

Abstract
Objective
To determine whether distinct aquaporin-4 (AQP4)-IgG lineages play a role in neuromyelitis optica spectrum disorder (NMOSD) pathogenesis, we profiled the AQP4-IgG polyclonal serum repertoire and identified, quantified, and functionally characterized distinct AQP4-IgG lineages circulating in 2 patients with NMOSD.
Methods
We combined high-throughput sequencing and quantitative immunoproteomics to simultaneously determine the constituents of both the B-cell receptor (BCR) and the serologic (IgG) anti-AQP4 antibody repertoires in the peripheral blood of patients with NMOSD. The monoclonal antibodies identified by this platform were recombinantly expressed and functionally characterized in vitro.
Results
Multiple antibody lineages comprise serum AQP4-IgG repertoires. Their distribution, however, can be strikingly different in polarization (polyclonal vs pauciclonal). Among the 4 serum AQP4-IgG monoclonal antibodies we identified in 2 patients, 3 induced complement-dependent cytotoxicity in a model mammalian cell line (p < 0.01).
Conclusions
The composition and polarization of AQP4-IgG antibody repertoires may play an important role in NMOSD pathogenesis and clinical presentation. Here, we present a means of coupling both cellular (BCR) and serologic (IgG) antibody repertoire analysis, which has not previously been performed in NMOSD. Our analysis could be applied in the future to clinical management of patients with NMOSD to monitor disease activity over time as well as applied to other autoimmune diseases to facilitate a deeper understanding of disease pathogenesis relative to autoantibody clones.
Neuromyelitis optica spectrum disorder (NMOSD) is an inflammatory disease of the CNS most frequently characterized by optic neuritis, transverse myelitis, or brainstem syndromes.1 Extensive experimental evidence supports a critical role for B-cell dysregulation in NMOSD with B-cell secretion of IgG autoantibodies targeting a water channel protein aquaporin-4 (AQP4) on astrocyte foot processes in the CNS.2-4 Indeed, 40%–90% of patients with NMOSD, depending on geographic location and ethnicity, are seropositive for autoantibodies against AQP4 (AQP4-IgG).5
Human AQP4 is expressed as 2 major isoforms, M1 and M23, produced by alternative splicing.6 Both isoforms are expressed on astrocytes and together form supramolecular aggregates in plasma membranes called orthogonal arrays of particles (OAPs).7 The size of the OAP depends on the ratio of M1: M23 AQP4.8 In patients with NMOSD, AQP4–IgG binds to the OAPs on astrocytes, which can activate the classical complement pathway, causing complement-dependent cytotoxicity (CDC).9 The anaphylatoxins and opsonins released during complement activation recruit inflammatory cells to the lesion site, promote eosinophil and neutrophil degranulation, and enhance antibody-dependent cell-mediated cytotoxicity. The astrocytes and the neighboring cells get injured, which leads to demyelination, neuronal loss, and subsequent neurologic deficits.10-12
Since the identification of AQP4-IgG as a class of pathogenic autoimmune antibodies (Abs) in NMOSD,2,13 multiple assays have been used to measure or detect AQP4-IgG en masse at the bulk level in body fluids.14,15 However, the AQP4-IgG abundance (titer) cannot be consistently correlated with disease activity, severity, or response to therapy,4,16,17 nor can it be used confidently to predict the risk of a relapse.18 Low-titer patients experience the same disease course as medium-titer and high-titer AQP4-IgG antibody patients with NMOSD.19 These observations suggest that rather than the sheer abundance of AQP4-IgG, it may instead be the underlying molecular diversity of the constituents, which comprise the AQP4-IgG titer that might be different between patients and during various stages of disease. Discovery of such additional stratifying biomarkers is critical because it could aid in prognostication and the design of effective antibody-based therapies for NMOSD.
We have developed a platform for the high-resolution proteomic analysis of immunoglobulin proteins (referred to as Ig-seq) coupled with high-throughput sequencing of B-cell receptor RNA transcripts (BCR-seq) to quantitatively profile the antibody repertoire associated with viral infection, vaccination, and solid tumor.20-22 Here, we applied the BCR-seq/Ig-seq technology to study the AQP4–IgG repertoire in patients with NMOSD at the molecular level to determine the sequence, relative concentrations, and function of the individual AQP4–IgG Abs that comprise the polyclonal Abs in the serum of 2 patients with NMOSD.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
All subjects provided written informed consent before study enrollment. The Research Ethics Committees of UT Austin and of UT Southwestern approved this study.
Patients With NMOSD and Sample Acquisition
NMOSD patient samples were obtained from the University of Texas Southwestern Medical Center Neurosciences Biorepository on approval of the use of human specimen by IRB at UTSWMC. The patients met the 2006 diagnostic criteria for NMOSD and contained 2%–6% circulating CD27hi plasmablasts in their peripheral blood as previously documented.23
Patient 1, a 47-year-old man with a history of ulcerative colitis presented with longitudinal transverse myelitis. His clinical evaluation led to the diagnosis of NMOSD. He was started on rituximab at presentation and remained stable until 2 years later, when he was unable to get his planned dose of rituximab and developed optic neuritis. The patient was found to be seronegative for both AQP4-IgG and MOG by ELISA. The patient was in remission at the time of research blood draw and had completed steroids and PLEX approximately 5 months prior. He had not yet started rituximab.
Patient 2, a 55-year-old woman with a history of Sjogren and Raynaud presented with transient vision loss and previous episode of left-sided optic neuritis. She was treated with IV steroids, and her vision returned to normal. The patient was diagnosed with NMOSD based on clinical presentation and positive AQP4-IgG and has been stable on CellCept. The patient had not yet started CellCept at the time of research blood draw.
Peripheral blood mononuclear cells (PBMCs) were extracted using a Ficoll gradient and split into 2 portions, one for BCR heavy-chain-variable (VH) genes sequencing and the other for heavy-chain-variable: light-chain-variable sequence (VH:VL) pairing. Serum was also acquired from these patients.
Cell-Based Assay for AQP4-IgG Detection
AQP4-IgG was detected using a visual fluorescence-observation cell-based assay according to the manufacturer's instructions (Kronus Inc., NJ). Briefly, the samples were diluted and incubated with biochips containing immobilized nontransfected or AQP-4 transfected cells, then washed and incubated with FITC-labeled secondary antibody to detect AQP4-IgG. The images were taken at 20X objective on a Nikon TS microscope.
Human AQP4 Expression and Protein Purification
Recombinant human AQP4 isoform M1 human AQP4 (hAQP4) was produced in P. pastoris and purified as described previously.24 Briefly, P. pastoris cells expressing hAQP4 (His-tagged) were lysed with a Bead Beater. The cell membrane was collected and solubilized. After centrifugation, the supernatant was collected and loaded on a Ni-NTA HisTrap HP column (GE Healthcare) and cycled for 2 hours. The column was washed, and His-tagged hAQP4 was eluted in fractions with buffer containing 300 mM imidazole. Protein fractions were analyzed by SDS-PAGE, and fractions containing hAQP4 were pooled (figure 1A). The hAQP4 protein was concentrated and stored at −80°C.
Figure 1. Human AQP4 Production, Experimental Design, and AQP4-IgG Detection by Cell-Based Assay.

(A) Typical elution profile of hAQP4 from a nickel affinity chromatography column, visualized by stain-free imaging technology. Individual fraction from the elution is loaded in each lane. The hAQP4-containing fractions, 3–14, were pooled and concentrated. (B) Experimental design: For each neuromyelitis optica spectrum disorder patient, both B cell receptor sequences (BCR-seq) and immunoglobulin sequences (Ig-seq) were acquired. Peripheral B cells were isolated and half was used for constructing VH library as a custom database for heavy chain peptide identification, the other half for VH:VL pairing to acquire the endogenous light chain sequences for the heavy chains. Serum AQP4-IgG were purified by affinity chromatography using AQP4-immobilized column and analyzed by LC-MS/MS. The repertoire was analyzed and recombinant mAbs were expressed for functional characterization. (C) A cell-based, immunofluorescence assay confirmed the presence of AQP4-IgG in select human serum and/or plasma samples. A FITC-labeled secondary antibody was used to detect the presence of AQP4-IgG in human serum or plasma samples. Patient 2 plasma tested positive for AQP4-IgG while patient 1 exhibited negative result. AHC = healthy control; P1 = Patient 1; P2 = Patient 2; NEG = negative control; POS = positive control; QP4 = aquaporin-4. All images were taken at 20× magnification using a Nikon T2 microscope.
VH Library Preparation
The PBMCs were washed with PBS and resuspended in TRIzol reagent (Thermo Fisher). RNA was extracted using RNeasy kit (Qiagen), and cDNA was synthesized from 500 ng total RNA using SuperScript II (Invitrogen). IgG, IgA, and IgM heavy chain repertoires were amplified using a multiplex primer set, as previously described.25 PCR products were concentrated and gel purified for sequencing by 2 × 300 paired-end Illumina MiSeq.
VH:VL Pairing and Sequencing
Paired VH:VL sequencing of single B cells from patients with NMOSD was carried out as previously described.26 Briefly, total B cells were isolated from PBMCs using the Human Memory B Cell Isolation Kit (Miltenyi Biotec) with an LD column. The B cells were loaded into a syringe at 2 × 105 cells/mL. All the reagents required for overlap extension (OE) RT-PCR was loaded into a second syringe. The syringes were connected to a Y junction and simultaneously expel into the emulsification oil. The emulsion was aliquoted into 96-well plates at 100 μL/well, and OE RT-PCR was performed. VH:VL amplicons were then extracted from the emulsions, amplified using nested PCR, and sequenced by 2 × 300 Illumina MiSeq, as previously described.25
Ig-Seq Sample Preparation and Mass Spectrometry
Total AQP4-IgG was purified from the serum sample of patients with NMOSD using Protein G Plus agarose (Thermo Fisher Scientific) and digested into F(ab')2 with IdeS. The IdeS was removed by binding to Strep-Tactin resin (IBA Lifesciences). The recombinant hAQP4 protein was immobilized on NHS-activated agarose resins (Pierce) and packed into a chromatography column (Clontech). The F(ab')2 sample was applied to the hAQP4 affinity column in gravity mode. The AQP4-enriched F(ab')2 was eluted with 2% formic acid in fractions and neutralized with 1M Tris, pH 8. The protein-containing elutes were pooled and prepared for liquid chromatography-tandem mass spectrometry (LC–MS/MS), as previously described.20 IgG peptides were separated using reversed-phase ultrahigh pressure nanoliter flow liquid chromatography and analyzed online by nanoelectrospray ionization tandem mass spectrometry using an Orbitrap Fusion Tribrid Mass Spectrometer (Thermo Scientific). MS1 scans were collected in the Orbitrap at a resolution of 60,000 A °, and the ions with > +1 charge were fragmented by collision-induced dissociation with up to 20 MS2 spectra collected per MS1.
LC-MS/MS Data Analysis
SEQUEST (Proteome Discoverer 1.4, Thermo Scientific) with previously described settings27,28 was used to search the spectra against a patient-specific protein database constructed from the full-length VH and VL sequences, Ensembl human protein-coding sequences, and common contaminants (maxquant.org). PSMs were filtered with a percolator (Proteome Discoverer 1.4) at a false discovery rate of <1% and filtered for average mass deviations <1.5 parts per million. Peptides mapping to the CDR-H3 region of unique IgG antibody clones were grouped, and peptide abundance was derived from the extracted-ion chromatogram (XIC) peak area, using peak area values of the respective precursor ions, generated by the Precursor Ions Area Detector node in Proteome Discoverer 1.4, as previously described.27 The XIC peak areas were averaged across 3 replicate injections for each sample; the eluate and flowthrough abundances were thus compared. Relative abundances were multiplied by the patient-specific AQP4-IgG titer to calculate the amount of AQP4-specific lineages in serum.
VH:VL Pairing Analysis
Raw 2 × 300 MiSeq reads from the paired VH:VL library were trimmed based on sequence quality29 and submitted to MiXCR for gene annotation.30 Productive VH and VL reads were split by isotype and paired using a custom Python script. Sequences with ≥2 reads were grouped into lineages by clustering the CDR-H3 region on 90% nucleotide identity.
Yeast Display Library
Yeast display was used to identify a compatible VL sequence to complement the lineage 1121 VH sequence isolated from NMOSD patient 2. The VH1121 sequence was purchased as a gBlock (Integrated DNA Technologies) and cloned into pCTCON2-Fab-κ and pCTCON2-Fab-λ vectors. The VL library was amplified using a multiplex primer set the same way as the preparation of VH library (above). S. cerevisiae competent cells were prepared and electroporated with the purified plasmids and the VL library.31 After induced with galactose, the cells were incubated with 1 µM hAQP4 (His-tagged), washed, and incubated with FITC-labeled anti-His Tag monoclonal antibody (mAb) (Thermo Fisher Scientific) and RPE-labeled anti-FLAG mAb (ProZyme). The cells were then sorted with flow cytometry for 5 times to select for clones that exhibited high affinity to hAQP4. Cells sorted from each round were subjected to plasmid separation and sequencing and aligned by BioEdit for analysis. One VL sequence (κ) was selected to pair with VH1121 and expressed as recombinant mAb (P2_κ).
Recombinant Antibody Expression and Purification
Selected antibody sequences were purchased as gBlocks and cloned into a customized pcDNA3.4 vector and then transfected into Expi293F cells (Invitrogen). The recombinant Abs were purified with Protein G Plus agarose affinity chromatography as described previously.21
Flow Cytometry for Binding Study
U87 cell line expressing human AQP4 isoform M23 (U87-AQP4) was kindly provided by Dr. Jeffrey L. Bennett (University of Colorado). The mAbs were diluted at different concentrations and incubated with U87-AQP4 cells on ice. The samples were washed and incubated with FITC-conjugated F(ab')2 fragment goat antihuman IgG (Jackson ImmunoResearch) before analyzing by flow cytometry. 10,000 events/sample were collected.
Western Blot
The hAQP4 protein and U87-AQP4 cell pellets were denatured and electrophoresed on Bolt 4%–12% Bis-Tris Plus gel (Invitrogen) and then transferred to PVDF membrane. The membranes were stained with Ponceau S, marked and cut into stripes for blotting with different antibodies. After blocking, the membranes were incubated with mAbs at 1.5 µg/mL, washed, and incubated with horseradish peroxidase conjugated rabbit anti-human IgG (Sigma). The membranes were stained with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). The images were developed by GBOX F3 gel doc system (Syngene).
ELISA
The antigen-antibody interaction was measured by ELISA as previously described.21 Isotype-matched irrelevant mAb was used as negative control. All ELISA assays were performed in triplicate.
CDC Assay
Chinese hamster ovary (CHO) cells stably expressing the M23 isoform of AQP4 (CHO-AQP4, the kind gift of Dr. Jeffrey L. Bennett) were used, as described.32 CHO-AQP4 cells were grown to 90% confluence and subcultured in cell-imaging plates. The cells were washed and incubated for 1 hour with the following conditions: 6 replicates per condition—medium alone, medium + serum (as complement source), irrelevant mAb (isotype control) + serum, P1 mAb + serum, P1 mAb + complement-depleted serum (CDS), P21 mAb + serum, P21 mAb + CDS, P22 mAb + serum, P22 mAb + CDS, P2_κ mAb + serum, and P2_κ mAb + CDS. Treatment medium contained 5% serum and 5 μg/mL mAb for each experimental condition. Cells were washed and treated with a 50 μL mixture of 1 μM green calcein AM (Invitrogen) and 1 μM propidium iodide (BD Pharmingen) for 15 minutes. The plate was then immediately imaged using a fluorescent inverted microscope. Images were taken near the center of each well and quantified using ImageJ. Statistical analysis was performed using Kruskal-Wallis ANOVA, followed by pairwise comparisons using the Mann-Whitney U test.
Data Availability
Data from this study are available from the authors on request.
Results
NMOSD Patient AQP4–IgG Repertoire Analysis
A high-throughput platform was used for the identification and subclassification of AQP4–IgG in the serum of patients with NMOSD (figure 1B). For Ig-seq proteomics, total IgG purified from patient serum was subjected to affinity chromatography using recombinant hAQP4, and the eluted AQP4–IgG was analyzed by high-resolution LC–MS/MS. For BCR-seq, patient B cells were split into 2 portions, one portion was used to decipher B cell immunoglobulin heavy-chain-variable (VH) genes to build a patient-specific Ab database, against which high confidence peptides obtained from Ig-seq were matched. The second portion of B cells was used for heavy-chain-variable: light-chain-variable sequence (VH:VL) pairing on a single cell basis as previously described.26 The combination of IgG peptide sequencing and matching VH:VL DNA sequencing allowed us to identify with molecular-level resolution the circulating AQP4–IgG in patients with NMOSD and to characterize recombinant AQP4–IgG mAbs.
We identified a total of 11,035 BCR VH:VL lineages derived from natively paired sequencing, 8293 VH genes from bulk B-cell sequencing, and 1,218 hAQP4-specific IgG peptides from NMOSD Patient 1. Comparably, a total of 670 BCR VH:VL lineages, 15,727 VH genes, and 1,317 hAQP4-sepcific IgG peptides were identified from NMOSD patient 2 (table 1). For our analysis, AQP4-specific IgG clones were defined by 2 rules21: (1) five-fold higher enrichment of IgG-matching peptides in the elution fractions compared with flowthrough fractions following affinity chromatography and (2) high-confidence peptides covering the CDR-H3 region. As we have previously shown, the Ig-seq technique can identify on average > 70% of total antigen-specific serum-antibodies at femtogram concentrations.27 Patient 1 had previously tested seronegative for AQP4-IgG by ELISA. As cell-based assay (CBA) carries a higher sensitivity, we repeated AQP4-IgG testing by CBA (figure 1C) and the patient was again noted to be seronegative. In applying Ig-seq, we found AQP4-IgG clearly present in patient 1's serum. The AQP4-IgG peptides detected by LC–MS/MS mapping to BCR lineage 4152 from NMOSD Patient 1 are illustrated in figure 2A. Multiple peptides were observed in the VH region, which were unique to this lineage as measured by the elution/flowthrough ratio. Peptides containing CDR-H1, CDR-H2, and CDR-H3 were all identified in the pulldowns. These results indicate that AQP4-IgG was successfully identified by Ig-seq.
Table 1.
Summary of B-Cell Receptor-Seq/Ig-Seq Productive Reads on NMOSD Patient Samples

Figure 2. The AQP4-IgG Serum Repertoire of NMOSD Patients.

(A) A representative BCR lineage of Patient 1 showing IgG peptides identified by Ig-seq. Green lines indicate peptides that are unique to the lineage, and numbers refer to the ratio of a peptide's abundance in the elution vs the flow-through chromatography fractions. Red indicates the CDR-H1, CDR-H2, and CDR-H3 regions. (B) Relative abundance of AQP4-IgG lineages in Patient 1 (B.a) and Patient 2 (B.b). Each bar represents AQP4-binding IgG clones mapping to a BCR CDR-H3 antibody lineage. Red *, VH:VL matched and recombinant mAb was generated. Blue *, VH:VL paired identified from yeast display library. (C) AQP4-IgG V-gene somatic hypermutation rates for NMOSD Patient 1 and Patient 2. (D) V-gene usage in the serologic repertoire of NMOSD patients. AQP4 = aquaporin-4; BCR = B-cell receptor; NMOSD = neuromyelitis optica spectrum disorder.
Typically, antibodies that share sequence similarity in the CDR-H3 region and target the same antigen tend to recognize the same epitope. In this study, high confidence CDR-H3 peptides were identified and grouped into the same lineage based on ≥90% amino acid identity. Relative quantification of the AQP4-IgG lineages was determined by their corresponding LC peak intensities of CDR-H3 peptides. The composition and relative quantities of serum AQP4-IgG repertoire at the clonotypic level were plotted as a bar chart, with each bar representing an individual IgG lineage and the y axis showing its relative abundance (figure 2B), also summarized in tables 2 and 3. The clonotypic distributions of the repertoire between Patient No. 1 and 2 were strikingly different: patient 1's AQP4-IgG repertoire was composed of multiple major IgG lineages, whereas Patient 2 displayed an oligoclonal and highly polarized repertoire that was dominated by 2 lineages contributing more than 90% of the antigen-specific peptide intensity by mass spectrometry.
Table 2.
The Serum Aquaporin-4-IgG Profile of Neuromyelitis Optica Spectrum Disorder Patient 1

Table 3.
The Serum Aquaporin-4-IgG Profile of Neuromyelitis Optica Spectrum Disorder Patient 2

The median somatic hypermutation (SHM) rates were similar between both patients; Patient 1 was observed to have an SHM rate of 8.7%, and Patient 2 6.45% (figure 2C), which is comparable with both the naturally occurring and the vaccine-induced human norovirus Ab SHM rate.22 The AQP4–IgG serologic repertoire in Patient 1 was predominantly comprised of antibodies using the immunoglobulin-heavy variable gene 3–30 (IGHV3-30), IGHV1-69, and IGHV6-1, whereas in Patient 2, IGHV2-5 dominated (figure 2D), likely because the 2 major lineages of Patient 2 are both encoded by IGHV2-5 gene in their heavy chain variable regions (tables 2 and 3).
Recombinant mAbs Derived From Circulating AQP4-IgG Bind to AQP4 Autoantigen
Next, we expressed and characterized 3 recombinant mAbs for which both VH and VL sequences were detected by paired VH:VL sequencing, including one clone from Patient 1 (mAb P1) and 2 clones from Patient 2 (mAbs P21 and P22). The binding of these mAbs to AQP4 was examined by Western blot under denaturing conditions (figure 3A). Both purified recombinant hAQP4 protein (figure 1A) and lysate of U87 cell line expressing human AQP4 (U87-AQP4) were used as antigen sources. All 3 mAbs recognized hAQP4 at a concentration of 0.5–1 µg/mL, whereas binding to U87-AQP4 lysate varied; mAbs P1 and P21 showed significant binding, whereas P22 did not. The relative weight of U87-AQP4 lysate proteins bound by P1 and P21 mAbs differed from the molecular weight of pure hAQP4 protein, which we suspect is likely because of various degrees of denaturation when using a pure protein vs a whole-cell lysate. In addition, P1 and P21 bound differentially to U87-AQP4 cell lysate when compared with the positive control mAb, which may be related to their epitope specificity as the positive control mAb targets the C-terminus of AQP4, whereas P1, P21, and P22 are capable of binding to the extracellular domains of AQP4 (flow cytometry data below). The binding activity of these mAbs to hAQP4 was further confirmed by ELISA (figure 3B). The 3 mAbs exhibited differential relative binding capacity in the order P1>P21 > P22, whereas the isotypic control mAb did not bind AQP4, as expected.
Figure 3. Serum IgG Clones Identified by B-cell Receptor-Seq/Ig-Seq Platform Bind to AQP4 When Expressed as Recombinant mAbs.

(A) Western blot of mAbs P1, P21, and P22 binding to recombinant human AQP4 and cell lysate of U87 cell line expressing AQP4 (1: hAQP4; 2: U87-AQP4). (B) Monoclonal Abs binding to hAQP4 was evaluated by ELISA. Values represent mean ± SD (n = 3). (C) Binding of the mAbs to the surface of U87-AQP4 analyzed by flow cytometry. 10,000 events/sample were collected and each sample was run in triplicates. Values represent mean ± SD (n = 3). (D) Histogram of mAb P1 binding to U87-AQP4 at different concentrations. Negative control mAb shown for comparison. AQP4 = aquaporin-4; mAb = monoclonal antibody.
To investigate whether the 3 mAbs bind to AQP4 in its native conformation, we incubated each mAb with the U87-AQP4 cell line at different concentrations and assayed binding by flow cytometry. All 3 mAbs bound to the extracellular domain of AQP4 expressed on the U87 cell surfaces. The relative binding strength of the 3 mAbs was P1>P21 > P22, in agreement with the ELISA results. Moreover, the binding was positively correlated to its concentration (r = 0.93) by the Pearson correlation test (figures 3C, 3D).
One very highly abundant serum IgG lineage in Patient 2 (lineage 1,121, figure 2B), for which the cognate VL sequence could not be found in the personalized VH:VL database, a compatible VL was identified by yeast surface display in which a library was constructed using the VH identified by Ig-seq (lineage 1,121) and combinatorially paired with VL-encoding cDNA from the patient's peripheral B cells. We screened out multiple VL which encoded highly similar CDR-L3 motifs and which used the same immunoglobulin kappa light chain variable and junctional gene segments, suggesting that screening had converged onto a VL that was likely to be clonally related to the cognate VH:VL pairs. One VL sequence displayed high AQP4-binding affinity when paired with the lineage 1121 VH gene and was selected for mAb construction and downstream characterization. This mAb (P2_κ) was verified binding to AQP4 by U87-AQP4 cell-binding assay (figure 3C).
MAbs Identified From Circulating AQP4-IgG Differ in Complement-Mediated Functional Activity
To further investigate the function of identified mAbs in mediating cell damage, we performed CDC assay on CHO-AQP4 cells using 4 mAbs: P1, P21, P22, and P2_κ (figure 4). We found that in the presence of complement, P1, P22, and P2_ κ mAbs exhibited significantly higher cytotoxicity compared with the isotypic control mAb (p < 0.01) and to the corresponding mAbs in the presence of complement-depleted serum (p < 0.01), suggesting that mAbs P1, P22, and P2_ κ were capable of causing damage to cells through CDC mechanism and were therefore pathogenic. In contrast, mAb P21 did not mediate cytotoxicity in this assay format.
Figure 4. Complement-Mediated Cytotoxicity Can Be Induced by Recombinant Anti-AQP4 mAbs.
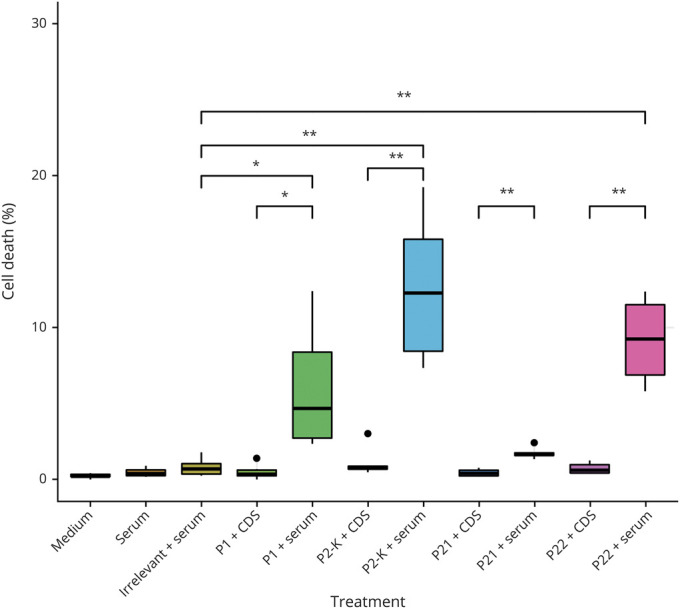
CHO cells expressing AQP4 (CHO-AQP4) were incubated with the recombinant mAbs (P1, P21, P22, or P2_κ) or irrelevant control mAb, in combination with serum as complement source or complement-depleted serum (CDS). Cell dyes were added and cell viability was visualized by fluorescent inverted microscope and quantified using ImageJ (n = 6 per group). The black dots represented outliers of replicates that were not included in the statistics. P1, P22, and P2_κ mAbs showed statistically significant (p < 0.01) killing of cells compared to irrelevant mAb control. AQP4 = aquaporin-4; CHO = Chinese hamster ovary; mAb = monoclonal antibody.
Discussion
The identification of AQP4-IgG as a diagnostic biomarker in NMOSD has been a milestone in differentiating NMOSD from other CNS autoimmune syndromes such as MS.2,13 However, the discordance between AQP4-IgG bulk serum titers and disease activity suggests that rather than the total AQP4-IgG abundance, it might be the patient-specific spectrum of distinct AQP4-IgG lineages that play a key role in NMOSD disease severity and prognosis. Using a synergistic combination of quantitative immunoproteomics and high-throughput B-cell sequencing with the preservation of the natural VH and VL pairing, we globally profiled the AQP4-IgG serum repertoire and identified specific clones of AQP4-IgG in patients with NMOSD. This finding together with differential pathogenicity of individual AQP4-IgG clones suggests that AQP4-IgG repertoire composition could play a differential role in NMOSD disease pathogenesis and presentation.
Previous studies have applied single-cell sorting and sequencing of BCR VH or VH:VL pairs to study the antibody repertoire in patients with NMOSD33-35; however, it is antibody proteins in plasma and CSF that are the primary effectors of the B-cell adaptive immune response. Thus, a deeper understanding of the composition of the serologic antibody response is essential for evaluating how humoral immunity may affect NMOSD disease outcomes. Here, using BCR-seq/Ig-seq proteomics, we have extrapolated the B-cell–centric approach of previous NMOSD studies to an examination of the circulating AQP4-IgG repertoire. Such an approach improves on the previous methodology in several novel ways: (1) more stringent molecular identification of constituent serum antibodies based on the CDR-H3 peptide interval, (2) quantitation of IgG antibody abundances, and (3) determination of IgG antibody-binding specificity and function. Compared with single-cell sequencing that has been traditionally used in the NMOSD field to establish transcriptome libraries,13,35 our method for VH:VL pairing accompanied by deep sequencing allows for greater depth of coverage of VH:VL pairs when starting with 105 peripheral blood B cells per run. Finally, BCR-seq/Ig-seq technology is highly sensitive for the identification of AQP4-specific mAbs, as exemplified by Patient 1, who was determined to be AQP4-IgG seronegative by clinical ELISA at the time of clinical presentation and subsequently by CBA (figure 1C). This patient was in fact found to possess a spectrum of AQP4-IgG by BCR-seq/Ig-seq (figure 2B). Of note, this patient had presented clinically with longitudinal transverse myelitis and optic neuritis at separate occasions and was MOG negative. A recombinant mAb identified from this patient bound to hAQP4 and triggered CDC in a cell-based assay (figures 3 and 4). These findings highlight shortcomings in the existing classification of NMOSD based on ELISA/CBA to determine AQP4-IgG positivity and warrant the development of more sensitive methods for the detection of AQP4-IgG in ostensibly seronegative patients with NMOSD.
Using the BCR-seq/Ig-seq platform, we identified spectra of AQP4-IgG clones circulating in NMOSD patient plasma and recombinantly expressed 3 mAbs for evaluation (figure 2B). All of the recombinant mAbs exhibited binding specificity to denatured AQP4 assayed by Western blot, both to hAQP4 and to U87-AQP4 cell lysate (figure 3A). For comparison, recombinant mAbs cloned from CSF plasma cells of a patient with NMOSD in a separate study failed to recognize denatured AQP4 protein in SDS-PAGE protein immunoblots.36 In yet another study, 2 recombinant mAbs cloned from CSF plasma cells of a patient with one demyelinating event and later diagnosed with NMOSD bound to AQP4 in a cell-based assay, but none of 10 recombinant mAbs cloned from peripheral B cells of patients with NMOSD bound to AQP4 in this same assay.23 These discrepancies may support the notion that a substantial portion of AQP4–IgG in CSF is generated by an intrathecal B cell population that may be different from the peripherally generated B cell clones.35 Moreover, we found that mAbs P1, P21, and P22 bind to hAQP4 with a decreasing relative affinity (P1>P21 > P22) in ELISA and flow cytometry assays (figure 3B and C) in contrast to their partially inverse capacity to mediate CDC (P22 > P1>P21) (figure 4). The lack of correlation between binding affinity and CDC function has also been observed by another group when examining the molecular determinants underlying CDC in NMO using recombinant mAbs to AQP4.37 The same study also found that antibody Fc-Fc interaction and formation of multimeric antibody complexes are critical for activation of the classical complement pathway.37
A limitation to our study is that as a pilot, this study was focused on 2 patients—one seronegative and one seropositive. This small number of patients in our study precludes generalization of the results to clinical significance at this time. However, the strength of our study is that it establishes the utility of BCR-seq/Ig-seq in patients with a strong clinical presentation of NMOSD and seronegative ELISA/CBA testing. Here, we show that BCR-Seq/Ig-Seq can confirm the presence of AQP4 IgG, which may be present at concentrations that may be too low for detection by more traditional ELISA and CBA. Our future direction is to expand our patient cohort at different NMOSD disease time points, during relapse and remission, and to help guide treatment decisions for seronegative patients with NMOSD.
Another potential but unlikely limitation of our study was the use of the M23 isoform of AQP4 in the model cell lines. Although capable of forming OAP, the M23-expressing cell line used in this study may not perfectly match the OAP pattern in astrocytes of patients with NMOSD, which is formed by the mixture of M1 and M23.38 OAP formation by AQP4 is essential in NMOSD pathogenesis and enhances CDC by the pathogenic AQP4-IgG.39 It remains unknown if the OAP size, shape, and composition vary among individuals or whether OAP differences may predispose some individuals to NMOSD or contribute to differential disease pathogenesis, which will be the focus of future studies.
In summary, we report the first proteomic analysis of the circulating AQP4-IgG repertoire in patients with NMOSD—at the mAb level—to identify, quantify, and functionally assay the polyclonal repertoire associated with this disease. Although this study was a snapshot at a single point in time, future studies will include multiple time points. The analysis of more patients with NMOSD will be required to establish whether the AQP4-IgG profile (lineage diversity, kinetics, turnover, or stability) can be correlated with disease severity and progression. Our identification of mAbs with differential ability to affect complement-mediated cell damage underscores the importance of scrutinizing individual IgG lineages rather than wholesale measurement of the AQP4-IgG titer en masse to better understand disease pathogenesis in NMOSD.
Given the heterogeneity of patients with NMOSD in AQP4-IgG milieu and the presence of other autoantibodies, such as MOG-IgG and GFAP-IgG,40 there is a compelling need in the NMOSD field to better define patients with NMOSD with a precise molecular-based diagnosis. Application of the BCR-seq/Ig-seq technology could help to better stratify patients into a specific antibody subtype NMOSD diagnosis and could guide future efforts in drug discovery of individualized blocking antibody treatments toward a more tailored approach to NMOSD patient care. In addition, our molecular-level subcategorization approach can be applied in the future as a paradigm for other autoimmune diseases to identify patient and disease-specific mAbs and their pathogenic roles, as well as provide opportunities for individualized treatments.
Acknowledgment
The authors thank William N. Voss for technical support and helpful discussions during the experiments and thank Yuri Tanno for assisting generation of monoclonal antibodies.
Glossary
- Abs
antibodies
- AQP4
aquaporin-4
- BCR
B-cell receptor
- BCR-seq
B-cell receptor sequencing
- CDC
complement-dependent cytotoxicity
- CDS
complement-depleted serum
- CHO
Chinese hamster ovary
- hAQP4
human AQP4
- LC–MS
liquid chromatography-tandem mass spectrometry
- mAb
monoclonal antibody
- NMOSD
neuromyelitis optica spectrum disorder
- OAP
orthogonal arrays of particle
- PBMC
peripheral blood mononuclear cell
- OE
overlap extension
Appendix. Authors
Contributor Information
Jie Li, Email: jieli@utexas.edu.
Sam A. Bazzi, Email: sbazzi@utexas.edu.
Florian Schmitz, Email: florian.schmitz@aquammodate.com.
Hidetaka Tanno, Email: hidetakatanno@utexas.edu.
Jonathan R. McDaniel, Email: jonrmcdaniel@gmail.com.
Chang-Han Lee, Email: bsoulchlee@gmail.com.
Chaitanya Joshi, Email: chaitanya.joshi@utsouthwestern.edu.
Jin Eyun Kim, Email: rlawlssus1@gmail.com.
Nancy Monson, Email: nancy.monson@utsouthwestern.edu.
Benjamin M. Greenberg, Email: benjamin.greenberg@utsouthwestern.edu.
Kristina Hedfalk, Email: kristina.hedfalk@gu.se.
Esther Melamed, Email: esther.melamed@austin.utexas.edu.
Study Funding
This work was supported by funding from the Owens Medical Research Foundation (E.M., G.I.), Dell Medical School startup funds (E.M.), NIH grant no. R01NS102417 (N.M.), and the Swedish Brain Foundation FO2018-0231 (K.H.). Sample acquisition costs were funded in part by the UT Southwestern CONQUER Program, and the Guthy-Jackson Charitable Foundation for NMO. H.T. was supported by UTHealth Innovation for Cancer Prevention Research Training Program Post-Doctoral Fellowship (Cancer Prevention and Research Institute of Texas grant no. RP160015). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Cancer Prevention and Research Institute of Texas.
Disclosure
B.M. Greenberg has received consulting fees from Alexion, EMD Serono, Novartis, Viela Bio, Roche, Greenwich Bio, Axon Advisors, Rubin Anders, and Abcam. He has received grant support from the NIH, NMSS, SRNA, Guthy-Jackson Charitable Foundation, PCORI, and Clene Nanomedicine. He serves as an unpaid board member of the Siegel Rare Neuroimmune Association. E. Melamed has received consulting fees from EMD Serono, Genentech, Teva, and Viela Bio and has received grant support from the NIH. J. Li, S.A. Bazzi, F. Schmitz, H. Tanno, J.R. McDaniel, C. Lee, J.E. Kim, N. Monson, K. Hedfalk, and G.C. Ippolito report no disclosures relevant to the manuscript. Go to Neurology.org/NN for full disclosures.
References
- 1.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202(4):473-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mader S, Brimberg L. Aquaporin-4 water channel in the brain and its implication for health and disease. Cells. 2019;8(2):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melamed E, Levy M, Waters PJ, et al. . Update on biomarkers in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. 2015;2(4):e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hyun JW, Jeong IH, Joung A, Kim SH, Kim HJ. Evaluation of the 2015 diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology. 2016;86(19):1772-1779. [DOI] [PubMed] [Google Scholar]
- 6.Lu M, Lee MD, Smith BL, et al. The human AQP4 gene: definition of the locus encoding two water channel polypeptides in brain. Proc Natl Acad Sci U S A. 1996;93(20):10908-10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang B, Brown D, Verkman AS. The mercurial insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J Biol Chem. 1996;271(9):4577-4580. [PubMed] [Google Scholar]
- 8.Rossi A, Moritz TJ, Ratelade J, Verkman AS. Super-resolution imaging of aquaporin-4 orthogonal arrays of particles in cell membranes. J Cell Sci. 2012;125(pt 8):4405-4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett JL, Owens GP. Neuromyelitis optica: deciphering a complex immune-mediated astrocytopathy. J Neuroophthalmol. 2017;37(3):291-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan T, Smith AJ, Verkman AS. Complement-dependent bystander injury to neurons in AQP4-IgG seropositive neuromyelitis optica. J Neuroinflammation. 2018;15(1):294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duan T, Smith AJ, Verkman AS. Complement-independent bystander injury in AQP4-IgG seropositive neuromyelitis optica produced by antibody-dependent cellular cytotoxicity. Acta Neuropathol Commun. 2019;7(1):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinson SR, McKeon A, Lennon VA. Neurological autoimmunity targeting aquaporin-4. Neuroscience. 2010;168(4):1009-1018. [DOI] [PubMed] [Google Scholar]
- 13.Bennett JL, Lam C, Kalluri SR, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66(5):617-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarius S, Wildemann B. Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol. 2013;23(6):661-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waters PJ, Pittock SJ, Bennett JL, Jarius S, Weinshenker BG, Wingerchuk DM. Evaluation of aquaporin-4 antibody assays. Clin Exp Neuroimmunol. 2014;5(3):290-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett JL. Finding NMO: the evolving diagnostic criteria of neuromyelitis optica. J Neuroophthalmol. 2016;36(3):238-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitley J, Woodhall M, Leite MI, Palace J, Vincent A, Waters P. Aquaporin-4 antibody isoform binding specificities do not explain clinical variations in NMO. Neurol Neuroimmunol Neuroinflamm. 2015;2(4):e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chanson JB, Alame M, Collongues N, et al. Evaluation of clinical interest of anti-aquaporin-4 autoantibody followup in neuromyelitis optica. Clin Dev Immunol. 2013;2013:146219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiao Y, Fryer JP, Lennon VA, et al. Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology. 2013;81(14):1197-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J, Boutz DR, Chromikova V, et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nat Med. 2016;22(12):1456-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDaniel JR, Pero SC, Voss WN, et al. . High-throughput discovery of antitumor antibodies mined from sentinel lymph node B cells and circulating IgG of breast cancer patients HHS Public Access. Cancer Immunol Immunother. 2018;67(5):729-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindesmith LC, McDaniel JR, Changela A, et al. Sera antibody repertoire analyses reveal mechanisms of broad and pandemic strain neutralizing responses after human norovirus vaccination. Immunity. 2019;50(6):1530-e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rivas JR, Ireland SJ, Chkheidze R, et al. . Peripheral VH4+ plasmablasts demonstrate autoreactive B cell expansion toward brain antigens in early multiple sclerosis patients. Acta Neuropathol. 2017;133(1):43-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Öberg F, Sjöhamn J, Conner MT, Bill RM, Hedfalk K. Improving recombinant eukaryotic membrane protein yields in Pichia pastoris: the importance of codon optimization and clone selection. Mol Membr Biol. 2011;28(6):398-411. [DOI] [PubMed] [Google Scholar]
- 25.McDaniel JR, DeKosky BJ, Tanno H, Ellington AD, Georgiou G. Ultra-high-throughput sequencing of the immune receptor repertoire from millions of lymphocytes. Nat Protoc. 2016;11(3):429-442. [DOI] [PubMed] [Google Scholar]
- 26.Tanno H, McDaniel JR, Stevens CA, et al. A facile technology for the high-throughput sequencing of the paired VH:VL and TCRβ:TCRα repertoires. Sci Adv. 2020;6(17):eaay9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lavinder JJ, Wine Y, Giesecke C, et al. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc Natl Acad Sci U S A. 2014;111(6):2259-2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boutz DR, Horton AP, Wine Y, Lavinder JJ, Georgiou G, Marcotte EM. Proteomic identification of monoclonal antibodies from serum. Anal Chem. 2014;86(10):4758-4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolotin DA, Poslavsky S, Mitrophanov I, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. 2015;12(5):380-381. [DOI] [PubMed] [Google Scholar]
- 31.Benatuil L, Perez JM, Belk J, Hsieh CM. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel. 2010;23(4):155-159. [DOI] [PubMed] [Google Scholar]
- 32.Phuan PW, Zhang H, Asavapanumas N, et al. C1q-targeted monoclonal antibody prevents complement-dependent cytotoxicity and neuropathology in in vitro and mouse models of neuromyelitis optica. Acta Neuropathol. 2013;125(6):829-840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kowarik MC, Astling D, Gasperi C, et al. . CNS Aquaporin-4-specific B cells connect with multiple B-cell compartments in neuromyelitis optica spectrum disorder. Ann Clin Transl Neurol. 2017;4(6):369-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chihara N, Aranami T, Oki S, et al. . Plasmablasts as migratory IgG-producing cells in the pathogenesis of neuromyelitis optica. PLoS One. 2013;8(12):e83036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kowarik MC, Dzieciatkowska M, Wemlinger S, et al. The cerebrospinal fluid immunoglobulin transcriptome and proteome in neuromyelitis optica reveals central nervous system-specific B cell populations. J Neuroinflammation. 2015;12:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owens GP, Ritchie A, Rossi A, et al. Mutagenesis of the aquaporin 4 extracellular domains defines restricted binding patterns of pathogenic neuromyelitis optica IgG. J Biol Chem. 2015;290(19):12123-12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soltys J, Liu Y, Ritchie A, et al. Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J Clin Invest. 2019;129(5):2000-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crane JM, Bennett JL, Verkman AS. Live cell analysis of aquaporin-4 M1/M23 interactions and regulated orthogonal array assembly in glial cells. J Biol Chem. 2009;284(51):35850-35860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phuan PW, Ratelade J, Rossi A, Tradtrantip L, Verkman AS. Complement-dependent cytotoxicity in neuromyelitis optica requires aquaporin-4 protein assembly in orthogonal arrays. J Biol Chem. 2012;287(17):13829-13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol Neuroinflamm. 2015;2(1):e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data from this study are available from the authors on request.