

Keywords: hypoxic-ischemic brain damage, in vitro, in vivo, microglia, NADPH, pyroptosis, ROS, TIGAR
Abstract
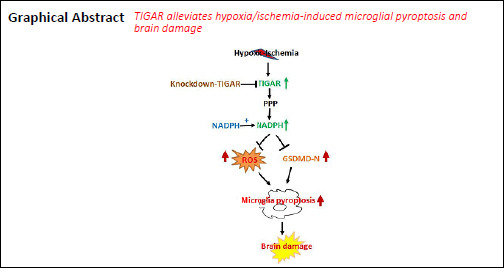
Our previous studies have demonstrated that TP53-induced glycolysis and apoptosis regulator (TIGAR) can protect neurons after cerebral ischemia/reperfusion. However, the role of TIGAR in neonatal hypoxic-ischemic brain damage (HIBD) remains unknown. In the present study, 7-day-old Sprague-Dawley rat models of HIBD were established by permanent occlusion of the left common carotid artery followed by 2-hour hypoxia. At 6 days before induction of HIBD, a lentiviral vector containing short hairpin RNA of either TIGAR or gasdermin D (LV-sh_TIGAR or LV-sh_GSDMD) was injected into the left lateral ventricle and striatum. Highly aggressively proliferating immortalized (HAPI) microglial cell models of in vitro HIBD were established by 2-hour oxygen/glucose deprivation followed by 24-hour reoxygenation. Three days before in vitro HIBD induction, HAPI microglial cells were transfected with LV-sh_TIGAR or LV-sh_GSDMD. Our results showed that TIGAR expression was increased in the neonatal rat cortex after HIBD and in HAPI microglial cells after oxygen/glucose deprivation/reoxygenation. Lentivirus-mediated TIGAR knockdown in rats markedly worsened pyroptosis and brain damage after hypoxia/ischemia in vivo and in vitro. Application of exogenous nicotinamide adenine dinucleotide phosphate (NADPH) increased the NADPH level and the glutathione/oxidized glutathione ratio and decreased reactive oxygen species levels in HAPI microglial cells after oxygen/glucose deprivation/reoxygenation. Additionally, exogenous NADPH blocked the effects of TIGAR knockdown in neonatal HIBD in vivo and in vitro. These findings show that TIGAR can inhibit microglial pyroptosis and play a protective role in neonatal HIBD. The study was approved by the Animal Ethics Committee of Soochow University of China (approval No. 2017LW003) in 2017.
Chinese Library Classification No. R453; R741; Q255
Introduction
Hypoxic-ischemic brain damage (HIBD) constitutes one of the major causes of high neonatal morbidity and neurological sequelae in fetuses and newborn infants worldwide (Psychogios et al., 2017; Jiang et al., 2020). Therapeutic hypothermia is widely used and is the gold standard of neonatal HIBD treatment (Chalak and Kaiser, 2007; Davies et al., 2019). However, this therapy has the shortcomings of a narrow therapeutic window and incomplete neuroprotection (Liu et al., 2017). Thus, further study is required to elucidate the underlying mechanism of HIBD and ultimately seek novel and potential therapeutic targets and approaches for HIBD treatment.
Pyroptosis, which is a novel type of proinflammatory programmed cell death, plays an important role in the pathological process of neurological diseases, such as Alzheimer’s disease and ischemic stroke (Ito et al., 2015; Barrington et al., 2017; Yagami et al., 2019). Pyroptosis is induced by the proteolytic activation of caspase-1 through cleaving the gasdermin D (GSDMD) N-terminal (GSDMD-N) (Kepp et al., 2010; Liu et al., 2018). GSDMD-N interacts with intimal lipids to form pores on the plasma membrane, which causes the cell membrane to rupture and release the pro-inflammatory mediator interleukin-1β (IL-1β) and ultimately leads to cell death (Sborgi et al., 2016; McKenzie et al., 2018; Xu et al., 2019b). Microglia, the resident innate immune cells in the central nervous system, are associated with the inflammatory response (Xu et al., 2019a; Xie et al., 2021). Microglial activation is considered to contribute to triggering neuroinflammation, reactive oxygen species (ROS) production, and cerebral injury via the release of pro-inflammatory cytokines (Arioz et al., 2019; Mo et al., 2020). Recent studies have shown that inflammasome-mediated microglial pyroptosis contributes to brain injury in neurological diseases including ischemic stroke, Parkinson’s disease, and traumatic brain injury (Sarkar et al., 2017; Lee et al., 2019; Poh et al., 2019). Inhibition of inositol-requiring enzyme 1α alleviates neuronal pyroptosis through the miR-125/NLR family pyrin domain containing 1 pathway and reduces neonatal rat hypoxic-ischemic brain injury (Huang et al., 2020). Recently, we found that inhibition of NLR family pyrin domain containing 3 (NLRP-3)/caspase-1/GSDMD axis-mediated microglial pyroptosis decreased brain injury in neonatal rats after HIBD (Lv et al., 2020), which implies that microglial pyroptosis plays a crucial role in neonatal HIBD. However, the underlying mechanisms that mediate microglial pyroptosis remain largely unclear.
TP53-induced glycolysis and apoptosis regulator (TIGAR) decreases fructose-2,6-bisphosphate levels in cells, subsequently leading to inhibition of glycolysis and a decrease in intracellular ROS levels (Bensaad et al., 2006). We previously found that TIGAR played a neuroprotective role in ischemic stroke by elevating the flux of the pentose phosphate pathway (PPP) (Li et al., 2014). Recently, we further demonstrated that TIGAR played a passive role in the astrocyte-mediated inflammatory response after ischemic stroke (Chen et al., 2018). Therefore, we hypothesized that TIGAR suppressed microglial pyroptosis and protected against neonatal HIBD by decreasing ROS. The present study tested this hypothesis in a neonatal rat hypoxic-ischemic brain injury model in vivo and in highly aggressively proliferating immortalized (HAPI) microglial cells in vitro.
Materials and Methods
Neonatal HIBD model
All animal experiments were conducted according to the institutional guidelines for animal care and use. Neonatal rats were freely fed by their mothers. The room temperature during feeding was 24 ± 2°C. The rats were maintained on a 12-hour light/dark cycle. The study protocol was approved by the Animal Ethics Committee of Soochow University of China (approval No. 2017LW003) in 2017. One hundred and fifty-three neonatal Sprague-Dawley rats (12–17 g, 7 days old) were purchased from Zhaoyan (Suzhou) New Drug Research Center Co., Ltd. (Suzhou, China, License No. SCXK (Su) 2018-0006). Neonatal rats were randomly divided into sham and HIBD groups. After viral infection, the HIBD group was divided into normal control, model, LV-sh_TIGAR, LV-sh_GSDMD, NADPH, and LV-sh_TIGAR + NADPH groups. The HIBD model was established according to a modified Vannucci method as described previously (Ding et al., 2015; Patel et al., 2015). Briefly, the left common carotid artery was permanently ligated in rats. After 1 hour of recovery with their mothers, the rats were subjected to hypoxia (8% O2/92 % N2) at 37°C for 2 hours and then returned to their mothers for free feeding. Sham-operated rats underwent the same surgery but were not ligated or subjected to hypoxia. Neonatal rats were euthanized at the indicated time point after hypoxic-ischemic brain injury.
Oxygen and glucose deprivation/reoxygenation
HAPI rat microglial cell lines were purchased from the Cell Bank of Enzyme-Linked Biotechnology Co., Ltd. (Shanghai, China). HAPI microglial cells were cultured in Dulbecco’s modified Eagle medium (Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Biological Industries, Kibbutz Beit-Haemek, Israel), 100 U/mL penicillin and 100 mg/mL streptomycin in a humidified atmosphere at 37°C with 5% CO2. The HAPI microglial cells were incubated with glucose-free Earle’s balanced salt solution and then placed in a chamber (ELECTROTEK, West Yorkshire, UK) with 95% N2 and 5% CO2 at 37°C for 2 hours. Control cells were incubated with glucose-free Earle’s balanced salt solution and then placed in normal culture conditions for 2 hours. Two hours after oxygen and glucose deprivation (OGD), the medium was replaced with normal culture medium (reoxygenation), and the cells were cultured at 37°C with 5% CO2 for 24 hours.
Viral infection and drug treatment in vitro and in vivo
For viral infection in vivo, 6 days prior to HIBD, a vector containing TIGAR short hairpin RNA (LV-sh_TIGAR; 1×109 transduction units/mL; Genechem, Shanghai, China) or GSDMD short hairpin RNA (LV-sh_GSDMD; 1 × 109 transduction U/mL; Genechem) was infused into the left lateral ventricle and striatum (2 μL/injection site) (Li et al., 2014) of the neonatal rats. Nicotinamide adenine dinucleotide phosphate (NADPH; 2.5 mg/kg; Beyotime, Nantong, China) was injected into the left lateral ventricle of the rat brain before HIBD modeling.
For viral infection in vitro, the HAPI rat microglial cells were treated with medium containing LV-sh_TIGAR (MOI = 100), LV-sh_GSDMD (MOI = 100), or LV-sh-scramble (normal control; MOI = 100; Genechem), and the medium was replaced with regular medium after 12 hours of viral treatment. The cells were cultured for 3 days and then subjected to OGD/reoxygenation (OGD/R). NADPH (10 μM; Beyotime) was added before OGD and was present in the regular medium used for reoxygenation at 24 hours.
In vitro cell death assay
Cell death was detected using a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) and a lactate dehydrogenase cytotoxicity assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s protocols.
Measurement of NADPH, glutathione, and ROS levels
The levels of NADPH and glutathione (GSH) in cultured HAPI microglial cells were measured with an NADPH/NADP+ kit (Beyotime) and a GSH/oxidized GSH (GSSG) kit (BioAssay Systems, San Francisco, CA, USA) following the manufacturer’s instructions. The ROS levels in HAPI microglial cells were detected using dihydroethidium staining (1 μM; Sigma, St. Louis, MO, USA) as previously described (Cao et al., 2017).
Western blot assay
Neonatal rats were deeply anesthetized with anhydrous 2-butanol and then sacrificed. Cortical tissues of the neonatal rat brain and HAPI microglial cells were lysed into homogenate in lysis buffer, and the extracted proteins (20 μg) were separated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred to nitrocellulose membranes (Millipore, Burlington, MA, USA). Membranes were incubated with mouse anti-TIGAR (1:1000; Cat# sc-166290; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-Caspase-1 (1:1000; Cat# ab207802; Abcam, Cambridge, UK), rabbit anti-GSDMD (1:1000; Cat# ab219800; Abcam), anti-GSDMD-N (1:1000; Cat# ab215203; Abcam), rabbit anti-IL-1β (1:1000; Cat# ab9787; Abcam), or mouse anti-β-actin (1:10 000; Cat# ab008; MULTI SCIENCES, Hangzhou, China) overnight at 4°C. The membranes were then incubated with peroxidase-conjugated rabbit/mouse secondary antibodies (1:5000; Cat# 111-035-003/115-035-003; Jackson, Lancaster, PA, USA), and bands were detected with an AmerSham Imager 600 (GE, Boston, MA, USA) and analyzed using ImageJ analysis software (National Institutes of Health, Bethesda, MD, USA).
2,3,5-Triphenyltetrazolium chloride staining
Cerebral lesion infarctions were measured 24 hours after HIBD by 2,3,5-triphenyltetrazolium chloride staining (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) as described previously (Cao et al., 2017). The rats were sacrificed under deep anesthesia with anhydrous 2-butanol. The brain was removed immediately and sliced into parallel 2-mm-thick slices, immersed in 2,3,5-triphenyltetrazolium chloride solution for 5 minutes, and fixed in 4% paraformaldehyde solution. The cerebral infarction lesion was measured as a percentage of the total hemisphere.
Hematoxylin and eosin staining and immunohistochemistry
Twenty-four hours after HIBD, the rat brains were removed and soaked in 20% and 30% sucrose for gradient dehydration. Brain sections (10 μm) were cut with a paraffin slicer (SLEE, Mainz, Germany) and then stained with hematoxylin and eosin. Briefly, the brain sections were immersed in xylene and alcohol and were then stained with hematoxylin. After washing with tap water, the sections were stained with eosin and sealed with neutral gum.
Brain sections (4 μm) were subjected to immunohistochemistry. The brain sections were immersed in xylene, anhydrous ethanol, and alcohol. The sections were then incubated with rabbit anti-GSDMD-N antibody (1:500; Cell Signaling Technology, Inc., Beverly, MA, USA) overnight at 4°C and were next incubated with HRP-labeled goat anti-rabbit IgG (1:200; Cat# GB23303; Servicebio, Wuhan, China). Finally, the brain sections were stained with the chromogenic agent 3,3-diaminobenzidine.
The number of viable surviving cells was measured as the number of cells per millimeter (1-mm) length in brain slices via light microscopy (BX40; Olympus, Tokyo, Japan) by two investigators who were blinded to the experimental conditions. Digitized images were obtained using a light microscope and quantified using ImageJ analysis software.
Electron microscope observation
Twenty-four hours after HIBD, neonatal rats in each group were anesthetized with anhydrous 2-butanol, and the cerebral cortex was removed and cut into 1 mm 3pieces. The pieces were quickly placed into a pre-cooled 2.5% glutaraldehyde solution and stored overnight in a refrigerator at 4°C. The brain tissue was fixed with a mixture of 4% paraformaldehyde solution and 0.5% glutaraldehyde, processed by embedding, sectioning, and staining as previously described (Zhang et al., 2019), and then detected using transmission electron microscopy (Olympus).
Statistical analysis
Statistical analysis was performed with Prism 5 (GraphPad Software, San Diego, CA, USA). All data are presented as the mean ± standard error of the mean (SEM) and evaluated with one-way analysis of variance with Bonferroni’s multiple-comparisons post hoc test. A value of P < 0.05 indicates statistical significance.
Results
TIGAR exerts a protective effect in neonatal HIBD models in vivo and in vitro
First, to explore whether TIGAR expression was induced in the neonatal rat cortex by HIBD in vivo, we detected the TIGAR level after HIBD. As shown in Figure 1A, TIGAR expression was increased in the neonatal rat cortex after HIBD, reaching a peak level at 24 hours (P < 0.05, P < 0.01, or P < 0.001). Next, to assess the role of TIGAR in neonatal HIBD rats, we performed LV-sh_TIGAR-mediated knockdown of TIGAR in neonatal rat brains. The results showed that TIGAR knockdown by LV-sh_TIGAR decreased TIGAR expression (P < 0.01) and markedly exacerbated the brain lesion fraction 24 hours after HIBD compared with the model group (P < 0.001; Figure 1B and C). Meanwhile, TIGAR expression was increased in OGD/R-induced HAPI microglial cells (P < 0.05 or P < 0.001; Figure 1D). Moreover, TIGAR knockdown by LV-sh_TIGAR not only significantly decreased TIGAR expression in HAPI microglial cells (P < 0.001; Figure 1E) but also aggravated cell viability and lactate dehydrogenase release (P < 0.01 or P < 0.001; Figure 1F and G). These results suggested that TIGAR exhibited a protective role in neonatal HIBD.
Figure 1.

TIGAR exerts a protective effect on neonatal HIBD models in vivo and in vitro.
(A) Quantification of TIGAR expression in neonatal rat cortex 24 hours after HIBD. (B) The expression of TIGAR after LV-sh_TIGAR transfection in the neonatal rat cortex. (C) Quantification of the infarct area in neonatal rats detected using 2,3,5-triphenyltetrazolium chloride staining 24 hours after HIBD. Red indicates normal tissue and white indicates ischemic tissue. (D) Expression of TIGAR in HAPI microglial cells 24 hours after OGD/R. (E) TIGAR protein expression after LV-sh_TIGAR transfection in HAPI microglial cells. (F) Cell viability was detected using a CCK-8 kit 24 hours after OGD/R. (G) Cell death was detected by LDH release 24 hours after OGD/R. Increased LDH release results in increased toxicity to cells and increased cell death. Data are expressed as the mean ± SEM (A, B, D–G: n = 3 animals/group; C: n = 6 animals/group). *P < 0.05, **P < 0.01, ***P < 0.001, vs. sham group; #P < 0.05, ###P < 0.001, vs. control group; §§P < 0.01, §§§P < 0.001 (one-way analysis of variance followed by Bonferroni’s multiple-comparisons post hoc test). CCK-8: Cell Counting Kit-8; Con: control; HIBD: hypoxic-ischemic brain damage; LDH: lactate dehydrogenase; LV-sh_TIGAR: vector containing short hairpin RNA of TIGAR; Model: HIBD; NC: LV-sh-scramble; OD: optical density; OGD(/R): oxygen and glucose deprivation(/reoxygenation); TIGAR: TP53 induced glycolysis and apoptosis regulator; HAPI: highly aggressively proliferating immortalized.
Pyroptosis is activated and increased by TIGAR knockdown in the cerebral cortex and HAPI microglial cells after neonatal HIBD modeling
To explore the effect of TIGAR on HIBD-induced activation of pyroptosis in vivo, we first examined the expression of pyroptosis-related proteins in the neonatal cortex after HIBD. The levels of caspase-1, GSDMD-N, and IL-1β were markedly increased after HIBD and peaked 24 hours after HIBD (P < 0.05, P < 0.01, or P < 0.001; Figure 2A). Meanwhile, GSDMD knockdown in neonatal brains by injecting LV-sh_GSDMD significantly reduced infarction lesions 24 hours after HIBD compared with the model group (P < 0.001; Figure 2B). Moreover, the in vitro levels of caspase-1, GSDMD-N, and IL-1β in HAPI microglial cells were markedly increased 24 hours after OGD/R (P < 0.05, P < 0.01, or P < 0.001; Figure 2C), and the levels of these proteins were further elevated by knockdown of TIGAR by LV-sh_TIGAR 24 hours after OGD/R (P < 0.01 or P < 0.001; Figure 2D).
Figure 2.

TIGAR knockdown increases the activation of pyroptosis in the neonatal cortex and HAPI microglial cells of HIBD models in vivo and in vitro.
(A) Quantification of caspase-1, GSDMD-N, and IL-1β in neonatal rat cortex 24 hours after HIBD. (B) The lesion infarct was detected using 2,3,5-triphenyltetrazolium chloride staining 24 hours after HIBD. Red indicates normal tissue and white indicates ischemic tissue. (C) Quantification of caspase-1, GSDMD-N, and IL-1β in HAPI microglial cells 24 hours after OGD/R. (D) Quantification of TIGAR, caspase-1, GSDMD-N, and IL-1β in HAPI microglial cells with or without LV-sh_TIGAR treatment 24 hours after OGD/R. Data are expressed as the mean ± SEM (A, C, D: n = 3 animals/group; B: n = 6 animals/group). *P < 0.05, **P < 0.01, ***P < 0.001, vs. sham group; #P < 0.05, ##P < 0.01, ###P < 0.001, vs. control group; §§P < 0.01, §§§P < 0.001 (one-way analysis of variance followed by Bonferroni’s multiple-comparisons post hoc test). Con: Control; GSDMD-N: gasdermin D N-terminal; HAPI: highly aggressively proliferating immortalized; HIBD: hypoxic-ischemic brain damage; IL-1β: interleukin-1β; LV-sh_TIGAR: vector containing short hairpin RNA of TIGAR; Model: HIBD; NC: LV-sh-scramble; OGD(/R): oxygen and glucose deprivation(/reoxygenation); TIGAR: TP53 induced glycolysis and apoptosis regulator.
TIGAR knockdown increases microglial pyroptosis through increasing ROS in vitro
Next, we explored the underlying mechanism of TIGAR-mediated pyroptosis in HAPI microglial cells after OGD/R in vitro. The results showed that TIGAR knockdown with LV-sh_TIGAR significantly accelerated OGD/R-induced cell death and lactate dehydrogenase release, and the effects were alleviated by NADPH treatment (P < 0.01 or P < 0.001; Figure 3A and B). OGD/R markedly decreased the NADPH level and the GSH/GSSG ratio Figure 3C and D) and increased ROS levels Figure 3E). This effect was further aggravated by TIGAR knockdown with LV-sh_TIGAR (P < 0.05 or P < 0.001) but was reduced by NADPH treatment (P < 0.05, P < 0.01, or P < 0.001). Moreover, TIGAR knockdown by LV-sh_TIGAR observably elevated the increase in GSDMD-N and IL-1β levels induced by OGD/R (P < 0.01 or P < 0.001), while this was prevented by NADPH treatment (P < 0.001; Figure 3F).
Figure 3.

TIGAR knockdown promotes microglial pyroptosis by increasing reactive oxygen species in vitro.
HAPI microglial cells were treated with LV-sh_TIGAR and subjected to OGD/R. Exogenous NADPH was added to HAPI microglial cells before OGD. (A) Cell viability was detected using a CCK-8 kit 24 hours after OGD/R. (B) Cell death was detected by LDH release 24 hours after OGD/R. Increased LDH release results in increased toxicity to cells and increased cell death. (C, D) The levels of NADPH (C) and rGSH (D) in HAPI microglial cells 24 hours after OGD/R. (E) Reactive oxygen species production was detected using DHE staining (red) 24 hours after OGD/R. Scale bar: 100 μm. (F) Quantification of TIGAR, GSDMD-N, and IL-1β in HAPI microglial cells 24 hours after OGD/R. Relative protein expression is shown as the ratio of the optical density of the target protein to that of β-actin. Data are expressed as the mean ± SEM (n = 3/group). §P < 0.05, §§P < 0.01, §§§P < 0.001 (one-way analysis of variance followed by Bonferroni’s multiple-comparisons post hoc test). CCK-8: Cell Counting Kit-8; Con: Control; DHE: dihydroethidium; GSDMD-N: gasdermin D N-terminal; HIBD: hypoxic-ischemic brain damage; IL-1β: interleukin-1β; LDH: lactate dehydrogenase; LV-sh_TIGAR: vector containing short hairpin RNA of TIGAR; NADPH: nicotinamide adenine dinucleotide phosphate; NC: LV-sh-scramble; OGD(/R): oxygen and glucose deprivation(/reoxygenation); TIGAR: TP53 induced glycolysis and apoptosis regulator; HAPI: highly aggressively proliferating immortalized; rGSH: reduced glutathione.
TIGAR knockdown-aggravated neonatal brain injury after HIBD is suppressed by NADPH
We further investigated the influence of the PPP product NADPH on the protective effects of TIGAR in neonatal rat HIBD in vivo. The results showed that the lesion infarcts induced by TIGAR knockdown by LV-sh_TIGAR 24 hours after HIBD were markedly alleviated by NADPH administration (P < 0.05, P < 0.01, or P < 0.001; Figure 4A). Meanwhile, TIGAR knockdown by LV-sh_TIGAR obviously reduced the number of surviving cells in the neonatal rat cortex compared with the model group (P < 0.01), and this effect was blocked by NADPH administration 24 hours after HIBD (P < 0.05 or P < 0.01; Figure 4B). Moreover, electron microscopy showed that TIGAR knockdown by LV-sh_TIGAR aggravated the formation of membrane pores and cell membrane rupture compared with the model group; however, this effect was reduced by NADPH administration (Figure 4C). Furthermore, immunohistochemical staining showed that GSDMD-N immunopositivity was upregulated in the neonatal rat cortex 24 hours after HIBD, and this effect was further elevated by TIGAR knockdown by LV-sh_TIGAR (P < 0.01) but prevented by NADPH administration (P < 0.05; Figure 4D).
Figure 4.

Administration of NADPH reduces brain injury induced by TIGAR knockdown after neonatal HIBD.
Neonatal rat brains were injected with NC or LV-sh_TIGAR and then were subjected to HIBD after 7 days. NADPH (2.5 mg/kg) was injected at the onset of HIBD. (A) Quantitative analysis of the infarct lesion 24 hours after HIBD detected using 2,3,5-triphenyltetrazolium chloride staining. Red indicates normal tissue, and white indicates ischemic tissue. (B) Representative hematoxylin and eosin staining images and quantitative analysis of viable surviving cells in brain tissue 24 hours after HIBD. The number of viable surviving cells was calculated as the number of cells per mm (1-mm) length of the brain slices by light microscopy. Organelles are normal and neatly arranged, and the nuclei are clearly stained in the center of surviving cells. (C) Representative transmission electron microscopy images from the neonatal rat ischemic cortex 24 hours after HIBD. Red arrows indicate cell membrane rupture and membrane pore formation. (D) Quantitative analysis of GSDMD-N immunopositivity in brain tissue 24 hours after HIBD. Scale bars: 500 μm in B, 1 μm in C, 50 μm in D. Data are expressed as the mean ± SEM (A: n = 6 animals/group; B, D: n = 3 animals/group). §P < 0.05, §§P < 0.01 (one-way analysis of variance followed by Bonferroni’s multiple-comparisons post hoc test). GSDMD-N: Gasdermin D N-terminal; HIBD: hypoxic-ischemic brain damage; LV-sh_TIGAR: vector containing short hairpin RNA of TIGAR; Model: HIBD; NADPH: nicotinamide adenine dinucleotide phosphate; NC: LV-sh-scramble; PPP: pentose phosphate pathway; ROS: reactive oxygen species; TIGAR: TP53 induced glycolysis and apoptosis regulator.
Discussion
In the present study, we demonstrated the critical role of TIGAR in neonatal HIBD. Our studies found that TIGAR was increased in the HIBD-induced neonatal rat cortex in vivo and in OGD/R-induced HAPI microglial cells in vitro. TIGAR knockdown in the neonatal rat brain significantly increased the infarct lesion after HIBD, and HAPI microglial cells exposed to OGD/R exhibited significantly increased pyroptosis. The detrimental effects of TIGAR knockdown in neonatal rat HIBD were blocked by the NADPH administration. These studies suggest that TIGAR prevents microglial pyroptosis and protects against neonatal HIBD.
Microglia play a crucial role in mediating the inflammatory response after HIBD (Kumari et al., 2010; Shen et al., 2010). Pyroptosis, a novel type of programed cell death associated with inflammation, exerts an important role in the process of ischemic brain damage (She et al., 2019). We previously demonstrated that TIGAR suppressed the astrocyte-mediated inflammatory response by inhibiting activation of nuclear factor-κB (Chen et al., 2018), and inhibition of nuclear factor-κB transcription suppressed proinflammatory responses by decreasing activation of the NLRP-3 inflammasome (García et al., 2015). Recently, we found that inhibition of the NLRP-3/caspase-1/GSDMD axis reduced microglial pyroptosis and then alleviated neonatal HIBD (Lv et al., 2020). Therefore, we assumed that TIGAR might have a crucial role in microglial pyroptosis. In the present study, we found that OGD/R could induce the activation of microglial pyroptosis, as evidenced by the increased expression of caspase-1, GSDMD-N, and IL-1β in HAPI microglial cells, and these effects were reduced by TIGAR knockdown. These results imply that TIGAR prevents microglial pyroptosis in response to HIBD in vitro.
To elucidate whether TIGAR could play a protective role in HIBD, the present study explored the effect of TIGAR on neonatal rat HIBD in vivo. The results showed that TIGAR knockdown markedly increased the infarct lesion and pyroptosis after HIBD, which suggests that TIGAR protects against neonatal rat HIBD.
ROS are powerful initiators of neuroinflammation (Bok et al., 2017). Excessive intracellular ROS can trigger inflammatory responses and lead to cell death (Iizumi et al., 2016). TIGAR exhibits an anti-apoptotic effect by reducing the ROS levels through increasing the flux of the PPP (Bensaad et al., 2006). Previously, we found that TIGAR protected against ischemic brain injury by elevating the PPP flux and decreasing ROS levels (Li et al., 2014). Recently, we further demonstrated that NADPH (a metabolic product of the PPP) reduced ischemia/reperfusion-induced brain injury, dependent on its antioxidant and anti-inflammatory effects (Li et al., 2016). In the present study, TIGAR knockdown decreased the NADPH levels and GSH/GSSG ratio and increased the ROS levels in OGD/R-induced HAPI microglial cells. These effects were ameliorated by the administration of NADPH in vitro. Furthermore, NADPH administration also markedly blocked the detrimental effects of TIGAR knockdown on neonatal rat HIBD in vivo, which suggests that TIGAR reduces HIBD by increasing NADPH.
In conclusion, this study demonstrated that TIGAR suppresses microglial pyroptosis and protects against neonatal HIBD. The protective effect of TIGAR may be associated with an increase in NADPH. Thus, this study may provide a novel therapeutic target to treat neonatal HIBD.
Additional file:
Additional file 1 (447.8KB, pdf) : Original data of the experiment.
Footnotes
C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Kreuber L, Yu J, Song CP; T-Editor: Jia Y
Funding: This work was supported by the National Natural Science Foundation of China, Nos. 81872845 (to ML), 81771625 (to XF); the Natural Science Foundation of Jiangsu Province of China, No. BK20180207 (to ML); Jiangsu Provincial Medical Youth Talent of China, No. QNRC2016762(to ML); the Pediatric Clinical Center of Suzhou City of China, No. Szzx201504 (to XF); Postgraduate Research & Practice Innovation Program of Jiangsu Province of China, No. KYCX19_1998 (to LLT); Jiangsu Government Scholarship for Overseas Studies of China, No. JS-2017-127 (to ML); and the Fifth Batch of Gusu Health Talent Plan of China (to ML).
Conflicts of interest: The authors declare that they have no conflict of interests.
Financial support: This work was supported by the National Natural Science Foundation of China, Nos. 81872845 (to ML), 81771625 (to XF); the Natural Science Foundation of Jiangsu Province of China, No. BK20180207 (to ML); Jiangsu Provincial Medical Youth Talent of China, No. QNRC2016762(to ML); the Pediatric Clinical Center of Suzhou City of China, No. Szzx201504 (to XF); Postgraduate Research & Practice Innovation Program of Jiangsu Province of China, No. KYCX19_1998 (to LLT); Jiangsu Government Scholarship for Overseas Studies of China, No. JS-2017-127 (to ML); and the Fifth Batch of Gusu Health Talent Plan of China (to ML). The funders had no roles in the study design, conduction of experiment, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional review board statement: The study was approved by the Animal Ethics Committee of Soochow University (approval No. 2017LW003) in 2017.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
References
- 1.Arioz BI, Tastan B, Tarakcioglu E, Tufekci KU, Olcum M, Ersoy N, Bagriyanik A, Genc K, Genc S. Melatonin attenuates LPS-induced acute depressive-like behaviors and microglial NLRP3 inflammasome activation through the SIRT1/Nrf2 pathway. Front Immunol. 2019;10:1511. doi: 10.3389/fimmu.2019.01511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrington J, Lemarchand E, Allan SM. A brain in flame; do inflammasomes and pyroptosis influence stroke pathology. Brain Pathol. 2017;27:205–212. doi: 10.1111/bpa.12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 4.Bok S, Kim YE, Woo Y, Kim S, Kang SJ, Lee Y, Park SK, Weissman IL, Ahn GO. Hypoxia-inducible factor-1α regulates microglial functions affecting neuronal survival in the acute phase of ischemic stroke in mice. Oncotarget. 2017;8:111508–111521. doi: 10.18632/oncotarget.22851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao L, Zhang D, Chen J, Qin YY, Sheng R, Feng X, Chen Z, Ding Y, Li M, Qin ZH. G6PD plays a neuroprotective role in brain ischemia through promoting pentose phosphate pathway. Free Radic Biol Med. 2017;112:433–444. doi: 10.1016/j.freeradbiomed.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 6.Chalak L, Kaiser J. Neonatal guideline hypoxic-ischemic encephalopathy (HIE) J Ark Med Soc. 2007;104:87–89. [PubMed] [Google Scholar]
- 7.Chen J, Zhang DM, Feng X, Wang J, Qin YY, Zhang T, Huang Q, Sheng R, Chen Z, Li M, Qin ZH. TIGAR inhibits ischemia/reperfusion-induced inflammatory response of astrocytes. Neuropharmacology. 2018;131:377–388. doi: 10.1016/j.neuropharm.2018.01.012. [DOI] [PubMed] [Google Scholar]
- 8.Davies A, Wassink G, Bennet L, Gunn AJ, Davidson JO. Can we further optimize therapeutic hypothermia for hypoxic-ischemic encephalopathy. Neural Regen Res. 2019;14:1678–1683. doi: 10.4103/1673-5374.257512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding X, Sun B, Huang J, Xu L, Pan J, Fang C, Tao Y, Hu S, Li R, Han X, Miao P, Wang Y, Yu J, Feng X. The role of miR-182 in regulating pineal CLOCK expression after hypoxia-ischemia brain injury in neonatal rats. Neurosci Lett. 2015;591:75–80. doi: 10.1016/j.neulet.2015.02.026. [DOI] [PubMed] [Google Scholar]
- 10.García JA, Volt H, Venegas C, Doerrier C, Escames G, López LC, Acuña-Castroviejo D. Disruption of the NF-κB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-a and blocks the septic response in mice. FASEB J. 2015;29:3863–3875. doi: 10.1096/fj.15-273656. [DOI] [PubMed] [Google Scholar]
- 11.Huang J, Lu W, Doycheva DM, Gamdzyk M, Hu X, Liu R, Zhang JH, Tang J. IRE1α inhibition attenuates neuronal pyroptosis via miR-125/NLRP1 pathway in a neonatal hypoxic-ischemic encephalopathy rat model. J Neuroinflammation. 2020;17:152. doi: 10.1186/s12974-020-01796-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iizumi T, Takahashi S, Mashima K, Minami K, Izawa Y, Abe T, Hishiki T, Suematsu M, Kajimura M, Suzuki N. A possible role of microglia-derived nitric oxide by lipopolysaccharide in activation of astroglial pentose-phosphate pathway via the Keap1/Nrf2 system. J Neuroinflammation. 2016;13:99. doi: 10.1186/s12974-016-0564-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, Morita R. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun. 2015;6:7360. doi: 10.1038/ncomms8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang LJ, Xu ZX, Wu MF, Dong GQ, Zhang LL, Gao JY, Feng CX, Feng X. Resatorvid protects against hypoxic-ischemic brain damage in neonatal rats. Neural Regen Res. 2020;15:1316–1325. doi: 10.4103/1673-5374.272615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kepp O, Galluzzi L, Zitvogel L, Kroemer G. Pyroptosis -a cell death modality of its kind. Eur J Immunol. 2010;40:627–630. doi: 10.1002/eji.200940160. [DOI] [PubMed] [Google Scholar]
- 16.Kumari R, Willing LB, Patel SD, Krady JK, Zavadoski WJ, Gibbs EM, Vannucci SJ, Simpson IA. The PPAR-gamma, darglitazone, restores acute inflammatory responses to cerebral hypoxia-ischemia in the diabetic ob/ob mouse. J Cereb Blood Flow Metab. 2010;30:352–360. doi: 10.1038/jcbfm.2009.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee SW, de Rivero Vaccari JP, Truettner JS, Dietrich WD, Keane RW. The role of microglial inflammasome activation in pyroptotic cell death following penetrating traumatic brain injury. J Neuroinflammation. 2019;16:27. doi: 10.1186/s12974-019-1423-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Sun M, Cao L, Gu JH, Ge J, Chen J, Han R, Qin YY, Zhou ZP, Ding Y, Qin ZH. A TIGAR-regulated metabolic pathway is critical for protection of brain ischemia. J Neurosci. 2014;34:7458–7471. doi: 10.1523/JNEUROSCI.4655-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li M, Zhou ZP, Sun M, Cao L, Chen J, Qin YY, Gu JH, Han F, Sheng R, Wu JC, Ding Y, Qin ZH. Reduced nicotinamide adenine dinucleotide, a pentose phosphate pathway product, might be a novel drug candidate for ischemic stroke. Stroke. 2016;47:187–195. doi: 10.1161/STROKEAHA.115.009687. [DOI] [PubMed] [Google Scholar]
- 20.Liu G, Li ZG, Gao JS. Hypothermia in neonatal hypoxic-ischemic encephalopathy (HIE) Eur Rev Med Pharmacol Sci. 2017;21:50–53. [PubMed] [Google Scholar]
- 21.Liu W, Chen Y, Meng J, Wu M, Bi F, Chang C, Li H, Zhang L. Ablation of caspase-1 protects against TBI-induced pyroptosis in vitro and in vivo. J Neuroinflammation. 2018;15:48. doi: 10.1186/s12974-018-1083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lv Y, Sun B, Lu XX, Liu YL, Li M, Xu LX, Feng CX, Ding X, Feng X. The role of microglia mediated pyroptosis in neonatal hypoxic-ischemic brain damage. Biochem Biophys Res Commun. 2020;521:933–938. doi: 10.1016/j.bbrc.2019.11.003. [DOI] [PubMed] [Google Scholar]
- 23.McKenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO, Lu JQ, Branton WG, Power C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc Natl Acad Sci U S A. 2018;115:E6065-E6074. doi: 10.1073/pnas.1722041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mo Y, Sun YY, Liu KY. Autophagy and inflammation in ischemic stroke. Neural Regen Res. 2020;15:1388–1396. doi: 10.4103/1673-5374.274331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel SD, Pierce L, Ciardiello A, Hutton A, Paskewitz S, Aronowitz E, Voss HU, Moore H, Vannucci SJ. Therapeutic hypothermia and hypoxia-ischemia in the term-equivalent neonatal rat: characterization of a translational preclinical model. Pediatr Res. 2015;78:264–271. doi: 10.1038/pr.2015.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poh L, Kang SW, Baik SH, Ng GYQ, She DT, Balaganapathy P, Dheen ST, Magnus T, Gelderblom M, Sobey CG, Koo EH, Fann DY, Arumugam TV. Evidence that NLRC4 inflammasome mediates apoptotic and pyroptotic microglial death following ischemic stroke. Brain Behav Immun. 2019;75:34–47. doi: 10.1016/j.bbi.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Psychogios MN, Knauth M, Bshara R, Schregel K, Tsogkas I, Papageorgiou I, Maier I, Liman J, Behme D. Computed tomography perfusion-based selection of endovascularly treated acute ischaemic stroke patients - Are there lessons to be learned from the pre-evidence era. Neuroradiol J. 2017;30:138–143. doi: 10.1177/1971400916689372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarkar S, Malovic E, Harishchandra DS, Ghaisas S, Panicker N, Charli A, Palanisamy BN, Rokad D, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinsons Dis. 2017;3:30. doi: 10.1038/s41531-017-0032-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P, Hiller S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–1778. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.She Y, Shao L, Zhang Y, Hao Y, Cai Y, Cheng Z, Deng C, Liu X. Neuroprotective effect of glycosides in Buyang Huanwu Decoction on pyroptosis following cerebral ischemia-reperfusion injury in rats. J Ethnopharmacol. 2019;242:112051. doi: 10.1016/j.jep.2019.112051. [DOI] [PubMed] [Google Scholar]
- 31.Shen H, Hu X, Liu C, Wang S, Zhang W, Gao H, Stetler RA, Gao Y, Chen J. Ethyl pyruvate protects against hypoxic-ischemic brain injury via anti-cell death and anti-inflammatory mechanisms. Neurobiol Dis. 2010;37:711–722. doi: 10.1016/j.nbd.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie WJ, Xia TJ, Zhou QY, Liu YJ, Gu XP. Role of microglia-mediated neuronal injury in neurodegenerative diseases. Zhongguo Zuzhi Gongcheng Yanjiu. 2021;25:1109–1115. [Google Scholar]
- 33.Xu P, Zhang X, Liu Q, Xie Y, Shi X, Chen J, Li Y, Guo H, Sun R, Hong Y, Liu X, Xu G. Microglial TREM-1 receptor mediates neuroinflammatory injury via interaction with SYK in experimental ischemic stroke. Cell Death Dis. 2019a;10:555. doi: 10.1038/s41419-019-1777-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu XE, Liu L, Wang YC, Wang CT, Zheng Q, Liu QX, Li ZF, Bai XJ, Liu XH. Caspase-1 inhibitor exerts brain-protective effects against sepsis-associated encephalopathy and cognitive impairments in a mouse model of sepsis. Brain Behav Immun. 2019b;80:859–870. doi: 10.1016/j.bbi.2019.05.038. [DOI] [PubMed] [Google Scholar]
- 35.Yagami T, Yamamoto Y, Koma H. Pathophysiological roles of intracellular proteases in neuronal development and neurological diseases. Mol Neurobiol. 2019;56:3090–3112. doi: 10.1007/s12035-018-1277-4. [DOI] [PubMed] [Google Scholar]
- 36.Zhang DM, Zhang T, Wang MM, Wang XX, Qin YY, Wu J, Han R, Sheng R, Wang Y, Chen Z, Han F, Ding Y, Li M, Qin ZH. TIGAR alleviates ischemia/reperfusion-induced autophagy and ischemic brain injury. Free Radic Biol Med. 2019;137:13–23. doi: 10.1016/j.freeradbiomed.2019.04.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.