

SUMMARY
Upon pathogen detection, the innate immune system triggers signaling events leading to upregulation of pro-inflammatory and anti-microbial mRNA transcripts. RNA-binding proteins (RBPs) interact with these critical mRNAs and regulate their fates at the post-transcriptional level. One such RBP is ELAVL1. Although significant progress has been made in understanding how embryonic lethal vision-like protein 1 (ELAVL1) regulates mRNAs, its target repertoire and binding distribution within an immunological context remain poorly understood. We overlap four high-throughput approaches to define its context-dependent targets and determine its regulatory impact during immune activation. ELAVL1 transitions from binding overwhelmingly intronic sites to 3′ UTR sites upon immune stimulation of cells, binding previously and newly expressed mRNAs. We find that ELAVL1 mediates the RNA stability of genes that regulate pathways essential to pathogen sensing and cytokine production. Our findings reveal the importance of examining RBP regulatory impact under dynamic transcriptomic events to understand their post-transcriptional regulatory roles within specific biological circuitries.
In brief
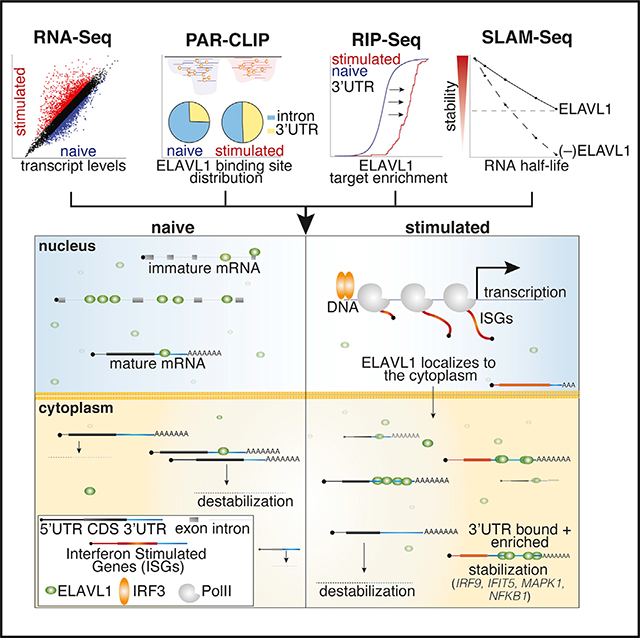
Rothamel et al. show that upon immune activation, the RNA-binding protein ELAVL1 accumulates in the cytoplasm and redistributes from introns to mRNA 3′ UTRs. 3′ UTR binding confers enrichment and transcript stability. Many top-ranking transcripts are interferon-stimulated genes (ISGs), indicating that ELAVL1 is a positive regulator of an innate immune response.
Graphical Abstract
INTRODUCTION
The need to rapidly control gene expression is of paramount importance to implement a robust but punctuated immune response. When a cell is exposed to an immunogenic stimulus, high levels of interferon-stimulated gene (ISG) transcripts are expressed, requiring the cell to orchestrate its translation while simultaneously preventing pathogenic (e.g., viral) RNA from using the same machinery (Liu and Qian, 2014; Piccirillo et al., 2014). ISGs encode for anti-viral, pro-inflammatory, and survival proteins, and their expression is essential in creating a heightened immunoreactive state (Hubel et al., 2019; Schneider et al., 2014). Just as crucial as the initiation of an inflammatory response are the processes that lead to its resolution; therefore, the cell must return to a basal state by limiting the activities of ISGs to prevent damage to the host tissue (Anderson, 2009; Khabar and Young, 2007; Rigby and Rehwinkel, 2015; Savan, 2014). Prolonged interferon-beta 1 (IFNB1) expression has been shown to increase susceptibility to many inflammatory diseases and is a hallmark of autoimmune diseases and cancer (Crow, 2015; Frangou et al., 2013; Reder and Feng, 2013). Emerging evidence indicates that RNA-binding proteins (RBPs) can affect the levels and translation rates of immune-specific transcripts to influence the intensity and duration of an immune response (Anderson, 2010; Hao and Baltimore, 2009; Kafasla et al., 2014; Mino and Takeuchi, 2013). RBPs facilitate RNA metabolism through the control of such processing events as splicing, subcellular localization, stability, and translation (Dreyfuss et al., 2002; Gerstberger et al., 2014; Keene, 2007; Lunde et al., 2007). RBPs act in trans by binding specific structural and/or sequence cis elements, often within the 3′ UTR of mRNAs, a highly trafficked region that is essential to many modes of post-transcriptional gene regulation (Gebauer et al., 2012).
A major strategy by which RBPs regulate mRNAs during immune-activated states is through managing their stability. RBPs such as tristetraprolin (TTP) and the cytotoxic granule-associated T cell-restricted intracellular antigen 1 (TIA1) function as negative regulators of cytokines facilitating increased transcript decay, which is essential for the resolution of inflammation (Herman et al., 2018; Meyer et al., 2018; Tiedje et al., 2016). Conversely, Embryonic Lethal Vision-Like Protein 1 (ELAVL1) has been reported to play a role in immunoregulation by antagonizing the effects of RBPs such as TTP (Kafasla et al., 2014). ELAVL1, also known as HuR (Szabo et al., 1991), binds uridine-(U) and adenyl-uridine-rich elements (AREs) (Chen and Shyu, 1995; López de Silanes et al., 2004), a common low-complexity cis element found throughout the transcriptome. Yet, ELAVL1 binds almost exclusively to cellular mRNAs within introns and the 3′ UTRs (Lebedeva et al., 2011; Mukherjee et al., 2011; Sedlyarov et al., 2016). ELAVL1 is ubiquitously expressed in most cell types and has three distinct and highly conserved RNA-binding domains belonging to the RNA-recognition motif (RRM) family. During steady-state conditions, ELAVL1 is predominantly found in the nucleus but can translocate to the cytoplasm via phosphorylation of Y200, S202, and S221, located in the hinge region of the protein between the second and third RRM. The phosphorylation of ELAVL1 at these residues is reported to occur as part of signal transduction events, including cellular response to immune agents and mitogen signal transduction events (Fan and Steitz, 1998; Grammatikakis et al., 2017).
Previous reports have shown that ELAVL1 is required to maintain the mRNA levels of AU-containing transcripts, including the immune-relevant transcripts IFNB1, COX-2, IL-8, and TGFB (Brennan and Steitz, 2001; Dixon et al., 2001; Fan et al., 2011; Fan and Steitz, 1998; Herdy et al., 2015; Levine et al., 1993). However, most of these studies do not examine whether these effects on mRNA levels are due to the direct binding of ELAVL1. In addition, many of these studies examine the regulatory impact of ELAVL1 on a singular target. Thus, how ELAVL1 prioritizes cellular targets and orchestrates its role in overall immunoregulation was not fully ascertained. Further complicating our understanding of the role of ELAVL1 in immunity and inflammation are the phenotypic outcomes reported in mouse models (Christodoulou-Vafeiadou et al., 2018; Katsanou et al., 2005). ELAVL1 knockouts (KOs) in murine cells have conflicting results depending on the cell type and opposing phenotypes depending on the pattern recognition receptor (PRR) agonist (Yiakouvaki et al., 2012).
With the advent of high-throughput sophisticated RBP-crosslinking and immunoprecipitation (CLIP) methods, such as Photo-Activable Ribonucleoside-enhanced-CLIP (PAR-CLIP), the ability to precisely capture the binding sites of RBPs in cells and study their direct effects has enabled a more molecular understanding of their function (Hafner et al., 2010). The targets and the regulatory impact of ELAVL1 on mRNA targets have been performed in HEK293 and HeLa cells (Lebedeva et al., 2011; Mukherjee et al., 2014). These landmark studies ushered in a broader appreciation for the post-transcriptional role that ELAVL1 can elicit on its targets, particularly for precursor mRNA (pre-mRNA) processing. However, these earlier reports examined ELAVL1 regulation of targets under steady-state conditions in cell types that do not reflect more specific biological processes for which ELAVL1 is implicated, making it difficult to extrapolate whether the reported direct targets contribute to the phenotypes associated with overexpression or KO of ELAVL1 in murine models. This is especially true for an RBP that binds to commonly occurring AREs, which obviates the effectiveness of using predictive analyses to identify its functional targets. Given its purported biological roles in cancers and immunoregulation, what remains lacking is a deeper understanding of how the targeting and binding distribution of ELAVL1 to RNA changes in response to a cellular signaling event that substantively alters the transcriptome and, by virtue, substrate pool of ELAVL1.
Here, we report a multidisciplinary analysis of the targeting and functional outcomes of ELAVL1 during a nucleic acid-induced innate immune response in THP-1 cells, a human monocytic cell line. We find that the mRNA targets of ELAVL1 in THP-1 cells share only 25% of the binding sites of previously published datasets. ELAVL1 largely transitions to binding the 3′ UTRs of mRNA transcripts upon innate immune activation, and this 3′ UTR shift is an absolute prerequisite for enrichment. The loss of ELAVL1 led to widespread destabilization of its enriched target transcripts. Specifically, we found that highly regulated targets had a 3-fold average reduction in their stabilities, losing 30%–80% of their original half-lives. Importantly, we found that among the most highly regulated targets were transcripts that encode for ISGs and their transcriptional regulators, suggesting that ELAVL1 contributes at multiple levels of a proinflammatory response. To date, this is the first report comparing how the targeting of an RBP changes between steady-state and innate immune conditions, thus providing a general framework for investigating RBPs as they govern dynamic transcriptomes.
RESULTS
Transcriptional landscape during an IRF3 innate immune response
To model a nucleic acid-induced innate immune transcriptional response, which would be analogous to viral infection, we stimulated THP-1 cells with cyclic GMP-AMP (cGAMP), the endogenous agonist of the immune adaptor stimulator of interferon genes (STING) (Cai et al., 2014; Ishikawa and Barber, 2008; Sun et al., 2013; Wu et al., 2013). cGAMP is a second messenger molecule produced by the PRR cGAMP synthase upon the detection of cytoplasmic dsDNA in the cytoplasm (Gao et al., 2013a, 2013b; Ablasser et al., 2013; Burdette et al., 2011). cGAMP’s activation of STING ultimately elicits an interferon regulatory factor-3 (IRF3)-dependent transcriptional response, upregulating of hundreds ISGs (Cai et al., 2014; Ishikawa and Barber, 2008; Ishikawa et al., 2009; Sato et al., 1998; Shae et al., 2019). Thus, exposing THP-1 cells to cGAMP allows for precise and robust activation of cells via IRF3, a major arm of the innate immune system, without confounding crosstalk effects through the stimulation of additional immune signaling pathways by other pathogen-associated molecular pattern molecules.
Against the transcriptomic background of an IRF3-driven innate immune response, we sought to characterize the post-transcriptional gene-regulatory role of ELAVL1. For our experimental design, we integrated four independent high-throughput datasets (Figure 1A). Using RNA sequencing (RNA-seq), we (1) performed gene expression analysis comparing the mRNA levels from naive and cGAMP-stimulated THP-1 cells. We next (2) identified the direct RNA targets of ELAVL1 in both cellular conditions using PAR-CLIP (Hafner et al., 2010). Using RNA immunoprecipitation (RIP) sequencing, we (3) quantified the relative enrichments of ELAVL1 targets (Keene et al., 2006; Ramanathan et al., 2019; Tenenbaum et al., 2000; Zhao et al., 2010). Finally, we (4) assessed the regulatory impact of ELAVL1 by measuring the RNA half-lives of its target mRNAs with thiol (SH)-linked alkylation for metabolic sequencing of RNA (SLAM-Seq) upon the loss of its expression during an immune-stimulated transcriptional state (Herzog et al., 2017). Together, these datasets will enable the identification of high-confidence ELAVL1-regulated targets during an innate immune response and provide insight into how signaling events alter the mRNA targets of an RBP during a changing substrate landscape.
Figure 1. RNA-seq and PAR-CLIP capture the context-dependent RNA substrates of ELAVL1.
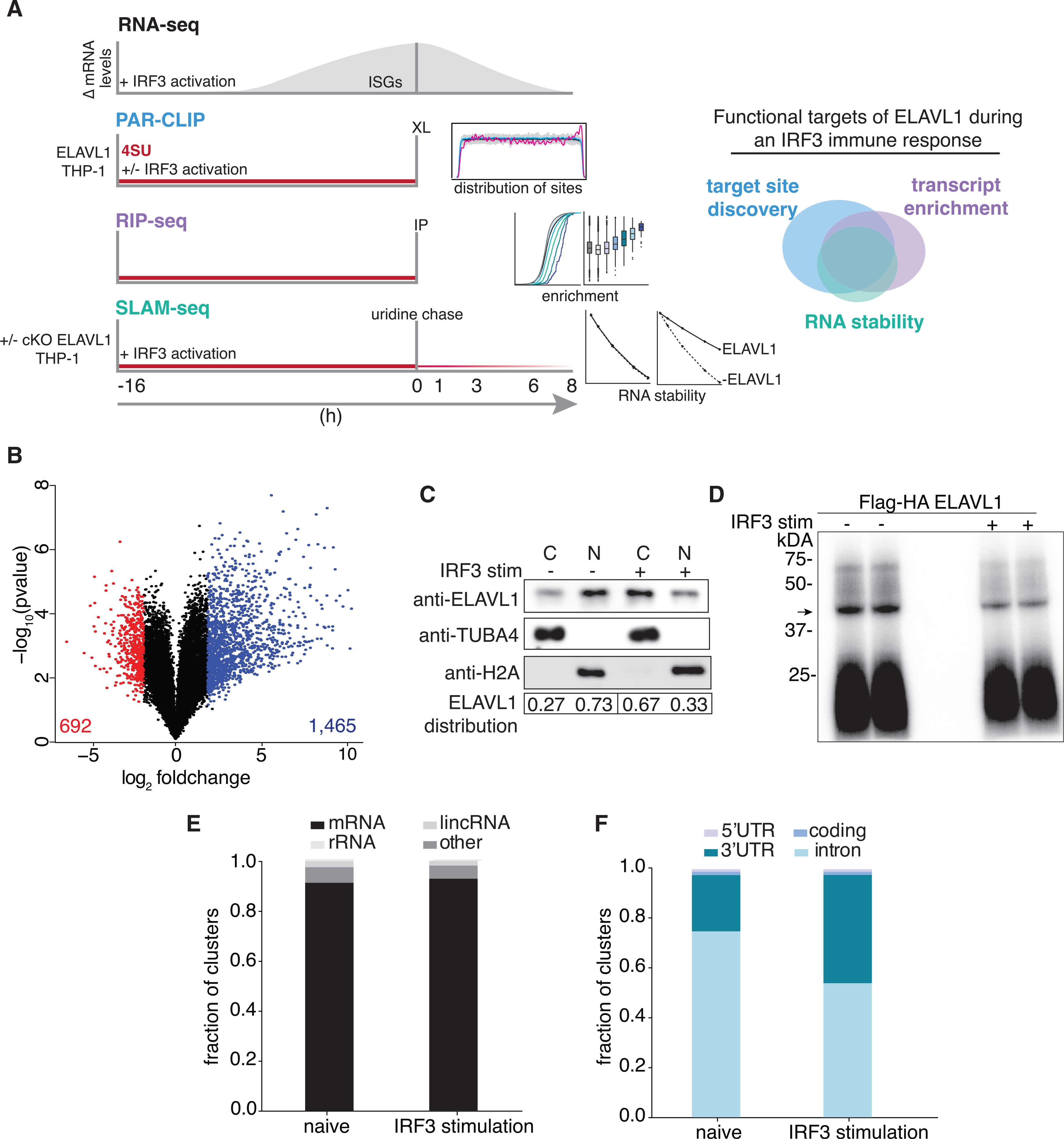
(A) Schematic of the experimental design used to define how ELAVL1 regulates its mRNA substrates during an innate immune response. The Venn diagram (right panel) illustrates how high-throughput datasets will be used to assess the functional targets of ELAVL1.
(B) Volcano plot comparing the differential mRNA levels between naive and stimulated THP-1 cells. The red and blue represent the downregulated and upregulated transcripts in response to cGAMP stimulation, respectively.
(C) Immunoblot showing the nuclear or cytoplasmic distribution of ELAVL1 during a naive and stimulated state. Tubulin and histone 2A are shown as localization controls. The quantitation is showing the percentage of total ELAVL1 in each compartment.
(D) Phosphorimage of 32P-RNA bound to ELAVL1 in the naive and stimulated THP-1 cells. Arrow denotes the ELAVL1 bound RNA.
(E) Distribution of ELAVL1 binding sites across indicated RNA categories.
(F) Distribution of binding sites across different mRNA transcript features for naive and stimulated PAR-CLIP samples.
To compare the mRNA levels that occur in THP-1 cells upon 16-h exposure to cGAMP, we performed gene expression profiling using RNA-seq (Figure 1B). The 16-h time point was selected based on our assessment of peak-level expression of ISGs, including IFNB1, which was previously identified as a direct target of ELAVL1 (Figure S1A) (Herdy et al., 2015; Takeuchi, 2015). We found that 2,157 (1,465 upregulated, 692 downregulated) genes were differentially regulated with 2-fold or greater change (adjusted p ≤ 0.005) upon stimulation (Figure 1B; Table S1). The changes in mRNA levels of several candidate genes were validated using quantitative real-time PCR (Figure S1B). Pathway analysis of the top (25%) upregulated genes indicated that these mRNAs are involved in the regulation of the innate immune response, apoptosis, and hematopoiesis (data not shown). Many of the upregulated genes (60%) are known ISGs, including the anti-viral effectors (OAS, MX1, ISG15), positive regulators of IFN response (IRFs, STAT1, JAK), and nucleic acid PRRs (TLR8, RIG-I, IFITs) (Hubel et al., 2019; Rusinova et al., 2013; Schneider et al., 2014; Schoggins et al., 2011).
Interestingly, ELAVL1 has been reported to translocate out of the nucleus into the cytoplasm by a mechanism that requires post-translational modification (Bidet and Garcia-Blanco, 2014; Grammatikakis et al., 2017; Lourou et al., 2019). To determine whether immune activation of THP-1 cells would lead to a change in ELAVL1 subcellular localization, we performed immunoblots on biochemically fractionated lysates. We observed an increase in the distribution of ELAVL1 in the cytoplasm upon IRF3-driven immune stimulation (Figure 1C). Furthermore, using an antibody that recognizes phosphorylated serine 221 on ELAVL1, we observed a slight increase (2-fold) in the activated state (Figure S1C). These data suggest that ELAVL1 in THP1 cells can be subject to signal transduction activity upon immune activation, leading to its post-translational modification and a marked change in its localization.
The binding site distribution and mRNA targets of ELAVL1 are cell type and context dependent
To identify the direct RNA targets of ELAVL1 during a naive and an IRF3-driven innate immune state (stimulated), we performed PAR-CLIP in THP-1 cells expressing FLAG and human influenza hemagglutinin (HA) epitope-tagged ELAVL1 (FLAG-HA ELAVL1) that were stimulated with cGAMP for 16 h or mock treated. A phosphorimage of the crosslinked and immunoprecipitated FLAG-HA ELAVL1 revealed one major band at approximately 40 kDa, the expected molecular mass of FLAG-HA ELAVL1, in the presence and absence of an immune stimulus (Figures 1D and S1D). RNA from this band was recovered and processed for small RNA-seq. Each cDNA library contained approximately 70 million reads with an average of ~10.7 and ~12.7 million unique reads for the naive and stimulated samples, respectively (Table S2). ~91% of the reads mapped to pre-mRNA with an 86.4% average T-to-C fraction across all samples from naive and stimulated conditions, altogether indicating high-quality recovery and crosslinking efficiencies of our PAR-CLIP procedure for isolating ELAVL1-bound RNA targets (Figure 1E). We identified 133,740 naive and 50,074 stimulated ELAVL1 distinct RNA binding sites that have ≥2 unique T-to-C positions and >20% T-to-C ratio. Although the greater than 2-fold difference in the total number of RNA binding sites between conditions was notable, this difference was not due to lower-complexity libraries for the stimulated samples because these libraries contained more unique reads mapped per cluster (Figure S1E). 106,081 (79%) and 40,681 (81%) of the clusters identified have RNA-seq expression data (>5 counts per million [CPMs]) and RIP-seq enrichment data for the RNA transcripts that correspond with the cluster (Table S3). Of those clusters, 98,689 (74%) and 38,607 (76%) of ELAVL1 binding sites map to pre-mRNA with high confidence in naive and stimulated samples, respectively.
To test the hypothesis that ELAVL1 has a distinct binding site distribution because of cell type, we compared the THP-1 naive binding sites with previously published PAR-CLIP data on ELAVL1 from HEK293 (Mukherjee et al., 2011). 29,820 clusters (30% of the THP-1 naive clusters and 25% of the HEK293 clusters) overlapped by at least 1 nt (Figure S1F). We find that ELAVL1 in naive THP-1 cells uniquely binds to 1,725 mRNAs even though 1,007 (58%) of these transcripts are expressed in HEK293 based on RNA-seq data (Mukherjee et al., 2011). Reactome pathway analysis revealed that the mRNAs bound by ELAVL1 in THP-1 cells are associated with nuclear factor κB (NF-κB) activation (R-HSA-933543), viral defense (R-HAS-168273), and toll-like receptor (TLR) signaling (R-HSA-5603041) (Figure S1G).
We found that a substantial fraction of ELAVL1 binding sites (74%) in THP-1 cells were located within the introns of pre-mRNAs in the naive state, as similarly observed in HEK293 cells (Mukherjee et al., 2011). However, upon immune stimulation, intronic binding was significantly depleted: only 52% of the clusters mapped to introns of target transcripts in the stimulated condition (Figure 1F; Table S2). Nearly 96% of the exonic binding sites mapped to the 3′ UTR in both conditions. The 3′ UTR binding sites were depleted proximal to the stop codon and enriched toward the most distal region of the transcript near the polyadenylation site (40 nt before the poly(A) site) (Figure S1H). Interestingly, when we compared the distribution of the binding sites along the 3′ UTR with previous ELAVL1 PAR-CLIPs (Lebedeva et al., 2011), we found that the enrichment of sites toward the poly(A) tail is specific for our dataset. The 3′ UTR binding site distribution for ELAVL1 in HeLa contains a similar depletion toward the stop codon but does not include the distinct enrichment of binding toward the poly(A) tail. Per previous reports, the HeLa binding sites are equally distributed across the distal region of the 3′ UTR (Lebedeva et al., 2011).
The most striking change observed between naive and stimulated conditions was the increase in the percentage of exonic sites (from 26% to 48%), indicating a profound shift in the binding site distribution of ELAVL1 upon immune stimulation (Figure 1F). This divergence in the total number of sites and the change in binding site specificity does not seem to be a result of a subsampling issue because there was a greater number of unique and total reads per cluster in the stimulated condition samples (Wang et al., 2015).
From our PAR-CLIP data, we observed a major shift in not only the number of ELAVL1 binding sites between the naive and stimulated states (106,081 to 40,681), but we also see a condition-specific difference in the proportion of sites that map to introns and the 3′ UTR. The change in the distribution of sites between the naive and stimulated cellular states gives insight into the context-dependent mRNA targeting of ELAVL1. From the RNA-seq data, we observed that the mRNA levels significantly change during an IRF3-driven immune response. Therefore, we wanted to understand how ELAVL1 mRNA targeting changes during a highly dynamic mRNA-substrate environment. Namely, we wanted to examine the proportion of ELAVL1 binding sites that are found on newly expressed stimulated-specific transcripts.
The mRNA target spectra of ELAVL1 differ in immune cell type
To investigate how ELAVL1 differentially targets mRNAs due to cellular condition, we determined the conservation of binding sites between the naive and stimulated states and found that 27,323 clusters overlapped by at least 1 nt (Figure 2A). This overlap comprises 27% and 70% of the naive and stimulated sites, respectively. At the transcript level, we found that the majority (5,051) of targets were bound in both conditions, although a notable number were uniquely found in the naive (1,289) or stimulated (444) states (Figure 2B; Table S4).
Figure 2. Innate immune stimulation pivots ELAVL1 binding toward 3′ UTR sites.
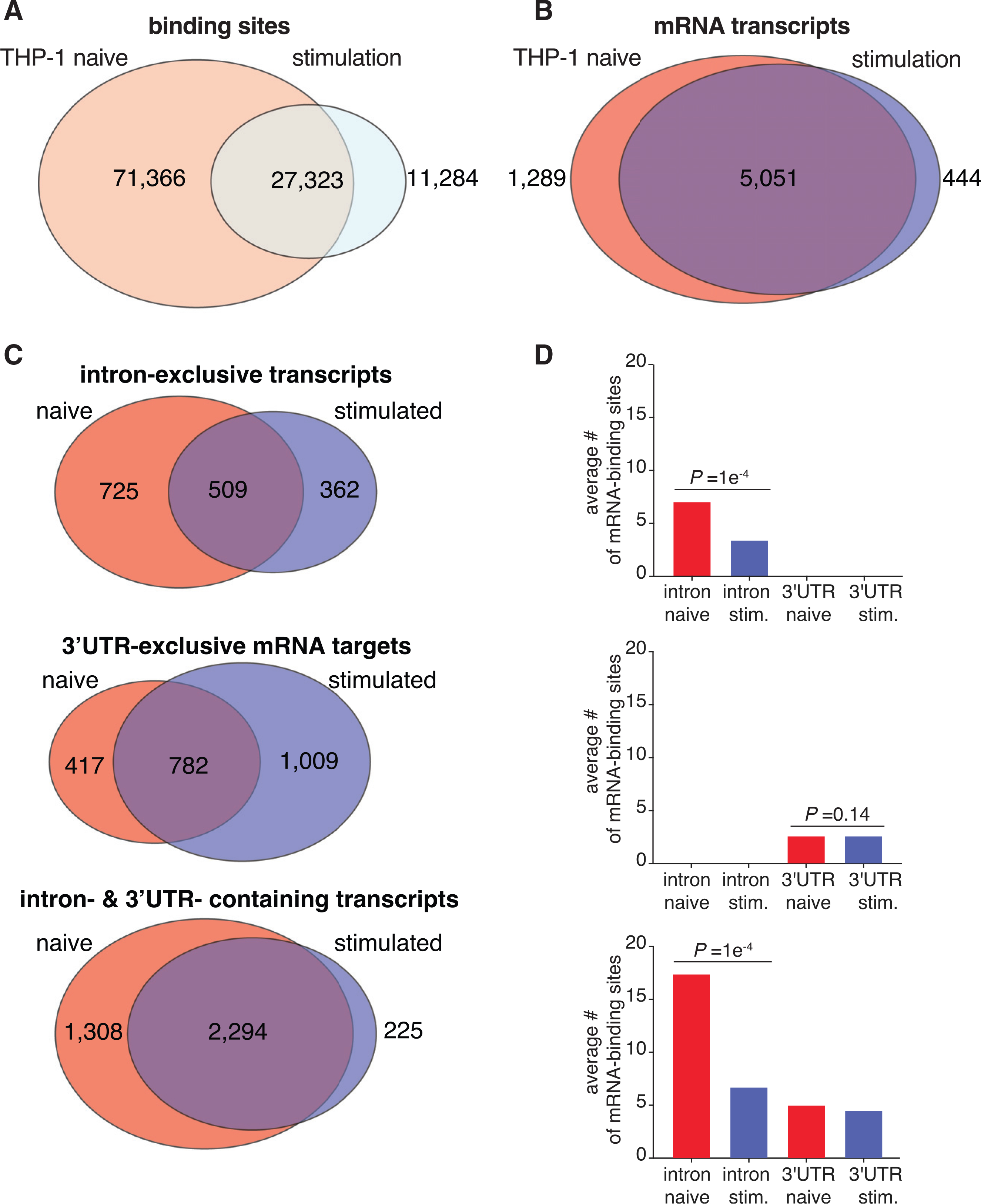
(A and B) Venn diagrams of the PAR-CLIP-defined ELAVL1 clusters (A) or bound transcripts (B) between the two states.
(C) Venn diagrams indicating the number of transcripts in naive and stimulated cells that are bound exclusively in the intron, 3′ UTR, or both.
(D) The average number of binding sites for the three mRNA location categories listed across conditions. p values were calculated using Student’s t test.
We wanted to parse these data further and investigate whether the mRNAs that were shared or uniquely bound were differentially targeted by ELAVL1. Of the 6,340 mRNAs bound by ELAVL1 in the naive state, >94% of transcripts contained sites that mapped to either intron-exclusive sites, 3′ UTR-exclusive sites, or both; <6% of transcripts contained sites within 5′ UTR and/or coding sequence (CDS). A similar proportion was observed for the transcripts bound in the stimulated state. Consequently, we focused our analyses on transcripts that bore a distribution of intron and 3′ UTR sites, which naturally divided into three populations: transcripts that contained ELAVL1 sites exclusively within (1) introns, (2) the 3′ UTR, or (3) both regions. In comparing the proportion and the absolute number of transcripts bound exclusively in the intron or 3′ UTR, we observed a significant shift in binding site distribution for the 3′ UTR in the stimulated state. Conversely, the naive state had nearly 50% more transcripts that were exclusively bound within introns (Figure 2C).
We next examined mRNAs that contained ELAVL1 binding sites within introns and the 3′ UTR because they represented the majority of the targets. Because these transcripts contained sites for both regions, we reasoned that a change in intra-transcript binding would give insight into how ELAVL1 binds RNA in a condition-specific manner. Previous work showed that the total number of ELAVL1 binding sites correlates with the extent of its regulation for that particular mRNA (Mukherjee et al., 2011). Therefore, we calculated the average number of ELAVL1 sites for each region, with the hypothesis that a change in intra-transcript binding site distribution would point to the region(s) of the mRNA that is relevant to its function during immune stimulation. In the stimulated state, we see a 2- to 3-fold reduction in the number of intronic binding sites for intron-exclusive transcripts and those that contained both intron and 3′ UTR sites (Figure 2D). Among the mRNA targets that contained both intron and 3′ UTR sites, the ratio of intron sites to 3′ UTR sites shifted from 3:1 to 1.3:1 upon stimulation. Corroborating the observation that the changes are due to the loss of intronic binding sites, we found no significant change in the average number of 3′ UTR sites upon immune stimulation. Altogether, our data show that immune stimulation leads to a loss of mRNA targets exclusively bound within introns, as well as the total number of intronic sites across all other transcripts, resulting in a net increase in the proportion of 3′ UTR-bound ELAVL1 targets.
Given the condition-dependent shift in the distribution of the binding sites from intron to 3′ UTR, we examined whether there was a change in motif usage by ELAVL1. Motif analysis of the binding sites in both conditions located in 3′ UTR and introns revealed a UUUUUU- and AUUUUA-rich RNA recognition element. Similar results were obtained from previous PAR-CLIPs from HeLa and HEK293, indicating that ELAVL1 does not change sequence-specific binding site preference in different cell types or conditions (Mukherjee et al., 2011). These data suggest that other factors, such as RNA secondary structure and competition with other RBPs, influence the ability of ELAVL1 to bind to its RNA targets.
PAR-CLIP and RIP-seq define enrichment criteria for ELAVL1 during an innate immune response
We assessed the quantitative changes in substrate binding by ELAVL1 as cells transitioned from the naive to the stimulated state, given the rapid changes to the transcriptome. ELAVL1 functions through its targeting of AREs within mRNAs. By quantifying its association to targets, we can discern the binding properties that drive the relative enrichment differences caused by an innate immune response. Therefore, we performed RIP-seq for ELAVL1 (Keene et al., 2006; Tenenbaum et al., 2000; Zhao et al., 2008, 2010). RNAs that co-immunoprecipitated with ELAVL1 were recovered and sequenced. A total of 3,459 and 3,406 PAR-CLIP-identified mRNA targets in naive and stimulated samples, respectively, were enriched over the IgG background (Figures 3A, 3B, and S2A; Table S4). Overlapping the enriched targets from both conditions, we found that 2,114 mRNAs were enriched in a context-independent manner; 1,345 mRNAs were specific to the naive state, while 1,292 were specific to the stimulated state (Figure S2B). We next discerned whether the enriched targets in the stimulated-specific state represented just the upregulated mRNAs. Surprisingly, we found that a majority (61%) of the 1,292 transcripts were already expressed in the naive state, and their expression values did not significantly change upon immune stimulation. The mean expression value for these existing transcripts was 10.7 CPMs (all transcripts mean = 9.2 CPMs). These data indicate that the differences in the target repertoire between the two cellular states are not solely due to changes in the transcriptional output of the mRNAs. ELAVL1 competes for binding sites with other post-transcriptional regulatory elements whose expression and activities are also dependent on the cellular context (Dassi, 2017; Lu et al., 2014; Srikantan et al., 2012; Young et al., 2012).
Figure 3. ELAVL1 RIP-seq and PAR-CLIP define transcript enrichment criteria during immune stimulation.
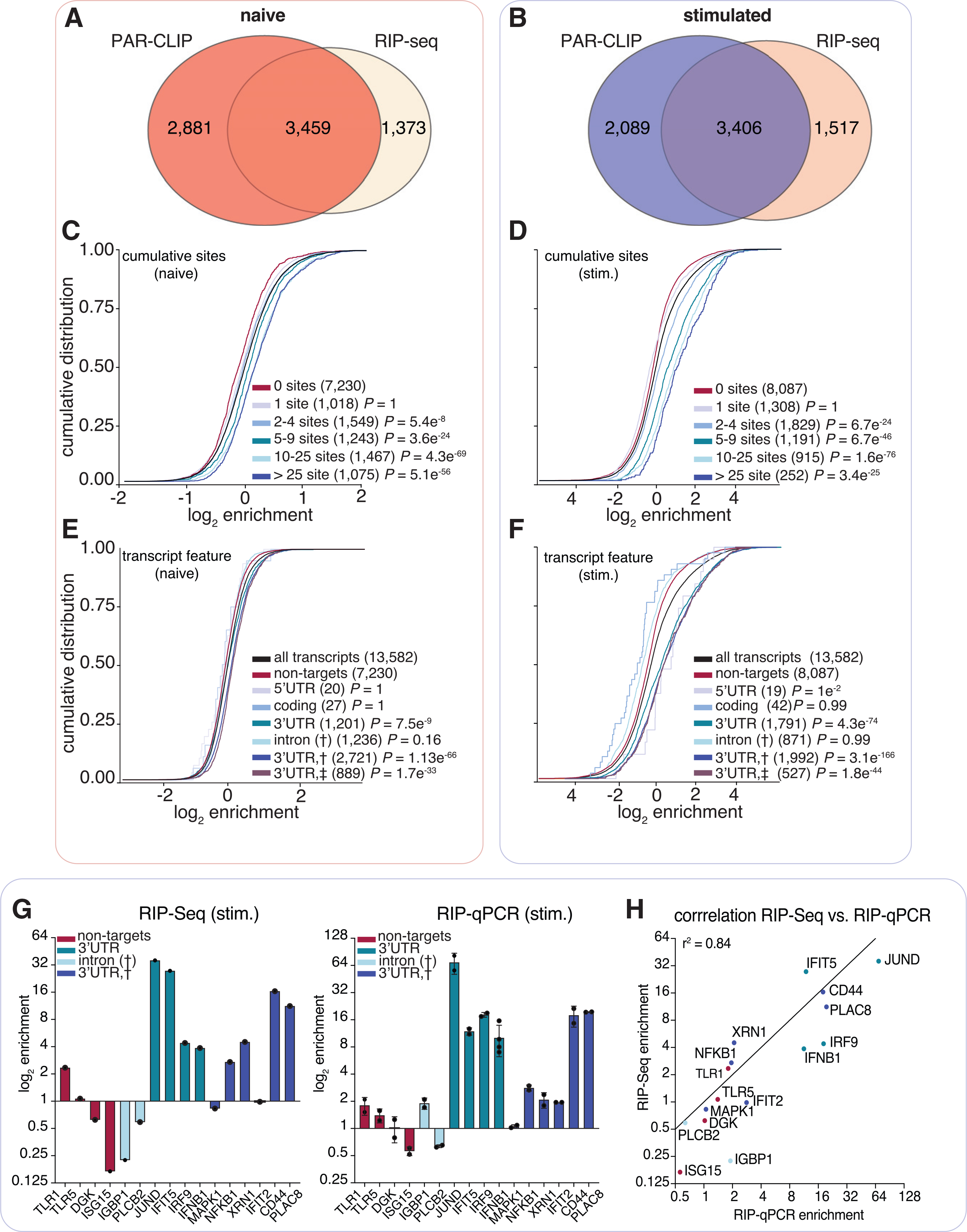
(A and B) Venn diagram of the transcripts defined as targets using PAR-CLIP and RIP-seq data in the (A) naive and (B) IRF3-stimulated states.
(C and D) Cumulative distribution fraction analyses for the targets of ELAVL1 in (C) naive or (D) stimulated conditions on the basis of the number of total binding sites indicated.
(E and F) Cumulative distribution fraction analyses for ELAVL1 targets in (E) naive or (F) stimulated cells. Transcripts were binned based on the indicated location of the ELAVL1 binding sites.
(G) Bar graphs of RIP-seq and RIP quantitative real-time PCR enrichment levels for transcripts listed. n = 2 (RIP-seq) and n = 3 (RIP-qPCR). Error bars represent mean ± SD.
(H) Correlation (Pearson correlation) plot of enrichment levels between RIP-seq and RIP quantitative real-time PCR.
Reactome pathway analysis on target genes that were either naïve specific or shared showed that they encoded for proteins involved in transcriptional regulation by TP53 (R-HSA-3700989), processing of capped mRNAs (R-HSA-72203), and cell-cycle checkpoints (R-HSA-69620) (Figures S2C and S2D; Table S5). These pathways comprise more ubiquitous cellular processes that are generalizable across cell types, often found as steady-state functions. By contrast, Reactome pathways enriched from targets specific to the stimulated state include TLR signaling (R-HSA-168181), NF-κB (R-HSA-975183), and mitogen-activated protein (MAP) kinase (MAPK) signaling (R-HSA-975138) (Figure S2E; Table S5). This observation is interesting because the majority of the targets that were enriched in the stimulated-specific state were expressed at similar levels in naive cells, but not enriched, thus suggesting that ELAVL1 associates with transcripts belonging to immune signaling pathways regardless of mRNA levels. Consistent with our observation, 71% (918/1,292) of these stimulated-specific enriched targets were also found as PAR-CLIP targets in a HEK293 cell, yet only 122 were enriched; pathway analysis of these 122 targets did not yield enrichment in the TLR, NF-κB, or MAPK signaling terms.
3′ UTR binding determines the level of enrichment to context-dependent mRNA targets
To test the hypothesis that the frequency and position of ELAVL1 binding sites influence levels of enrichment in THP-1 cells, we examined the cumulative distribution of ELAVL1 target enrichment (RIP-seq) based on PAR-CLIP binding site data. Independent of cellular state, transcripts with ≥2 binding sites showed significant enrichment compared with transcripts with no sites. Furthermore, an increase in the number of ELAVL1 binding sites showed a positive correlation with enrichment (Figures 3C and 3D). Interestingly, we saw that enrichment levels were more pronounced in the stimulated state. Overall, for transcripts that had ≥2 sites in the stimulated state, there was nearly a 200% increase in fold enrichment over non-targets, whereas targets (≥2 sites) in the naive state were only nominally enriched (10%) over non-targets.
From our PAR-CLIP data, we observed a net decrease in the total number of binding sites per transcript in the stimulated compared with naive state. However, we found that an increase in the number of sites per transcript correlated with greater enrichment, especially in the stimulated state. To reconcile these observations, we grouped transcripts based on the location of PAR-CLIP sites. We found that mRNA that contained at least one 3′ UTR binding site had the highest levels of enrichment compared with binding sites in other transcript regions (Figures 3E and 3F). These results were further validated and highly correlated with RIP quantitative real-time PCR data (r2 = 0.82) (Figures 3G and 3H).
A majority of 3′ UTR-bound transcripts also contained additional intronic sites, and previous reports showed that intronic binding contributed to enrichment (Lebedeva et al., 2011; Mukherjee et al., 2011). Therefore, we tested whether increasing numbers of intron-bound sites led to greater enrichment but found no correlation (Figures 4A and 4B). Enrichment was dependent solely on the number of 3′ UTR sites in both naive and stimulated states. Of note, the 3′ UTR-bound transcripts in the stimulated state were five times more enriched over non-3′ UTR-bound targets compared with the same populations in the naive condition (Figures 4C and 4D). We also observed that the fractional occupancy of 3′ UTR sites per transcript was significantly favored in the stimulated state (Figure 4E). This might explain the increase in the 3′ UTR-specific enrichment in the stimulated state compared with the naïve condition.
Figure 4. ELAVL1-mRNA enrichment is exclusively dependent on 3′ UTR association and intensifies upon immune stimulation.
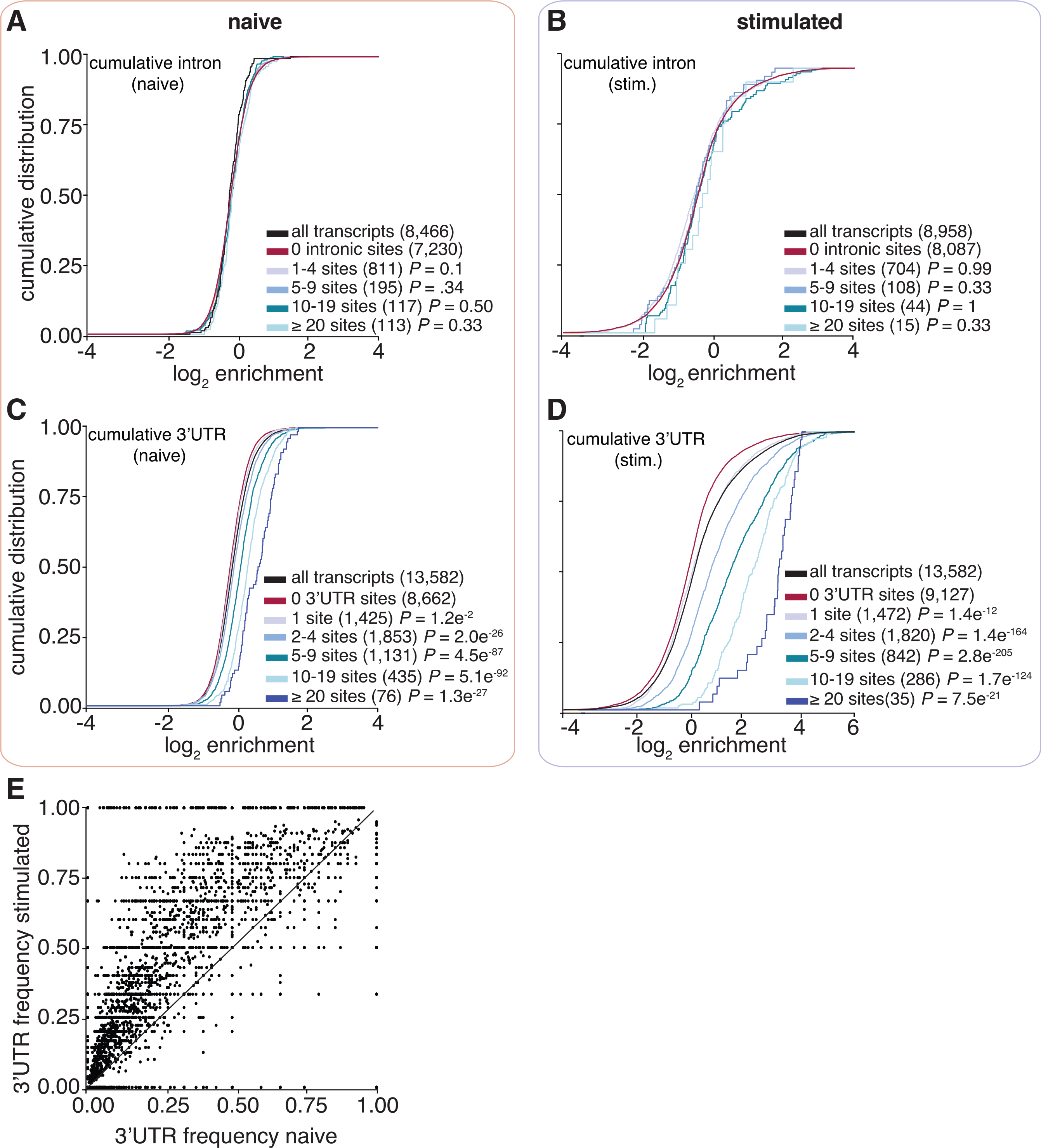
(A–D) Cumulative distribution function analyses were used to determine whether (A and B) intronic binding versus (C and D) 3′ UTR binding confers greater transcript enrichment in naive and stimulated states.
(E) A scatterplot shows the fraction of 3′ UTR sites over the total number of binding sites. The x axis represents the naive state, and the y axis represents the stimulated state.
To assess whether the targets of ELAVL1 would be similarly enriched in other human monocytic cells, we examined the target spectra and protein localization of ELAVL1 in U937 cells. This cell line has been previously used to study how ELAVL1 regulates an inflammatory response (Sahlberg et al., 2013). We found that the expression of ISGs in response to cGAMP activation correlated well (r2 = 0.79) in U937 and THP-1 cells (Figure S3A). However, when investigating the localization and protein expression levels of ELAVL1 across the two cell types, we unanticipatedly found that in U937 cells, the predominant form of ELAVL1 is approximately 12 kDa smaller than the full-length ELAVL1 (36 kDa), and that the majority of the protein was found to be cytoplasmic independent of stimulation (Figure S3B). Previous publications have reported the existence of a caspase-dependent cleaved form of ELAVL1 of similar mass (Mazroui et al., 2008; Sahlberg et al., 2013; Talwar et al., 2011; von Roretz et al., 2013). The caspase-dependent cleaved form was reported to be localized to the cytoplasm and missing its third RRM and dimerization domain. We next transiently transfected a FLAG-HA ELAVL1 to assess whether U937 cells were genetically expressing a truncated ELAVL1 or if the smaller molecular weight form of ELAVL1 is more consistent with that reported by previous investigators. Immunoblot analysis of the expression in U937 cells indicated that the recombinant form was also truncated, despite encoding for a full-length protein (Figure S3C). We anticipated that the truncated form of ELAVL1 would not behave similarly to the full-length protein. Accordingly, immunoprecipitation of FLAG-HA ELAVL1 in U937 cells followed by quantitative real-time PCR analysis showed no significant enrichment of highly enriched THP-1 validated targets (IFIT2, IFNB1, IFIT5, IRF9, JUND) (Figure S3D).
From the integration of RIP-seq and PAR-CLIP datasets, we observed that an increasing number of 3′ UTR sites correlates with greater enrichment in THP-1 cells. Importantly, mRNAs bound by ELAVL1 in the immune-stimulated state showed significantly higher enrichment levels, demonstrating a stronger association and potentially indicating a more profound level of post-transcriptional gene regulation. However, the binding and enrichment of a transcript with an RBP is not directly equivalent to its regulatory fate, given that any single mRNA is potentially subject to other post-transcriptional factors that may impose a stronger regulatory effect. In most reported cases, the primary function of ELAVL1 is through the stabilization of its target transcripts (Herdy et al., 2015; Srikantan et al., 2012; Takeuchi, 2015; Turner and Díaz-Muñoz, 2018). ELAVL1 competes over AREs on transcripts, opposing negative post-transcriptional regulators like RNA-induced silencing machinery or TTP, which can recruit deadenylase complexes (Fu and Blackshear, 2017). Consequently, to understand the functional impact of our enriched ELAVL1 targets, we utilized SLAM-seq to quantitatively measure transcript decay rate (Herzog et al., 2017).
ELAVL1 stabilizes a subset of 3′ UTR targets involved in innate immune signaling
By overlapping SLAM-seq results with our RIP-seq and PAR-CLIP datasets, we were able to precisely identify the consequences of ELAVL1 absence on the stabilities of its target transcripts during an innate immune response (Figures 5A and 5B). Slam-seq uses 4SU to metabolically label nascent RNA transcripts, which is subsequently chased with unlabeled uridine. Thiol-alkylation of RNA generates chemical adducts that induce reverse transcriptase-dependent deoxycytosine substitutions at 4SU positions during cDNA library preparation (T-to-C substitutions). The ratio of unlabeled to T-to-C-containing reads across all the time points is used to calculate RNA half-life for each expressed transcript (Herzog et al., 2017; Neumann et al., 2019). Here, we performed SLAM-seq during an IRF3-driven innate immune response. ELAVL1-WT (wild-type) and ELAVL1-KO THP-1 cells were labeled with 4SU for 16 h, followed by washes and a uridine chase. Cells were collected at various time points after the chase, and extracted RNAs were processed for high-throughput sequencing. We determined the half-lives of nearly ~5,000 transcripts that were shared across ELAVL1-WT and -KO datasets and found their median half-lives to be 6.7 and 6.4 h, respectively (Figure 5C; Table S6), indicating comparable global RNA stability independent of condition type. Importantly, the difference in the median half-lives between targets and non-targets is greater when ELAVL1 is present (1.8 h), in comparison with the median half-life difference when it is knocked out (0.7 h) (Figure 5D). These data indicate that the half-lives of ELAVL1 target transcripts are more similar to non-targets upon its KO.
Figure 5. Analysis of transcriptome-wide mRNA stability in the absence of ELAVL1.
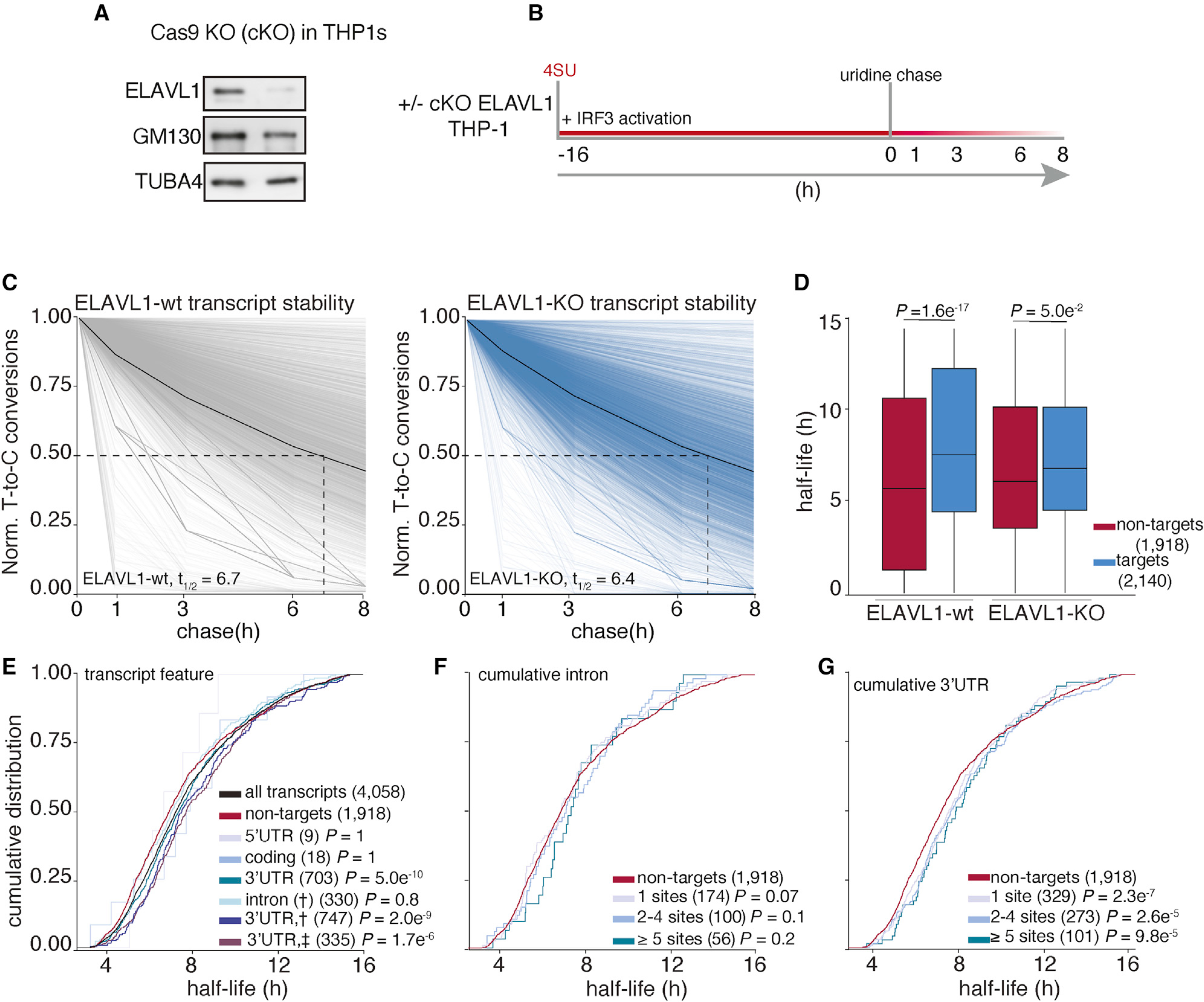
(A) Immunoblot staining for endogenous ELAVL1-WT and ELAVL1-KO THP-1 cells.
(B) Schematic of the SLAM-seq experiment setup. THP-1 cells ( ± ELAVL1KO) were stimulated, and 4SU was labeled for 16 h before wash and uridine chase. Time points for SLAM-seq were 0, 1, 3, 6, and 8 h after uridine chase.
(C) The decay of T-to-C conversions was determined by fitting the data to a single-exponential decay model to derive mRNA half-lives (dotted line). Graphs show that the RNA stabilities over time. The solid black line indicates the median T-to-C conversion rate over time; the dotted black line denotes median half-life (t1/2), as indicated per condition.
(D) Boxplots showing the median half-lives of non-targets (red) and targets (blue) in the ELAVL1-WT and ELAVL1-KO cells.
(E) Cumulative distribution fraction analyses of the RNA half-lives. Transcripts were grouped by the location of the ELAVL1 binding sites.
(F and G) Cumulative distribution fraction analyses were used to determine whether the RNA half-lives of targets are affected by the number of (F) intronic or (G) 3′ UTR binding sites.
In the ELAVL1-WT cells, we observed that transcripts with at least one 3′ UTR binding site had statistically significantly longer RNA half-lives (t1/2 = 7.5 h) than non-targets (t1/2 = 5.7 h), whereas 5′ UTR-, coding-, or intronic-bound targets were not significant (Figure 5E). We noticed that transcripts that have binding sites either in the intron and 3′ UTR (t1/2 = 7.6 h) or intron and 3′ UTR plus another location (t1/2 = 7.5 h) tended to have slightly longer RNA half-lives than transcripts bound exclusively in the 3′ UTR (t1/2 = 7.4 h). Similar to our analysis of transcript enrichment, comparing the intron or 3′ UTR exclusively bound transcripts, we found that only the presence and increase in the number of 3′ UTR sites conferred greater stability (Figures 5F and 5G). Interestingly, targets with >10 3′ UTR binding sites exhibited half-lives 3 h longer than non-targets (t1/2 = 8.9 versus 5.7 h). In looking at the change in target transcript half-lives between KO and WT, no feature other than 3′ UTR conferred any significant stability effect (Figure S4A).
To assess whether target transcripts that were enriched based on 3′ UTR content were specifically stabilized by ELAVL1, we examined their behaviors when ELAVL1 was knocked out. The half-lives of only the enriched 3′ UTR-containing ELAVL1 transcripts were significantly reduced in the absence of ELAVL1 (Figure 6A). The half-lives of the non-enriched transcripts bound by ELAVL1 were not as affected by the absence of ELAVL1, suggesting that the decay rates of these transcripts are either partially or completely independent of ELAVL1 regulation, despite being bound. Moreover, transcripts with an increasing number of ELAVL1 sites have the greatest decrease in half-lives, further supporting this 3′ UTR stability signature (Figure 6B). Among the most 3′ UTR-enriched targets (n = 1,141), we noted that a substantial fraction (50%) are ISGs, and collectively they had a greater change in half-life in the KO compared with enriched transcripts that are not ISGs (Figure 6C). ISGs, such as NFKB1, IRF9, IFIT5, and MAPK1, are less stable in the absence of ELAVL1, in contrast with ISGs for which we had zero evidence of ELAVL1 association or enrichment (Figure 6D). The change in the mRNA half-lives of targets and non-targets of ELAVL1 were confirmed by shutting off transcription (actinomycin D) and measuring mRNA decay using quantitative real-time PCR. Transcription inhibition coupled with quantitative real-time PCR validated the SLAM-seq data showing that the half-lives of ISGs with 3′ UTR binding were significantly affected because of the absence of ELAVL1 (Figures 6E, S4B, and S4C).
Figure 6. The RNA half-lives of highly enriched ELAVL1 target ISGs are the most affected by its loss.
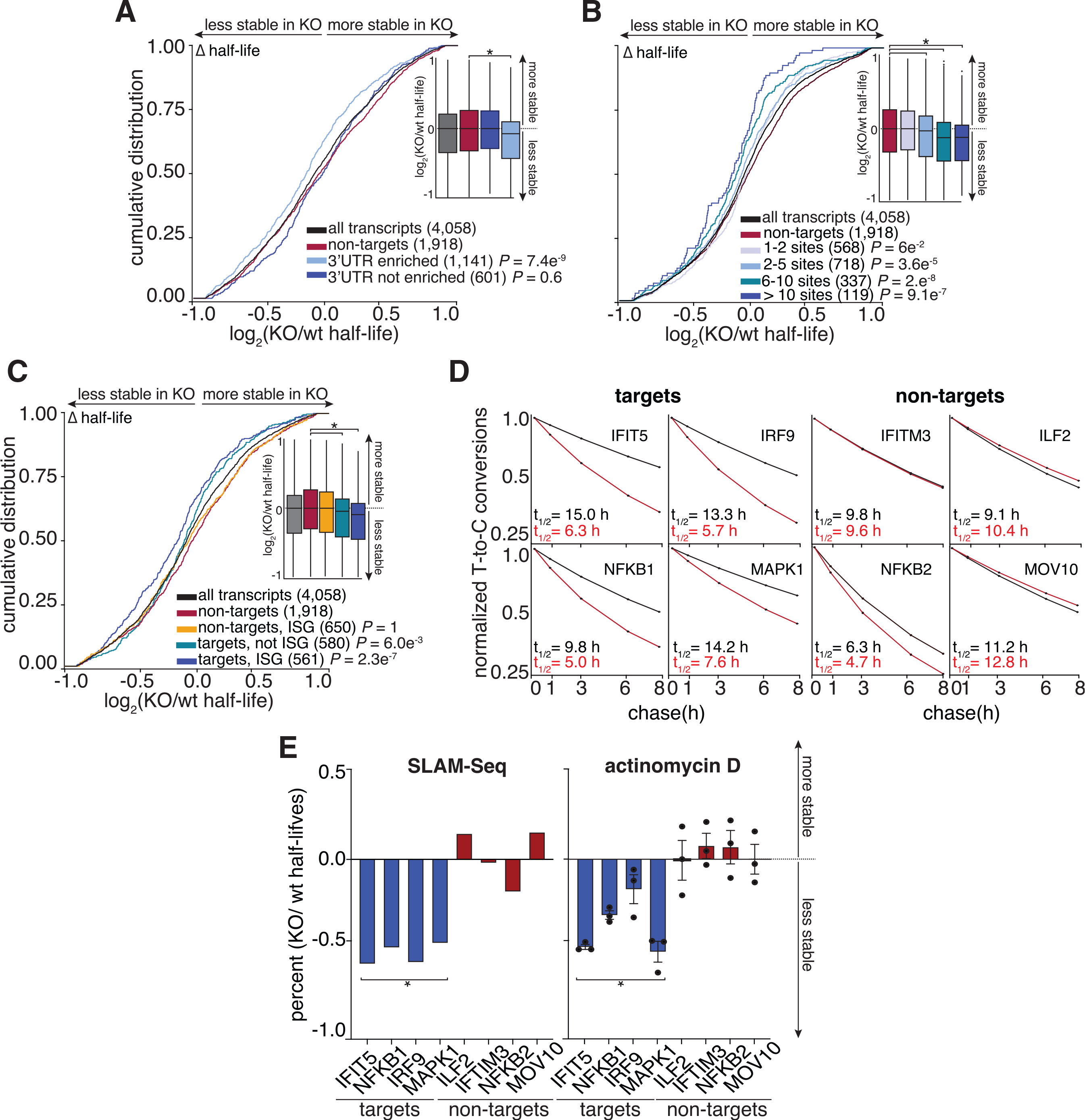
(A and B) Cumulative distribution analyses of the log2 fold change in half-life (KO/WT), binning transcripts based on enrichment. Insets show a boxplot of the log2 fold change (KO/WT) in half-life based on indicated groupings.
(C) Cumulative distribution analysis of the log2 fold change of half-life (KO/WT) to test if transcripts classified as ISGs are more affected by the loss of ELAVL1 than non-ISGs.
(D) Transcript stability plots and the calculated RNA half-lives of the indicated ISG mRNA targets or non-targets are shown; ELAVL1-WT (black) or ELAVL1-KO THP-1 cells (red).
(E) Bar plots validating the change in the half-lives (KO/WT) of the listed transcripts caused by the loss of ELAVL1 measured by SLAM-seq and actinomycin D experiments. n = 2 (SLAM-Seq) and n = 3 (actinomycin D), error bars represent mean ± SD. *p < 0.05.
To further establish a direct link between ELAVL1 binding and mRNA stability, we generated reporter plasmids expressing the luciferase open reading frame (ORF) followed by the WT or the mutant 3′ UTR of IRF9 (Figure S4D). The IRF9 3′ UTR was used because it contains two distinct 3′ UTR binding sites and is an enriched, functional target validated through multiple independent experiments. THP-1 cells were transfected with both plasmids and 24 h later stimulated with cGAMP. The transfected and activated cells were then treated with actinomycin D to inhibit transcription, followed by taking time-resolved samples to measure mRNA decay by quantitative real-time PCR using primers that can distinguish between the WT and mutant 3′ UTRs. We found that the mRNA fused with the mutant 3′ UTR showed a statistically significant decrease in half-life from 3.4 to 2.6 h (Figure S4E), a 24% drop in transcript stability that is consistent with our quantitative real-time PCR analysis of the endogenous IRF9 transcript (Figure S4B).
Pathway analysis showed that the ELAVL1-regulated targets (n = 409), which we defined as 3′ UTR-enriched targets whose half-life decreased by 1.5-fold change in the absence of ELAVL1, encode for proteins associated with endocytosis, transcriptional dysregulation in cancer, and multiple innate immune signaling pathways (Figure 7A; Table S7). Of the immune-relevant pathways, ELAVL1 regulated components of interleukin-17 (R-HSA-448424), TNF pathway (R-HSA-75893), and TLR signaling (R-HSA-168164). Other signaling pathways, such as MAPK (R-HSA-450294) and apoptosis (R-HSA-109606) terms, were also enriched. MAPK signaling, along with NF-κB and IRFs, are important for generating an immunoreactive state in the presence of a pathogen (Arthur and Ley, 2013). In the case of TLR signaling, ELAVL1 binds directly and stabilizes the adaptor protein (TRAF6), a kinase involved in integrating upstream PRR activity (TAK1) and the transcription factor itself (NF-κB), which are all positive regulators of the pathway. Overall, we found that these innate immune pathways form a network containing regulators of ISG expression (STAT3, MAPK, IRF9, FOS, NF-κB, RIPK2) (Figure 7B) (Gilchrist et al., 2012; Mostafavi et al., 2016). Thus, our data indicate that ELAVL1 stabilizes the transcripts of ISGs and ISG regulators critical for a cell to mount an immunoreactive state.
Figure 7. Canalization of ELAVL1 function toward the post-transcriptional regulation of immunologic pathways by IRF3 stimulation.
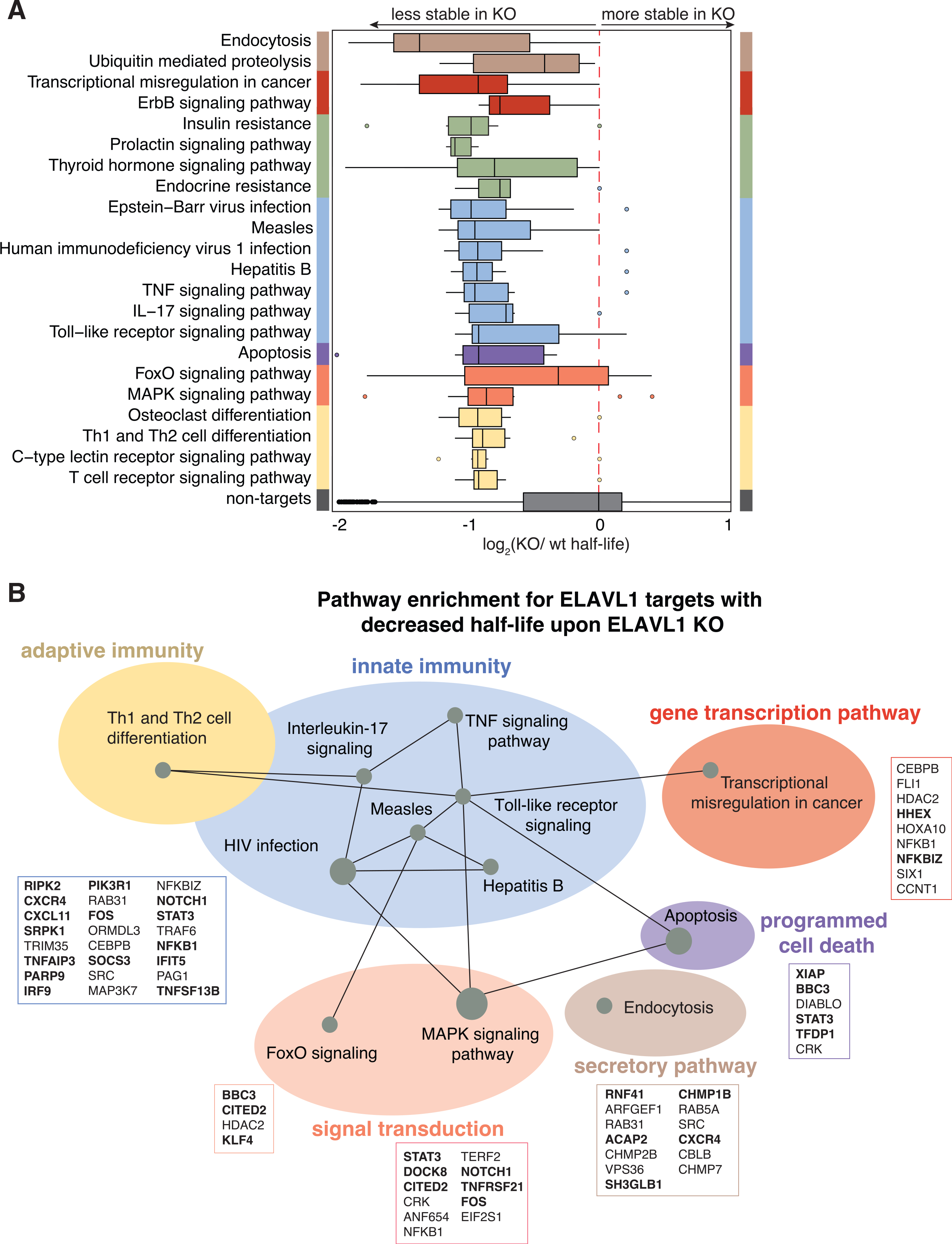
(A) Boxplot grouping transcripts based on the top enriched Reactome pathway terms of the ELAVL-regulated targets protein components.
(B) Pathways of functional targets of ELAVL1. Each node represents a specific pathway, and colored circles represent closely related pathways. Connections represent shared genes between pathways. Boxes show specific mRNA transcripts and ISGs (bolded) in each pathway that are targeted by ELAVL1.
Of note, we found that a surprising number of the highly regulated transcripts were already bound by ELAVL1 in the THP-1-naive state. However, their enrichment level (based on rank) was significantly higher upon immune stimulation. This increase in enrichment was concomitant with higher 3′ UTR fractional occupancy, reinforcing the importance of a transition to 3′ UTR binding. Only ~10% of our ELAVL1-regulated targets were found as enriched or even affected by the knockdown of ELAVL1 in HEK293 cells, underscoring the importance of examining ELAVL1 targets under changing transcriptomic contexts.
DISCUSSION
We present a multi-layered analysis of high-throughput transcriptomics of the targeting and functional outcomes of the RBP ELAVL1 during an innate immune response. We find that ELAVL1 largely transitions to binding the 3′ UTRs of mRNA transcripts during an innate immune response. 3′ UTR binding is an absolute prerequisite for enrichment, and KO of ELAVL1 led to widespread destabilization of its enriched transcripts. Specifically, we found that highly regulated targets had a 3-fold average reduction in their stabilities, losing 30%–80% of their original half-lives. Importantly, ELAVL1-regulated targets encode for ISGs and their transcriptional regulators, suggesting that ELAVL1 contributes at multiple levels of a pro-inflammatory response.
Two other ELAVL1 binding site reports performed in HeLa and bone marrow-derived macrophages (BMDMs) show that ELAVL1 mostly binds the 3′ UTR of mRNA targets (Lebedeva et al., 2011; Sedlyarov et al., 2016). In both of these reports, investigators immunoprecipitated ELAVL1 using endogenous antibodies. In our hands, we found that endogenous antibodies obstructed RNA-protein interactions. Therefore, we used anti-FLAG antibodies and compared our results with the previous PAR-CLIP work performed in HEK293 cells, which used the same antibodies. We also observed a high proportion of ELAVL1 binding occurring at intronic sites, particularly during the naive state. Nonetheless, our data did not show a link between ELAVL1 intronic sites and an increase in mature mRNA stability (Lebedeva et al., 2011; Mukherjee et al., 2011). Those previous reports used microarrays that could detect introns and pre-mRNAs to discover an intron-dependent role of ELAVL1 for stabilizing pre-mRNAs. In contrast, our approach with SLAM-seq was geared toward measuring the RNA decay rates of mature transcripts. Although intuitively changes in pre-mRNA levels would be predicted to contribute to the eventual levels of mature transcripts, we did not observe an intron-dependent effect on mature mRNAs. It is conceivable that our approach was insufficiently sensitive to detect intronic-dependent stability effects or that the two ELAVL1-dependent mechanisms are distinct.
One of the strongest signatures we were able to identify was a shift in the distribution of binding sites in favor of the 3′ UTRs, and we speculate that this change is most likely due to the subcellular relocalization of ELAVL1 to the cytoplasm upon immune stimulation. As previously reported, ELAVL1 can be post-translationally modified by kinases and methylases, which can affect its nucleocytoplasmic localization and RNA binding site distribution (Grammatikakis et al., 2017). Future studies using subcellularrestricted forms of ELAVL1 (e.g., phospho-ablated or phospho-mimetic) could add to the granularity of our understanding of how ELAVL1 transitions from binding intronic versus 3′ UTR sites and its movement out of the nucleus. However, the subcellular localization of ELAVL1 can only partially explain our work because signal transduction and associated transcriptomic differences between naive and stimulated states also play a role in defining the ELAVL1-regulated mRNAs (Abe et al., 2012; Rabani et al., 2011). ELAVL1 likely competes with other post-transcriptional factors and might also be affected by viral RNAs that act as competitors to titrate it away from cellular targets (Barnhart et al., 2013; Dassi, 2017; Hentze et al., 2018; Kim et al., 2020; Lu et al., 2014). Therefore, when characterizing the function of a given RBP and its biological role, it is important to consider how cellular context-specific factors can have a profound impact on how a target enriches with an RBP and the extent of its regulation.
Because transient associations can be identified between potential RNA targets and RBPs by CLIP methodologies, there is a need to quantify enrichment levels as a measure of interaction affinity. The stoichiometry of interactions between the limited number of RBP molecules and its changing RNA substrate pool makes it difficult to predict enrichment and post-transcriptional regulatory impact, especially when using information gleaned strictly from steady-state data and for RBPs with a preference for highly redundant recognition sequences (Ascano et al., 2012). High-throughput discovery methods, such as RNAcompete and RNA Bind-n-Seq, show that most RBPs tested have convergent RNA recognition sequences (Dominguez et al., 2018; Lambert et al., 2014; Ray et al., 2017). For 27 different RBPs (including ELAVL1), the preferred 6-mer binding site overlapped with the top-ranked 6-mer site of at least one other RBP, despite having distinct RNA-binding domains (Dominguez et al., 2018). This observation underscores that many RBPs have similar and short binding motif preferences of low complexity (Adinolfi et al., 2019; Nussbacher and Yeo, 2018). Although these studies are valuable in discovering the primary and flanking sequence preferences of RBPs, these methods often analyze a single RBP in isolation and do not include competitor RBPs and miRNAs that will influence the RNA-substrate structure or the availability of a particular RNA binding site. Therefore, relying on sequence motifs and predicted RNA structure is insufficient to delineate the specific binding sites and the functional RNA targets of RBPs in specific cells and contexts. In recognizing these limitations, we integrated multiple high-throughput sequencing datasets to differentiate between RNA transcripts that are simply “sampled” versus bona fide targets that are regulated during an immune signaling event.
A majority of targets exhibited a reduction in their half-lives upon loss of ELAVL1. We termed these mRNAs ELAVL1-regulated transcripts and performed pathway analysis to understand the cellular pathways that ELAVL1 could regulate during an immune response. Many of them encode innate immune signaling components that positively drive the expression of ISGs. These genes reinforce upstream signaling events triggered by PRRs, leading to enhanced cytokine and interleukin production. We speculate that ELAVL1 regulates these central signaling components to sensitize the cell for dealing with an infection, allowing the cell to quickly integrate incoming pathogen- or damage-associated molecular pattern-triggered signaling. ELAVL1 regulates the mRNA of several biological processes within immune signaling pathways, from adaptor protein (TRAF6) to transcription factor (NF-κB, IRF9). ELAVL1 also regulates components of endocytosis, which plays a role in cytokine signaling and TLR receptor trafficking (Kurgonaite et al., 2015; Lund and DeLotto, 2011). These transcripts were strongly regulated by ELAVL1 upon immune stimulation, despite many of these targets already being sampled by the RBP under naive states. Interestingly, we also observed that the half-lives of nearly 100 highly enriched transcripts increased in the absence of ELAVL1. This suggests that ELAVL1 can also destabilize targets. Together, our data support the idea of an “RNA regulon” where an RBP can differentially regulate distinct groups of functionally related mRNAs (Keene, 2007; Simone and Keene, 2013). Ultimately, the final fate of an mRNA will be subject to its combination of bound RBPs.
With the increased interest in understanding the function of RBPs in gene regulation, numerous laboratories have undertaken essential and broad surveys of the target spectra of RBPs, largely under steady-state conditions (Castello et al., 2016; Darnell, 2010; Hafner et al., 2010; König et al., 2010; Van Nostrand et al., 2016, 2020; Ule et al., 2003). But it is important to recognize that RBPs represent a broad class of gene regulators that are post-translationally modified and are sensitive to cellular signaling events and changing transcriptomes. In examining the contribution of ELAVL1 to immune stimulation, we provide a general framework for studying other RBPs subject to analogous changes to a dynamic transcriptome or signal transduction event. This is especially relevant during host-pathogen interactions when substrate RNAs compete for post-transcriptional gene regulation by cellular proteins, leading to dramatic changes in the balance of host versus pathogenic transcript binding with limited trans-acting factors.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Dr. Manuel Ascano, manuel.ascano@vanderbilt.edu.
Materials availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Manuel Ascano (manuel.ascano@vanderbilt.edu). All plasmids and stable cell lines generated in this study are available without restrictions from the Lead Contact and/or through Addgene.
Data and code availability
All code used for sequencing analysis and figure generation is accessible at https://github.com/Ascano-Lab. All sequencing data are deposited to National Center for Biotechnology Information via Sequence Read Archive (SRA); BioProject ID: PRJNA659987.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture
Human THP-1 s monocytes (male) were cultured in RPMI (GIBCO) with 10% fetal bovine serum (FBS from Peal Serum), 100 μg/ml streptomycin (GIBCO), 100 U/ml penicillin (GIBCO), 25 μg/ml blasticidin, and 100 μg/ml hygromycin (Invivogen).
Plasmid construction
For the cloning of the lentiviral expression construct, the coding sequence of ELAVL1 was PCR amplified from THP-1 cDNA, introducing attB-sites for the Flip-IN-recombinase system. The PCR product was gel purified and then recombined (LR clonase) into the pLenti-CMVtight-Flag-HA-DEST-Blast plasmid. The Flag-HA-tag lentiviral inducible expression vector pLenti CMVtight Blast Flag-HA-DEST was constructed by insertion of Flag-HA-tag from pFRT_TO_DEST Flag-HA (#26361, Addgene) into the plasmid pLenti-CMVtight-Blast-DEST (w762–1) (#26434, Addgene).
For the luciferase reporter plasmid, the wt and mutant 3′UTRs of IRF9 were custom-ordered from IDT (MiniGene) and cloned into the firefly luciferase 3′UTR plasmid (#12178, Addgene) with SacI (5′ end) and XbaI (3′end) and T4 DNA ligation (NEB).
METHOD DETAILS
Lentiviral production and generation of inducible expressing Flag-HA ELAVL1
For lentiviral production, HEK293T cells were first cultured in 15 cm plates in high glucose DMEM (GIBCO) supplemented with 10% FBS. They were then transfected using Lipofectamine 2000 (Invitrogen) according to manufacture suggestion with 9 μgs of lentiviral vector and 9 μg viral particle packaging vectors, 6.75 μg psPAX2 (12260, Addgene) and 2.25 μg pMD2.G(12259, Addgene). 48 hours after transfection, the viral particle-containing supernatant was collected and spun-down at 3000 g for 15 mins. The supernatant was then concentrated and purified by layering the supernatant over a 20% sucrose cushion in TNE buffer (50 mM Tris-HCl [pH 7.2], 0.1 M NaCl, and 1 mM EDTA) and ultra-centrifuged at 25,000 g for 4 hours in a Beckman SW32Ti rotor. Viral pellets were then resuspended in fresh DMEM media and filtered through a 0.45 μm syringe filter unit (Millex-HV). For viral transduction, THP-1-rtTA cells were spuninoculated (800 g for 2 hours at 32°C) with an MOI ~100. Two days after viral inoculation, cells were moved into selection media 25 μgs/ml blasticidin and 100 μg/ml hygromycin. Expression of ELAVL1 was then verified via immunoblot using both ELAVL1 endogenous antibody or anti-HA antibody.
RNA-sequencing and library prep
RNA from 1 × 106 THP-1cells were collected at the indicated time point and were washed with 1 × PBS. For stimulated samples, we activated cells for 16 hours with the 70 nM (EC50) of encapsulated cGAMP (Shae et al., 2019). Washed cells were then resuspended in 1 mL of TRizol. RNA was extracted following the manufacture’s protocol. Total RNA was converted into cDNA and sequenced using NEBNext DNA Library Prep Kit for Illumina on the Illumina NovaSeq6000 platform using PE150 at the Vanderbilt Technologies for Advanced Genomics (VUMC VANTAGE). Fastq files were pre-processed with trim-galore with the default settings (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) to remove any adaptor contamination and then aligned to the human genome (Genocode, hg19) with STAR mapper (Dobin et al., 2013). DeSeq2 (Love et al., 2014) was used to calculate differential expressed genes.
PAR-CLIP
PAR-CLIP was performed as previously described (Garzia et al., 2017; Hafner et al., 2010) with minor adjustments. In brief, 3–5 × 109 THP-1 cells were doxycycline induced and labeled with 100 μM 4SU 16 hours before harvesting and UV365nm irradiation. Stimulated THP-1 cells were additionally treated with EC50 cyclic GMP-AMP 16 hours before harvest. After crosslinking, THP-1 cells were lysed using NP-40 lysis buffer (50 mM HEPES [pH 7.5], 150 mM KCl, 2 mM EDTA, 1 mM NaF, 2% (v/v) NP-40, 0.5 mM DTT, Roche EDTA-free protease inhibitor) and incubated with Dyna-protein G beads (Invitrogen) coupled with anti-FLAG M2 antibody (Sigma) for 2 hours at 4°C. Beads were washed with high-salt buffer and then underwent CIP and T4 PNK mediated 5′ end RNA radiolabeling with [γ−32P]-ATP. Flag-tagged ELAVL1 crosslinked to RNA was then was resolved on a 4%–20% Bis-Tris, NuPage gradient gel (Invitrogen). The band corresponding to ELAVL1 protein was cut out. The protein: RNA complex was then electroeluted out of the gel and treated with proteinase K (Roche). RNA was then size-selected and underwent both 3′ (MultiplexDX Inc.) and 5′ adaptor (Illumina compatible) ligation and was reverse transcribed into cDNA. cDNA library was sequenced on the NextSeq Illumina platform at Hudson Alpha.
Defining binding sites
PARpipe (https://github.com/ohlerlab/PARpipe) (Mukherjee et al., 2019) with PARalyzer (Corcoran et al., 2011; Mukherjee et al., 2014) was used to define crosslinking sites from the PAR-CLIP data. PARalyzer calculates the T-to-C fraction, which serves as a quality index that is calculated based on the frequency of a given uracil (thymidine) to be substituted with a cytosine. For groups of reads (> 5 unique reads), kernel density estimates were calculated for both reads with and without T-to-C conversions. Clusters (i.e., binding sites) were defined as a group of transcripts that had a higher kernel density for T-to-C converted reads over unmodified reads.
Motif analysis
For the 6-mer analysis, we counted the most frequent 6-mers from each unique PAR-pipe called clusters annotated as either intron or 3′UTR using BioStrings R/Bioconductor (Yu et al., 2012). To calculate the top enriched 6-mers for ELAVL1, we regressed the 6-mer frequency relative to a reference library of annotated 3′UTRs or introns (Mukherjee et al., 2019).
RIP-sequencing
Immunoprecipitation was performed as previously described in the PAR-CLIP method section without UV crosslinking or RNase treatment. An anti-IgG immunoprecipitation (IP) was used to subtract background RNA expression that is intrinsic of IPs. Following anti-FLAG and anti-IgG IP, beads were added to 1 mL TRizol (Ambion), and RNA was extracted following the manufacture’s protocol, and total RNA was submitted to the Vantage sequencing core.
Reactome analysis
Reactome analysis was performed using R/Bioconductor packages clusterProfiler with default settings. (Yu et al., 2012).
RT-qPCR
RNA collected and extracted using Trizol (Ambion). The concentration of total RNA was determined using NanoDrop 2000 (ThermoFisher). Equal amounts of total RNA for each sample were reverse transcribed using SuperScript III (ThermoFisher) with random hexamer. Real-time PCR reactions were done with FastSYBR Green Plus Master Mix (Applied Biosystems) and a StepOnePlus qPCR machine (Applied Biosystems). Target Ct values were normalized to TUBA1A Ct values and used to calculate ΔCt. Relative mRNA expression of target genes was then calculated using the ΔΔCt method (2ΔΔCt).
Nucleo-cytoplasmic fractionation
Harvested cells were washed with 1 × PBS and resuspended in hypotonic lysis buffer (10 mM HEPES, 60 mM KCl, 1 mM EDTA, 0.075% (v/v) NP-40, 1 mM DTT, and 1 mM EDTA-free Roche PMSF) and incubated on ice for 10 minutes. Cell lysis was then centrifuged at 4°C (15,000 g, 5 minutes) to pellet the nuclear fraction. The top cytoplasmic fraction was removed and placed into a new tube. Nuclear fraction pellet was washed twice in hypotonic lysis buffer without NP-40 and then lysed with hypertonic lysis buffer (20 mM Tris-Cl, 420 mM NaCl, 1.5 MgCl2, 0.2 mM EDTA, 1 mM EDTA-free PMSF (Roche), and 25% (v/v) glycerol). Nuclear lysis was then centrifuged at 4°C (20,000 g, 5 minutes), and the supernatant was collected as the nuclear fraction.
Antibodies and immunoblotting
The antibodies anti-ELAVL1 (ab170193) and anti-TUBA4A (ab7291) are from Abcam; anti-Histone 2A (JBW301) and anti-phospho HuR (Ser 221) (ABE265) are from Sigma. anti-GM130 (12480) is from Cell Signaling.
Samples were separated by SDS-PAGE. After electrophoresis, proteins were semi-dry transferred (Bio-Rad) to nitrocellulose membranes (Hybond-ECL, GE Life Science). Protein membranes were processed via a standard immunoblot protocol followed by enhanced chemiluminescent detection (Luminata Forte ECL, Millipore) using a chemiluminescence imaging system (ChemiDoc MP, Bio-Rad).
Generations of Cas9 sgRNA knockout in THP-1 monocytes
crRNAs were designed using the CRISPR design tool in Benchling [Biology Software] (2019) retrieved from https://www.benchling.com/crispr/ and ordered from IDT. To assemble Cas9/sgRNA RBPs, Alt-R crRNAs and trcRNAs were first reconstituted in Nuclease Free Duplex buffer (IDT). An equimolar ratio of crRNA and trcRNA were added to a nuclease-free tube and denatured by heating at 95°C for 5 minutes. The oligo duplex was then cooled at room temperature for 10 minutes prior to adding Cas 9 nuclease enzyme (IDT) to assemble RNPs (Vakulskas et al., 2018). Duplexed oligos and Cas9 were then incubated at room temperature for 20 minutes prior to THP-1 electroporation.
For electroporation, 2 × 106 THP-1 cells per sample were counted and washed twice with 1 × PBS. Assembled Cas9 RNPs and washed cells were then suspended in 100 μL of R buffer (NeonTransfection) and electroporated in 100 μL tip with the NeonTransfection unit (1600V, 10 ms, 2 pulses). Electroporate cells were then added to pre-warmed THP-1 media (RPMI + 10% FBS) without antibiotics and cultured in an incubator 37 C + 5% CO2 for 48 hours before changing the media.
SLAM-Seq
THP-1 (ELAVL1-wt and ELAVL1-KO) cells were seeded the day before the experiment at a density of 1 × 106 cells/ ml. For the pulse, cells were labeled with 100 μM 4SU and stimulated with 70 nM (EC50) cyclic GMP-AMP 16 hours before harvest. For the chase, cells were washed twice in 1x PBS and then incubated with RPMI + 10% FBS supplemented with 10 mM of uridine (Sigma). Cells were harvested, washed in 1x PBS, and added to TRIzol at the respective time points (0, 1, 3, 6, and 8 hours after the chase). Total RNA (~5 μgs per sample) was extracted and then treated with 10 mM iodoacetamide (Sigma) as described in Herzog et .al, 2017 and in Protocol Exchange (https://doi.org/10.1038/protex.2017.105). SLAM-Seq libraries were prepared using the Lexogen QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina (Cat. No. 015.24) following the manufacturer’s instructions. Libraries were sequenced using Illumina NextSeq 550 in a 75 bp single-end mode. SLAM-Seq libraries were analyzed as previously described in Herzog et al. (2017) and (Neumann et al., 2019). Briefly, read converged normalized T-to-C conversion rates were generated using the SLAM-DUNK pipeline. To calculate RNA half-lives, T-to-C conversion rates were normalized to chase onset (0-hour time point) and used to fit a first-order decay reaction in R using the min-pack. lm package (Elzhov, 2016).
Calculating RNA half-life with actinomycin D
THP-1 cells (ELAVL1-wt and ELAVL1-KO) were seeded the day before the experiment at a density of 1 × 106 cells/ ml and stimulated with 70 nM cyclic GMP-AMP 16 hours before polymerase II inhibition with actinomycin D (5 μg/ mL). Cells were harvested, washed in 1x PBS, and added to TRIzol at the respective time points (0, 1, 2, 3, 4, and 5 hours after transcription inhibition). The concentration of total RNA was determined using NanoDrop 2000 (ThermoFisher). Equal amounts of total RNA for each sample were reverse transcribed using SuperScript III (ThermoFisher) with random hexamer. Real-time PCR reactions were done with FastSYBR Green Plus Master Mix (Applied Biosystems) and a StepOnePlus qPCR machine (Applied Biosystems). Target Ct values were normalized to 18S Ct values and used to calculate ΔCt. Relative mRNA abundance was calculated to the start of transcription inhibition (0-hour time point*) (2−(ΔCt − ΔCt*)) for each gene. RNA half-lives were calculated using first-order decay in Prism 9.
For the luciferase-3′UTR half-life reporter, THP-1 cells (1 × 106 cells/ ml) were seeded in a 6-well plate and transfected with wt and mutant 3′UTR luciferase plasmids using Lipofectamine LTX PLUS following the manufacture’s protocol. After 24-hour transfection, cells were stimulated with 70 nM cyclic GMP-AMP 16 hours before actinomycin D (5 μg/ mL) treatment.
QUANTIFICATION AND STATISTICAL ANALYSIS
The significant differences between cumulative distribution fractions were calculated using Wilcoxon.Test, and P.values were adjusted using the Bonferroni method to the number of observations. The Student’s t test was used for statistical analysis (e.g., the difference in the number of binding sites between two conditions). Number of biological replicates of assays are defined within the corresponding figure. Error bars shown in the Figures represent means ± SD.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-GM130 | Cell Signaling | Cat#12480; RRID:AB_2797933 |
| Rabbit monoclonal anti-HuR / ELAVL1 | abcam | Cat#ab170193; RRID:AB_2784506 |
| Mouse monoclonal anti-TUBA4A | abcam | Cat#ab7291; RRID:AB_2241126 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat#F1804; RRID:AB_262044 |
| Mouse monoclonal anti-HA | BioLegend | Cat#901502; RRID:AB_2565007 |
| Rabbit monoclonal anti-ELAVLI P221 | Sigma-Aldrich | Cat#ABE265 |
| Mouse monoclonal IgG1 Isotype Control | Cell Signaling | Cat#5415; RRID:AB_10829607 |
|
Bacterial and virus strains | ||
| One Shot TOP10 Chemical Competent E.coli | ThermoFisher | Cat#C404010 |
| STBL3 Chemical Competent E.coli | ThermoFisher | Cat#C737303 |
|
Chemicals, peptides, and recombinant proteins | ||
| Actinomycin D | Sigma | Cat#A9415 |
| proteinase K | Sigma | Cat#3115879001 |
| RNaseT1 | Thermo Scientific | Cat#EN0542 |
| CIP | NEB | Cat#M0525S |
| T4 PNK | NEB | Cat#M0201S |
|
Critical commercial assays | ||
| Fast SYBR Green Master Mix | Thermo Scientific | Cat#4385612 |
|
Deposited data | ||
| PAR-CLIP data of ELAVL1 from THP-1 s | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157052 |
| RIP-Seq data of ELAVL1 from THP-1 s | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157052 |
| SLAM-Seq data | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157052 |
| RNA-Seq data from THP-1 s | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157052 |
| Database of annotated interferon-regulated genes | Rusinova et al., 2013 | http://www.interferome.org/interferome/home.jspx |
|
Experimental models: Cell lines | ||
| THP-1 | ATCC | TIB-202 |
| U937 | ATCC | CRL-1539.2 |
| 293T | ATCC | CRL-3216 |
|
Oligonucleotides | ||
| Oligomers used in study, please see Table S8 | This paper | N/A |
|
Recombinant DNA | ||
| pLenti CMVtight Blast DEST (w762–1) | (Campeau et al., 2009) | Addgene #26434 |
|
Software and algorithms | ||
| GO analysis (clusterProfiler package for R) | Yu et al., 2012, | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| ggplot2 package for R | (Wickham, 2016) | https://ggplot2.tidyverse.org |
| UpSetR package for R | (Conway et al., 2017) | |
| DeSeq2 package for R | Love et al., 2014 | https://github.com/mikelove/DESeq2 |
| PARalyzer | Corcoran et al., 2011; Mukherjee et al., 2014 | https://ohlerlab.mdc-berlin.de/software/PARalyzer_85/ |
| SLAM-DUNK | Neumann et al., 2019 | https://github.com/t-neumann/slamdunk |
| Trim-galore | (Martin, 2011) | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Benchling | Benchling [Biology Software] 2019 | https://www.benchling.com/ |
| Graph pad Prism 8 | Graph Pad software | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
ELAVL1 changes its mRNA binding distribution during innate immunity
ELAVL1 stabilizes the mRNAs of interferon-stimulated genes (ISGs)
RNA half-lives of ISG targets decrease in the absence of ELAVL1
ACKNOWLEDGMENTS
We thank Dr. Kelly Barnett for computational help, Monica Bomber for technical help with the generation of the ELAVL1-KO, and the Vanderbilt Technologies for Advanced Genomics (VANTAGE) core for sequencing. We would like to thank Jessalyn Baljo, Dr. Daniel Shae, and Dr. John Wilson for the encapsulated cGAMP. Finally, we thank members of the Ascano laboratory for their support, collegiality, and critical review of the manuscript. This work was supported by the National Institutes of Health grant 1R35GM119569-01 (to M.A.); Vanderbilt University Department Biochemistry start-up funds (to M.A.); University of Colorado Anschutz Medical Campus RNA Bioscience Initiative (N.M.); Boettcher Foundation Webb-Waring Early Career Investigator Award AWD-103075 (N.M.); Chemistry-Biology Interface training grant 5T32GM065086-14 (to K.R.); Chemical Biology of Infectious Disease training grant 5T32AI11254-02 (to S.A.); Cellular, Biochemical and Molecular Sciences Training Program 5T32GM008554 (to S.L.); and Clinical and Translation Science Award no. UL1 TR002243 (to K.R.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109178.
REFERENCES
- Abe K, Ishigami T, Shyu A-B, Ohno S, Umemura S, and Yamashita A (2012). Analysis of interferon-beta mRNA stability control after poly(I:C) stimulation using RNA metabolic labeling by ethynyluridine. Biochem. Biophys. Res. Commun 428, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner KP, Ludwig J, and Hornung V (2013). cGAS produces a 2′−5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adinolfi M, Pietrosanto M, Parca L, Ausiello G, Ferre F, Helmer-Citterich M, et al. (2019). Discovering sequence and structure landscapes in RNA interaction motifs. Nucleic Acids Research. 10.1093/nar/gkz250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P (2009). Post transcriiptional regulons coordinate the initiation and resolution of inflammation. Nature Reviews Immunology. 10.1038/nri2685. [DOI] [PubMed] [Google Scholar]
- Anderson P (2010). Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat. Rev. Immunol 10, 24–35. 10.1038/nri2685. [DOI] [PubMed] [Google Scholar]
- Arthur JSC, and Ley SC (2013). Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol 13, 679–692. [DOI] [PubMed] [Google Scholar]
- Ascano M, Hafner M, Cekan P, Gerstberger S, and Tuschl T (2012). Identification of RNA-protein interaction networks using PAR-CLIP. Wiley Interdiscip. Rev. RNA 3, 159–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhart MD, Moon SL, Emch AW, Wilusz CJ, and Wilusz J (2013). Changes in cellular mRNA stability, splicing, and polyadenylation through HuR protein sequestration by a cytoplasmic RNA virus. Cell Rep. 5, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidet K, and Garcia-Blanco MA (2014). Flaviviral RNAs: weapons and targets in the war between virus and host. Biochem. J 462, 215–230. [DOI] [PubMed] [Google Scholar]
- Brennan CM, and Steitz JA (2001). HuR and mRNA stability. Cell. Mol. Life Sci 58, 266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu Y-H, and Chen ZJ (2014). The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296. [DOI] [PubMed] [Google Scholar]
- Campeau E, Ruhl V, Rodier F, Smith C, L, Rahmberg B, Fuss J, Campisi J, Yaswen P, Cooper P, and Kaufman P (2009). A Versatile Viral System for Expression and Depletion of Proteins in Mammalian Cells. Plos One. 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Fischer B, Frese CK, Horos R, Alleaume A-M, Foehr S, Curk T, Krijgsveld J, and Hentze MW (2016). Comprehensive Identification of RNA-Binding Domains in Human Cells. Mol. Cell 63, 696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, and Shyu AB (1995). AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci 20, 465–470. [DOI] [PubMed] [Google Scholar]
- Christodoulou-Vafeiadou E, Ioakeimidis F, Andreadou M, Giagkas G, Stamatakis G, Reczko M, Samiotaki M, Papanastasiou AD, Karakasiliotis I, and Kontoyiannis DL (2018). Divergent Innate and Epithelial Functions of the RNA-Binding Protein HuR in Intestinal Inflammation. Front. Immunol 9, 2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway J, Lex A, and Gehlenborg N (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics. 10.1093/bioinformatics/btx364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, and Ohler U (2011). PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol. 12, R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ (2015). Type I interferonopathies: mendelian type I interferon upregulation. Curr. Opin. Immunol 32, 7–12. [DOI] [PubMed] [Google Scholar]
- Darnell RB (2010). HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 1, 266–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassi E (2017). Handshakes and Fights: The Regulatory Interplay of RNA-Binding Proteins. Front. Mol. Biosci 4, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, and Prescott SM (2001). Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J. Clin. Invest 108, 1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T, Bazile C, Lambert NJ, Van Nostrand EL, Pratt GA, et al. (2018). Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell 70, 854–867.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G, Kim VN, and Kataoka N (2002). Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol 3, 195–205. [DOI] [PubMed] [Google Scholar]
- Fan XC, and Steitz JA (1998). HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc. Natl. Acad. Sci. USA 95, 15293–15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzhov T (2016). R Interface to the Levenberg-Marquardt Nonlinear Least-Squares Algorithm Found in MINPACK, Plus Support for Bounds. CRAN. [Google Scholar]
- Fan J, Ishmael FT, Fang X, Myers A, Cheadle C, Huang S-K, Atasoy U, Gorospe M, and Stellato C (2011). Chemokine transcripts as targets of the RNA-binding protein HuR in human airway epithelium. J. Immunol 186, 2482–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangou EA, Bertsias GK, and Boumpas DT (2013). Gene expression and regulation in systemic lupus erythematosus. Eur. J. Clin. Invest 43, 1084–1096. [DOI] [PubMed] [Google Scholar]
- Fu M, and Blackshear PJ (2017). RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat. Rev. Immunol 17, 130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. (2013a). Cyclic [G(2′,5′)pA(3′,5′) p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, et al. (2013b). Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 154, 748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzia A, Meyer C, Morozov P, Sajek M, and Tuschl T (2017). Optimization of PAR-CLIP for transcriptome-wide identification of binding sites of RNA-binding proteins. Methods 118–119, 24–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer F, Preiss T, and Hentze MW (2012). From cis-regulatory elements to complex RNPs and back. Cold Spring Harb. Perspect. Biol 4, a012245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstberger S, Hafner M, and Tuschl T (2014). A census of human RNA-binding proteins. Nat. Rev. Genet 15, 829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist DA, Fromm G, dos Santos G, Pham LN, McDaniel IE, Burkholder A, Fargo DC, and Adelman K (2012). Regulating the regulators: the pervasive effects of Pol II pausing on stimulus-responsive gene networks. Genes Dev. 26, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammatikakis I, Abdelmohsen K, and Gorospe M (2017). Posttranslational control of HuR function. Wiley Interdiscip. Rev. RNA 8, e1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr., Jungkamp A-C, Munschauer M, et al. (2010). Transcriptome-wide identification of RNA-binding protein and micro-RNA target sites by PAR-CLIP. Cell 141, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, and Baltimore D (2009). The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol 10, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Castello A, Schwarzl T, and Preiss T (2018). A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell. Biol 19, 327–341. [DOI] [PubMed] [Google Scholar]
- Herdy B, Karonitsch T, Vladimer GI, Tan CSH, Stukalov A, Trefzer C, Bigenzahn JW, Theil T, Holinka J, Kiener HP, et al. (2015). The RNA-binding protein HuR/ELAVL1 regulates IFN-b mRNA abundance and the type I IFN response. Eur. J. Immunol 45, 1500–1511. [DOI] [PubMed] [Google Scholar]
- Herman AB, Vrakas CN, Ray M, Kelemen SE, Sweredoski MJ, Moradian A, Haines DS, and Autieri MV (2018). FXR1 Is an IL-19-Responsive RNA-Binding Protein that Destabilizes Pro-inflammatory Transcripts in Vascular Smooth Muscle Cells. Cell Rep. 24, 1176–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog VA, Reichholf B, Neumann T, Rescheneder P, Bhat P, Burkard TR, Wlotzka W, von Haeseler A, Zuber J, and Ameres SL (2017). Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 14, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel P, Urban C, Bergant V, Schneider WM, Knauer B, Stukalov A, Scaturro P, Mann A, Brunotte L, Hoffmann HH, et al. (2019). A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat. Immunol 20, 493–502. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, and Barber GN (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafasla P, Skliris A, and Kontoyiannis DL (2014). Post-transcriptional coordination of immunological responses by RNA-binding proteins. Nat. Immunol 15, 492–502. [DOI] [PubMed] [Google Scholar]
- Katsanou V, Papadaki O, Milatos S, Blackshear PJ, Anderson P, Kollias G, and Kontoyiannis DL (2005). HuR as a negative posttranscriptional modulator in inflammation. Mol. Cell 19, 777–789. [DOI] [PubMed] [Google Scholar]
- Keene JD (2007). RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet 8, 533–543. [DOI] [PubMed] [Google Scholar]
- Keene JD, Komisarow JM, and Friedersdorf MB (2006). RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc 1, 302–307. [DOI] [PubMed] [Google Scholar]
- Khabar KSA, and Young HA (2007). Post-transcriptional control of the interferon system. Biochimie 89, 761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Arcos S, Rothamel K, Jian J, Rose KL, McDonald WH, Bian Y, Reasoner S, Barrows NJ, Bradrick S, et al. (2020). Discovery of Wide-spread Host Protein Interactions with the Pre-replicated Genome of CHIKV Using VIR-CLASP. Mol. Cell 78, 624–640.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, and Ule J (2010). iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol 17, 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurgonaite K, Gandhi H, Kurth T, Pautot S, Schwille P, Weidemann T, and Bökel C (2015). Essential role of endocytosis for interleukin-4-receptor-mediated JAK/STAT signalling. J. Cell Sci 128, 3781–3795. [DOI] [PubMed] [Google Scholar]
- Lambert N, Robertson A, Jangi M, McGeary S, Sharp PA, and Burge CB (2014). RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol. Cell 54, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M, and Rajewsky N (2011). Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol. Cell 43, 340–352. [DOI] [PubMed] [Google Scholar]
- Levine TD, Gao F, King PH, Andrews LG, and Keene JD (1993). Hel-N1: an autoimmune RNA-binding protein with specificity for 3′ uridylate-rich untranslated regions of growth factor mRNAs. Mol. Cell. Biol 13, 3494–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, and Qian S-B (2014). Translational reprogramming in cellular stress response. Wiley Interdiscip. Rev. RNA 5, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López de Silanes I, Zhan M, Lal A, Yang X, and Gorospe M (2004). Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. USA 101, 2987–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourou N, Gavriilidis M, and Kontoyiannis DL (2019). Lessons from studying the AU-rich elements in chronic inflammation and autoimmunity. J. Autoimmun 104, 102334. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y-C, Chang S-H, Hafner M, Li X, Tuschl T, Elemento O, and Hla T (2014). ELAVL1 modulates transcriptome-wide miRNA binding in murine macrophages. Cell Rep. 9, 2330–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund VK, and Delotto R (2011). Regulation of Toll and Toll-like receptor signaling by the endocytic pathway. Small GTPases 2, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde BM, Moore C, and Varani G (2007). RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol 8, 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Mazroui R, Marco S, Clair E, Roretz C, Tenenbaum S, Keene J, Saleh M, and Gallouzi I (2008). Caspase-mediated cleavage of HuR in the cytoplasm contributes to pp32/PHAP-I regulation of apoptosis. J Cell Biol 180, 113–127. 10.1083/jcb.200709030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C, Garzia A, Mazzola M, Gerstberger S, Molina H, and Tuschl T (2018). The TIA1 RNA-Binding Protein Family Regulates EIF2AK2-Mediated Stress Response and Cell Cycle Progression. Mol. Cell 69, 622–635.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mino T, and Takeuchi O (2013). Post-transcriptional regulation of cytokine mRNA controls the initiation and resolution of inflammation. Biotechnol. Genet. Eng. Rev 29, 49–60. [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Yoshida H, Moodley D, LeBoité H, Rothamel K, Raj T, Ye CJ, Chevrier N, Zhang S-Y, Feng T, et al. ; Immunological Genome Project Consortium (2016). Parsing the Interferon Transcriptional Network and Its Disease Associations. Cell 164, 564–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M Jr., Tuschl T, Ohler U, and Keene JD (2011). Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol. Cell 43, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N, Jacobs NC, Hafner M, Kennington EA, Nusbaum JD, Tuschl T, Blackshear PJ, and Ohler U (2014). Global target mRNA specification and regulation by the RNA-binding protein ZFP36. Genome Biol. 15, R12–R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N, Wessels H-H, Lebedeva S, Sajek M, Ghanbari M, Garzia A, Munteanu A, Yusuf D, Farazi T, Hoell JI, et al. (2019). Deciphering human ribonucleoprotein regulatory networks. Nucleic Acids Res. 47, 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann T, Herzog VA, Muhar M, von Haeseler A, Zuber J, Ameres SL, and Rescheneder P (2019). Quantification of experimentally induced nucleotide conversions in high-throughput sequencing datasets. BMC Bioinformatics 20, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbacher J, and Yeo G (2018). Systematic Discovery of RNA Binding Proteins that Regulate MicroRNA Levels. Molecular Cell. 10.1016/j.molcel.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo CA, Bjur E, Topisirovic I, Sonenberg N, and Larsson O (2014). Translational control of immune responses: from transcripts to translatomes. Nat. Immunol 15, 503–511. [DOI] [PubMed] [Google Scholar]
- Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, et al. (2011). Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol 29, 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan M, Porter DF, and Khavari PA (2019). Methods to study RNA-protein interactions. Nat. Methods 16, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Ha KCH, Nie K, Zheng H, Hughes TR, and Morris QD (2017). RNAcompete methodology and application to determine sequence preferences of unconventional RNA-binding proteins. Methods 118–119, 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reder AT, and Feng X (2013). Aberrant type I interferon regulation in autoimmunity: opposite directions in MS and SLE, shaped by evolution and body ecology. Front. Immunol 4, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby RE, and Rehwinkel J (2015). RNA degradation in antiviral immunity and autoimmunity. Trends Immunol. 36, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, and Hertzog PJ (2013). Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41, D1040–D1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlberg AS, Ruuska M, Granfors K, and Penttinen MA (2013). Altered Regulation of ELAVL1/HuR in HLA-B27-Expressing U937 Monocytic Cells. PLoS One. 8, e70377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Tanaka N, Hata N, Oda E, and Taniguchi T (1998). Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett. 425, 112–116. [DOI] [PubMed] [Google Scholar]
- Savan R (2014). Post-transcriptional regulation of interferons and their signaling pathways. J. Interferon Cytokine Res 34, 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol 32, 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlyarov V, Fallmann J, Ebner F, Huemer J, Sneezum L, Ivin M, Kreiner K, Tanzer A, Vogl C, Hofacker I, and Kovarik P (2016). Tristetraprolin binding site atlas in the macrophage transcriptome reveals a switch for inflammation resolution. Mol. Syst. Biol 12, 868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, Ascano M, Kelley M, Johnson DB, Balko JM, and Wilson JT (2019). Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat. Nanotechnol 14, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone LE, and Keene JD (2013). Mechanisms coordinating ELAV/Hu mRNA regulons. Curr. Opin. Genet. Dev 23, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikantan S, Tominaga K, and Gorospe M (2012). Functional interplay between RNA-binding protein HuR and microRNAs. Curr. Protein Pept. Sci 13, 372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo A, Dalmau J, Manley G, Rosenfeld M, Wong E, Henson J, Posner JB, and Furneaux HM (1991). HuD, a paraneoplastic encephalomyelitis antigen, contains RNA-binding domains and is homologous to Elav and Sex-lethal. Cell 67, 325–333. [DOI] [PubMed] [Google Scholar]
- Takeuchi O (2015). HuR keeps interferon-b mRNA stable. Eur. J. Immunol 45, 1296–1299. [DOI] [PubMed] [Google Scholar]
- Talwar S, Jin J, Carroll B, Liu A, Boyd Gillespie B, and Palanisamy V (2011). Caspase-Mediated Cleavage of RNA-Binding Protein HuR Regulates C-Myc Protein Expression After Hypoxic Stress. J. Biol. Chem 286, 32333–32343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenbaum SA, Carson CC, Lager PJ, and Keene JD (2000). Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc. Natl. Acad. Sci. USA 97, 14085–14090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedje C, Diaz-Muñoz MD, Trulley P, Ahlfors H, Laaß K, Blackshear PJ, Turner M, and Gaestel M (2016). The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucleic Acids Res. 44, 7418–7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner M, and DÍaz-Muñoz MD (2018). RNA-binding proteins control gene expression and cell fate in the immune system. Nat. Immunol 19, 120–129. [DOI] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, and Darnell RB (2003). CLIP identifies Nova-regulated RNA networks in the brain. Science 302, 1212–1215. [DOI] [PubMed] [Google Scholar]
- Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, Bode NM, McNeill MS, Yan S, Camarena J, et al. (2018). A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med 24, 1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen J-Y, Cody NAL, Dominguez D, et al. (2020). A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Roretz C, Lian XJ, Macri AM, Punjani N, Clair E, Drouin O, Dormoy-Raclet V, Ma JF, and Gallouzi I-E (2013). Apoptotic-Induced Cleavage Shifts HuR From Being a Promoter of Survival to an Activator of Caspase-Mediated Apoptosis. Cell Death Differ. 20, 154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Xiao G, Chu Y, Zhang MQ, Corey DR, and Xie Y (2015). Design and bioinformatics analysis of genome-wide CLIP experiments. Nucleic Acids Res. 43, 5263–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2. Elegant Graphics for Data Analysis. Springer. [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiakouvaki A, Dimitriou M, Karakasiliotis I, Eftychi C, Theocharis S, and Kontoyiannis DL (2012). Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J. Clin. Invest 122, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LE, Moore AE, Sokol L, Meisner-Kober N, and Dixon DA (2012). The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol. Cancer Res. 10, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, and Lee JT (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, and Lee JT (2010). Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 40, 939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code used for sequencing analysis and figure generation is accessible at https://github.com/Ascano-Lab. All sequencing data are deposited to National Center for Biotechnology Information via Sequence Read Archive (SRA); BioProject ID: PRJNA659987.