017). CDH3 gene related hypotrichosis and juvenile macular dystrophy—A case with a novel mutation. American Journal of Ophthalmology Case Reports, 7, 129–133. 10.1016/j.ajoc.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri, S. , Akahori, M. , Sergeev, Y. , Yoshitake, K. , Ikeo, K. , Furuno, M. , Hayashi, T. , Kondo, M. , Ueno, S. , Tsunoda, K. , Shinoda, K. , Kuniyoshi, K. , Tsurusaki, Y. , Matsumoto, N. , Tsuneoka, H. , & Iwata, T. (2014). Whole exome analysis identifies frequent CNGA1 mutations in Japanese population with autosomal recessive retinitis pigmentosa. PLoS ONE, 9(9), e108721. 10.1371/journal.pone.0108721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri, S. , Yoshitake, K. , Akahori, M. , Hayashi, T. , Furuno, M. , Nishino, J. , Ikeo, K. , Tsuneoka, H. , & Iwata, T. (2013). Whole‐exome sequencing identifies a novel ALMS1 mutation (p. Q2051X) in two Japanese brothers with Alström syndrome. Molecular Vision, 19, 2393–2406. https://www.ncbi.nlm.nih.gov/pubmed/24319333 [PMC free article] [PubMed] [Google Scholar]
- Kjaer, K. W. , Hansen, L. , Schwabe, G. C. , Marques‐de‐Faria, A. P. , Eiberg, H. , Mundlos, S. , & Rosenberg, T. (2005). Distinct CDH3 mutations cause ectodermal dysplasia, ectrodactyly, macular dystrophy (EEM syndrome). Journal of Medical Genetics, 42(4), 292–298. 10.1136/jmg.2004.027821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuma, T. , Katagiri, S. , Hayashi, T. , Yoshitake, K. , Iejima, D. , Gekka, T. , Kohzaki, K. , Mizobuchi, K. , Baba, Y. , Terauchi, R. , Matsuura, T. , Ueno, S. , Iwata, T. , & Nakano, T. (2019). Novel biallelic loss‐of‐function KCNV2 variants in cone dystrophy with supernormal rod responses. Documenta Ophthalmologica, 138(3), 229–239. 10.1007/s10633-019-09679-6 [DOI] [PubMed] [Google Scholar]
- Leibu, R. , Jermans, A. , Hatim, G. , Miller, B. , Sprecher, E. , & Perlman, I. (2006). Hypotrichosis with juvenile macular dystrophy: Clinical and electrophysiological assessment of visual function. Ophthalmology, 113(5), 841–847.e843. 10.1016/j.ophtha.2005.10.065 [DOI] [PubMed] [Google Scholar]
- McCulloch, D. L. , Marmor, M. F. , Brigell, M. G. , Hamilton, R. , Holder, G. E. , Tzekov, R. , & Bach, M. (2015). ISCEV standard for full‐field clinical electroretinography (2015 update). Documenta Ophthalmologica, 130(1), 1–12. 10.1007/s10633-014-9473-7 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, L. , & Weis, W. I. (2009). Structure and biochemistry of cadherins and catenins. Cold Spring Harbor Perspectives in Biology, 1(3), a003053. 10.1101/cshperspect.a003053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura, Y. , Wajid, M. , Shapiro, L. , & Christiano, A. M. (2008). P‐cadherin is a p63 target gene with a crucial role in the developing human limb bud and hair follicle. Development, 135(4), 743–753. 10.1242/dev.006718 [DOI] [PubMed] [Google Scholar]
- Singh, M. S. , Broadgate, S. , Mathur, R. , Holt, R. , Halford, S. , & MacLaren, R. E. (2016). Hypotrichosis and juvenile macular dystrophy caused by CDH3 mutation: A candidate disease for retinal gene therapy. Scientific Reports, 6, 23674. 10.1038/srep23674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprecher, E. , Bergman, R. , Richard, G. , Lurie, R. , Shalev, S. , Petronius, D. , Shalata, A. , Anbinder, Y. , Leibu, R. , Perlman, I. , Cohen, N. , & Szargel, R. (2001). Hypotrichosis with juvenile macular dystrophy is caused by a mutation in CDH3, encoding P‐cadherin. Nature Genetics, 29(2), 134–136. 10.1038/ng716 [DOI] [PubMed] [Google Scholar]
- Takeuchi, T. , Hayashi, T. , Bedell, M. , Zhang, K. , Yamada, H. , & Tsuneoka, H. (2010). A novel haplotype with the R345W mutation in the EFEMP1 gene associated with autosomal dominant drusen in a Japanese family. Investigative Ophthalmology & Visual Science, 51(3), 1643–1650. 10.1167/iovs.09-4497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente, L. P. , Finzi, S. , Susanna, R., Jr. , & Young, T. L. (2017). Hypotrichosis with juvenile macular dystrophy: A case report with molecular study. Arquivos Brasileiros de Oftalmologia, 80(1), 49–51. 10.5935/0004-2749.20170013 [DOI] [PubMed] [Google Scholar]
- Wagner, H. (1935). Maculaaffektion, vergesellschaftet mit Haarabnormität von Lanugotypus, beide vielleicht angeboren bei zwei Geschwistern. Albrecht von Graefes Archiv für Ophthalmologie, 134, 74–81. [Google Scholar]
- Xu, L. , Overbeek, P. A. , & Reneker, L. W. (2002). Systematic analysis of E‐, N‐ and P‐cadherin expression in mouse eye development. Experimental Eye Research, 74(6), 753–760. 10.1006/exer.2002.1175 [DOI] [PubMed] [Google Scholar]
- Yang, X. , Chung, J. Y. , Rai, U. , & Esumi, N. (2018). Cadherins in the retinal pigment epithelium (RPE) revisited: P‐cadherin is the highly dominant cadherin expressed in human and mouse RPE in vivo. PLoS ONE, 13(1), e0191279. 10.1371/journal.pone.0191279 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.