

PURPOSE
Hepatocellular carcinoma (HCC) has well-defined environmental risk factors. In addition, epidemiologic studies have suggested hereditary risk factors. The goals of this study were to determine the rate of pathogenic and likely pathogenic (P/LP) germline variants in cancer predisposition genes in patients with HCC, possible enrichment of P/LP variants in particular genes, and potential impact on clinical management.
MATERIALS AND METHODS
A prospective study at a tertiary medical center enrolled 217 patients with a personal history of HCC. Multigene panel testing was performed for 134 cancer predisposition genes in all patients. The rate of P/LP variants was compared with population rates. A separate retrospective cohort included 219 patients with HCC who underwent testing at a commercial laboratory.
RESULTS
In the prospective cohort, P/LP germline variants were identified in 25 of 217 patients with HCC (11.5%). Four patients (1.8%) had P/LP variants in the highly penetrant cancer genes BRCA2 (n = 2), MSH6 (n = 1), and PMS2 (n = 1). In addition, multiple patients had P/LP variants in FANCA (n = 5) and BRIP1 (n = 4), which were significantly enriched in HCC compared with the general population. Detection of P/LP variants led to changes in clinical management in regard to therapy selection, screening recommendations, and cascade testing of relatives. In a separate retrospective analysis of 219 patients with HCC, 30 (13.7%) were positive for P/LP variants including 13 (5.9%) with highly penetrant genes APC (n = 2), BRCA1 (n = 1), BRCA2 (n = 6), MSH2 (n = 2), or TP53 (n = 2).
CONCLUSION
P/LP germline variants in cancer predisposition genes were detected in 11%-14% of patients with HCC. Inherited genetics should not be overlooked in HCC as there are important implications for precision treatment, future risk of cancers, and familial cancer risk.
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fourth most common cause of cancer mortality worldwide.1 The development of HCC is strongly linked to underlying chronic liver disease and cirrhosis, most commonly because of chronic infection with either hepatitis C virus (HCV) or hepatitis B virus (HBV).2 Other HCC risk factors include nonalcoholic fatty liver disease; excessive alcohol use; exposure to hepatotoxins; and rare genetic disorders such as hereditary hemochromatosis, hereditary tyrosinemia, and acute hepatic porphyrias.1,3 Analyses of HCC tumor tissues have revealed an array of somatic mutations, the most common being TERT promoter–activating mutations, CTNNB1-activating mutations, and TP53-inactivating mutations.2 HCC has not been linked with pathogenic and likely pathogenic (P/LP) variants in germline cancer predisposition genes to date.
CONTEXT
Key Objective
Hepatocellular carcinoma (HCC) has been linked to a number of environmental risk factors. An additional hereditary component has remained unexplored. The objective of this study was to determine the yield of testing for cancer predisposition genes in the germline of patients with HCC and to explore the potential implications for clinical management.
Knowledge Generated
Pathogenic and likely pathogenic variants in cancer predisposition genes were detected in the germline of 11%-14% of patients with HCC, with 1.8%-5.5% of these occurring in highly penetrant cancer genes. These findings affected the clinical treatment and risk reduction strategies for patients and their family members.
Relevance
Treating physicians should be aware of the possibility of germline variants in cancer predisposition genes in patients with HCC. Multigene panel genetic testing should be considered, at least for the patients who meet testing criteria on the basis of a family history of cancers.
Familial aggregation studies and case-control series have demonstrated that a hereditary component influences the risk of HCC, suggesting an interaction between genetic and environmental factors.4,5 A meta-analysis of studies that included 3,600 individuals with liver cancer concluded that a family history of liver cancer increased the risk of HCC by more than 2.5-fold.6
As the detection of inherited P/LP variants in cancer predisposition genes would have profound implications for surveillance and management of patients and their family members,7 it is important to know the yield of germline multigene panel testing in patients with HCC. The goals of this study were to determine the rate of P/LP variants in patients with HCC, the possible associations with particular cancer predisposition genes, and the potential impact on clinical management, including surveillance and precision therapy.
MATERIALS AND METHODS
Prospective Cohort (Penn cohort)
Study design
The prospective cohort is a cross-sectional study of patients seen in the hepatology, oncology, or radiology clinics at Penn Medicine (an academic hospital system in Philadelphia, PA) with a history of HCC diagnosed in their native liver. The diagnosis was corroborated by review of radiology and/or pathology reports by the board-certified gastroenterologists and hepatologists on the study team according to American Association for the Study of Liver Diseases guidelines (M.A.H. and K.J.W.).8 Patients were enrolled between April 30, 2019 and June 24, 2020. The study was approved by the University of Pennsylvania Institutional Review Board (IRB) (Protocol No. 832453).
Study population and data collection
Eligible patients were at least 18 years of age. Patients provided informed consent for genetic testing and submitted a saliva or blood sample. Patients were excluded if actively listed for solid organ transplantation at the time of enrollment, to eliminate any potential that genetic testing results could influence eligibility for transplantation. Questionnaires were used to collect data on ethnicity, history of liver disease risk factors, history of other primary cancers, and a multigenerational family history of cancer. Self-reported data were corroborated using the electronic health record.
Sample collection and genetic testing
Patient saliva or blood samples were sent to a commercial laboratory (Invitae, San Francisco, CA) for processing and testing. The DNA extracted from saliva or blood samples was tested using a Clinical Laboratory Improvement Amendments (CLIA)–certified next-generation sequencing–based panel of 134 cancer-associated genes (Appendix Table A1). Full-gene sequencing and deletion and duplication analysis, including coding exons and 10-20 base pairs of adjacent intronic sequence on either side of the coding exons, was performed to detect single-nucleotide variants, insertions and deletions < 15 bp in length, and exon-level deletions and duplications, as previously described.9 The variant interpretation was performed by Invitae scientific and clinical staff using a refinement of the American College of Medical Genetics (ACMG) criteria.10
Reporting of genetic test results
As the testing was CLIA-certified, the prospective study released P/LP results to patients and recorded the findings in the electronic health record. Variants of uncertain significance and normal results were not reported to patients. Patients were counseled on recommended follow-up in medical genetics or cancer genetics clinics for further management, as appropriate.
P/LP rate in genome aggregation database
A method adapted from Hu et al was used to estimate the P/LP carrier rate in the general population for BRIP1 and FANCA using the genome aggregation database (gnomAD v2.1.1), which includes more than 134,000 unrelated individuals from various international genome projects.11,12 P/LP variants were selected from whole-exome sequences with > 50× coverage, including all frameshift, stop gained, splice donor, and splice acceptor variants (BRIP1, n = 232; FANCA, n = 456), less those reported as benign, or likely benign or of uncertain significance by a majority of laboratories on ClinVar (BRIP1, n = 97; FANCA, n = 16), plus all missense variants reported as P/LP in ClinVar (BRIP1, n = 92; FANCA, n = 169). P/LP variants were then weighted on the basis of self-reported ancestry for a total of 202 P/LP variants of 115,244 individuals for BRIP1 and 535 P/LP variants of 111,718 individuals for FANCA.
Statistical analysis
Descriptive statistics were used to summarize the clinical and demographic characteristics. The rates of P/LP variants of BRIP1 and FANCA were compared with the population rates using the Fisher's exact test with P value reported.
Retrospective Comparison Cohort (Invitae cohort)
Under IRB approval, de-identified clinical information was retrospectively reviewed on a separate cohort of 219 consecutive participants who underwent genetic testing for HCC between December 2015 and September 2020 at a commercial laboratory (herein referred to as the Invitae cohort). HCC cases were determined from phenotypic data and International Classification of Diseases–10 codes reported by clinicians on the test requisition forms. Sample types for this cohort included germline DNA extracted from blood or saliva. Once extracted, DNA was processed and tested as described above.9 There was no overlap in cases between the prospective and retrospective cohorts.
RESULTS
Patient Characteristics: Prospective Cohort
Table 1 summarizes the demographic characteristics of study participants from the prospective cohort. A total of 232 patients enrolled in the study, and 217 (93.5%) patients completed testing.
TABLE 1.
Demographic and Clinical Features of 232 Enrolled Participants (Prospective Cohort)

The distribution of sex, history of underlying liver disease and transplantation status, Barcelona Clinic Liver Cancer stage,8 and ethnicity was similar between patients found to have P/LP variants compared with those who did not (Table 1). Of 18 patients who had small HCCs found incidentally on explant and completed genetic testing, all were negative for P/LP variants. Of the 25 patients with P/LP gene variants, one (4.0%) had a first- or second-degree relative with HCC versus 12 of 207 (6.3%) patients without any such variant. Considering family history of other cancers, 88.0% of patients with P/LP variants had a relative with cancer as compared with 77.1% of those without.
Spectrum of Pathogenic and Likely Pathogenic Variants in the Prospective Cohort
The cancer predisposition multigene panel used for the study comprises 134 genes linked to risks for a large variety of cancers (Appendix Table A1). This includes all 24 cancer predisposition genes recommended for reporting by the ACMG in patients undergoing whole-exome sequencing as they are considered highly penetrant and clinically actionable.13
Among the 217 consecutive patients undergoing testing in the prospective cohort, there were 28 heterozygous P/LP germline variants in cancer-associated genes identified in the germline of 25 patients (11.5%) (Table 2 and Fig 1A). Three patients had P/LP variants in two different genes.
TABLE 2.
Germline P/LP Variants in Cancer Predisposition Genes in Patients With HCC (prospective cohort)

FIG 1.
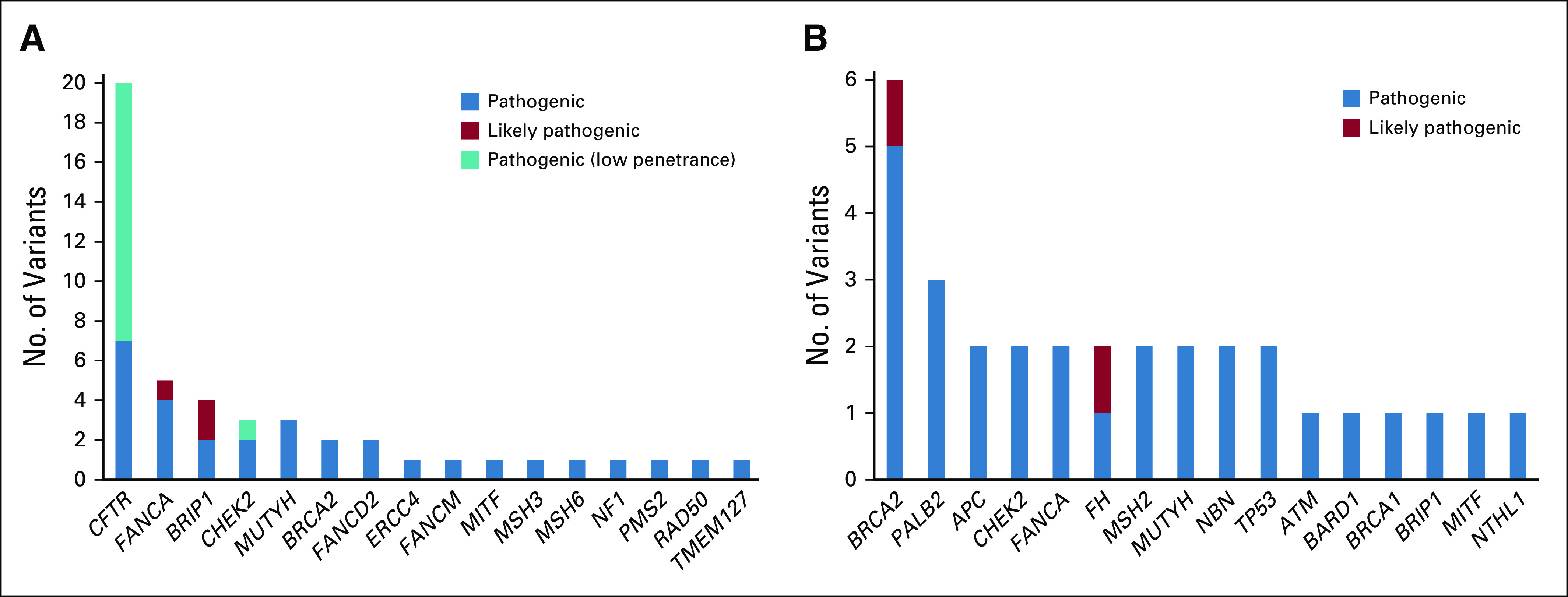
Distribution of P/LP counts for each gene with at least one P/LP variant identified in patients with HCC. (A) Prospective cohort: a consecutive series of patients with HCC undergoing multigene panel testing at a tertiary medical center. Pathogenic (low penetrance) indicates variants with sufficient evidence of pathogenicity but with an attenuated phenotype as compared with other P/LP variants of the gene. (B) Retrospective cohort: a consecutive series of patients with HCC completing testing at a commercial laboratory. HCC, hepatocellular carcinoma; P/LP, pathogenic and likely pathogenic.
There were an additional 20 heterozygous P/LP variants in CFTR, which is associated with autosomal recessive cystic fibrosis. This gene is included on the panel because of an association with chronic pancreatitis, which in turn is linked to pancreatic cancer. Since heterozygous CFTR variants occurred at population rates,14,15 and are implausibly involved in the pathogenesis of HCC, they were excluded from the analyses.
ACMG-reportable genes
Of the 217 patients completing testing, four (1.8%) had P/LP variants in highly penetrant genes recommended to be reported by the ACMG (Table 2).13 Two patients had BRCA2 pathogenic variants, which are causative of hereditary breast and ovarian cancer syndrome, and two patients had pathogenic variants in mismatch repair genes causative of Lynch syndrome, namely MSH6 and PMS2. Both patients with BRCA2 pathogenic variants had compensated cirrhosis and multifocal HCC at presentation (Barcelona Clinic Liver Cancer stage B and C), and one had a second primary cancer, renal cell carcinoma. A patient with PMS2-associated Lynch syndrome had an adrenal adenoma. All four patients with pathogenic variants in ACMG-reportable genes had first- or second-degree relatives with cancers and were slightly younger at HCC diagnosis with average age 56 (range 47-66) versus 61 years.
Variants in FANCA
Five patients (2.3% of total) had heterozygous P/LP variants in the gene FANCA (Table 2). Preliminary data support an association between variants in FANCA and prostate cancer.16-18 Interestingly, one patient with a FANCA pathogenic variant had prostate cancer in addition to HCC, and another patient had a first-degree relative with prostate cancer.
There was a significant enrichment of P/LP variants in FANCA among patients with HCC as compared with the rates in the general noncancer population on gnomAD11 (Fisher's exact P value = .0043), demonstrating a possible association in HCC for P/LP variants in FANCA.
Variants in BRIP1
Four P/LP variants (1.8% of total) were detected in BRIP1 (Table 2), a gene linked to ovarian cancer susceptibility.19,20 The rate of P/LP variants in BRIP1 reached significant enrichment in HCC as compared with individuals without cancer in gnomAD (Fisher's exact P value = .0007).
Other genes
A number of additional genes associated with cancer predisposition were identified in patients with HCC in the Penn cohort (Fig 1A and Table 2). There were three P/LP variants in CHEK2, which is associated with an autosomal dominant risk of breast, prostate, colon, and thyroid cancers.21 There were three heterozygous P/LP variants in MUTYH, a gene associated with autosomal recessive polyposis.13 Heterozygosity for MUTYH occurs in up to 2% of European populations22,23 and has been linked to a slightly increased risk of colon cancer.24
Additional heterozygous variants were identified in genes associated with autosomal recessive Fanconi anemia including FANCD2 (n = 2), FANCM, and ERCC4. There were single cases of P/LP variants in RAD50, NF1, TMEM127, and MITF, genes associated with autosomal dominant susceptibility to a variety of cancers.25-30 There was a heterozygous pathogenic variant in MSH3, which is linked to autosomal recessive polyposis.31
Impact on Clinical Management: Prospective Cohort
P/LP results and post-test genetic counseling were provided to patients. In several instances, the genetic testing results had an immediate impact on the clinical management of patients, particularly for those cured of HCC by liver transplantation. In one patient who had progression of HCC on standard therapy, the discovery of a germline BRCA2 variant prompted the use of the PARP inhibitor olaparib, as there is evidence that this class of drug can be used as a targeted therapy in BRCA1 and BRCA2 carriers with breast, ovarian, prostate, and pancreatic cancer.16,21,32 Unfortunately, this patient has progressive disease on olaparib. One patient with a pathogenic deletion of TMEM127, which is associated with autosomal dominant hereditary paraganglioma and pheochromocytoma syndromes, was recommended to undergo surveillance imaging with MRI.33 Two patients were recommended to undergo regular screening according to clinical guidelines for the management of Lynch syndrome. Multiple patients’ families were recommended to undergo cascade genetic testing.
Variants Detected in the Retrospective Cohort
The spectrum of variants among 219 patients with HCC referred for testing at a large-volume commercial laboratory (Invitae) was examined retrospectively. These patients underwent testing with a variety of cancer predisposition gene panels ranging in size between 1 and 154 genes. In contrast to the prospective cohort, the retrospective cohort had a majority of females (Table 3).
TABLE 3.
Demographic Features of 219 Patients With HCC Undergoing Genetic Testing at a Commercial Laboratory (Retrospective Cohort)

Among the 219 patients with HCC who underwent genetic testing, there were 31 P/LP variants in 30 patients (13.7%) (Fig 1B and Table 4). There were 13 (5.9%) patients with P/LP variants in ACMG-reportable cancer predisposition genes including BRCA2 (n = 6) and BRCA1 (n = 1) for an overall rate of BRCA1/2 of 2.7%, MSH2 (n = 2, associated with Lynch syndrome), TP53 (n = 2, associated with Li Fraumeni syndrome), and APC (n = 2, associated with familial adenomatous polyposis syndrome). The patients with APC P/LP variants likely had hepatoblastoma based on their early age of diagnosis of liver cancer (4 and 13 years) and the known association with APC.34 In addition to the ACMG-reportable genes, of 12 who underwent testing for FANCA, one patient was found to have two pathogenic variants (c.709+5G>A splice site and c.1304G>A, phase unknown). Of 190 patients tested for BRIP1, there was one pathogenic variant.
TABLE 4.
P/LP Variants in Cancer-Associated Genes in Patients With HCC (retrospective cohort)

In addition to the ACMG-reportable genes, there were a number of other variants in genes with potential for precision therapy, clinical treatment trial, and/or management implications including cascade family variant testing (Fig 1B).
DISCUSSION
In this study, which is the largest analysis of multigene panel testing in HCC, the overall rate of P/LP germline variants was 11%-14%, or more than one of nine patients. The rate was 1.8%-5.9% for highly penetrant cancer genes. Positive test results led to changes in clinical management for some patients and recommendations for cascade genetic testing in at-risk relatives.
Hereditary predisposition may have been overlooked in HCC because of clear environmental risk factors. Indeed, there were no distinguishing clinical features in patients harboring P/LP variants, and they had a variety of underlying acquired risk factors such as chronic HCV, HBV, and nonalcoholic fatty liver disease. This may help to explain the absence of established, clinical criteria–based recommendations for germline testing in patients with HCC.
P/LP variants in FANCA and BRIP1 were found in more than 4% of patients with HCC in the prospective cohort, representing a new potential association. Both FANCA and BRIP1 are involved in the homologous recombination DNA damage repair (HR-DDR) pathway, and biallelic inactivating mutations in these genes cause Fanconi anemia.35 Growing evidence also implicates monoallelic variants in predisposition to prostate cancer16-18 and ovarian cancer,19,20 respectively. Intriguingly, Oussalah et al22 identified a haplotype of three single-nucleotide polymorphisms in BRIP1 that was significantly associated with the risk of HCC in patients of European and African ancestry with cirrhosis because of viral hepatitis, supporting links for this gene and HCC. Our retrospective cohort had too small of a sample size to replicate the findings for BRIP1 and FANCA, and a larger study will be needed to evaluate the generalizability of these results.
A majority of P/LP variants identified in our study were in genes involved in HR-DDR pathways. Recent studies support a role for this pathway in HCC pathogenesis. In one molecular profiling study of solid tumor samples, somatic homologous variants in HR-DDR genes were found in 20.9% of 115 tested HCC samples.23 An analysis of data from the cancer genome atlas identified 15 cases of HCC with germline or somatic variants in genes related to the HR-DDR pathway.24 Among a sample of 214 patients with HCC, somatic and germline variants in HR-DDR genes were found in 42 and seven patients, respectively, the latter encompassing variants in BRCA1 (n = 2), ATM (n = 1), PMS2 (n = 2), BLM (n = 1), and RAD50 (n = 1).25 Together, this suggests that HR-DDR genes may be involved in the mechanism of HCC carcinogenesis. Examining tumor tissues for loss of heterozygosity in association with germline variants with HR-DDR or Lynch syndrome genes would further suggest that germline variants may be drivers of HCC development. Future studies can evaluate for loss of heterozygosity with paired germline-tumor analysis.
The links between HCC and HR-DDR genes are intriguing as the identification of alterations in these genes may provide therapeutic options. PARP inhibitors have shown efficacy in patients with breast, ovarian, prostate, and pancreatic cancers who have germline or somatic variants in BRCA1, BRCA2, CHEK2, and FANCA.16,21,32 A clinical trial is underway to study whether PARP inhibitors are effective in patients with germline defects in HR-DDR genes as part of a cancer-agnostic treatment approach for solid tumors (ClinicalTrials.gov identifier: NCT04171700). Other precision medicine opportunities include the use of PD-1 inhibitors in patients found to have Lynch syndrome.26,27
Future studies should examine the potential for an interaction between environmental factors and hereditary predisposition to HCC. It is plausible that individuals with germline defects in DNA repair genes may be more prone to the development of HCC in the setting of liver disease, given that chronic inflammation induces double-strand DNA breaks in liver cells.28 Interestingly, among a cohort of patients with Lynch syndrome in the United Kingdom, a region with relatively low prevalence of chronic viral hepatitis infection, the lifetime incidence of HCC was found to be 0.5%,29 whereas in a Chinese cohort, where there is a high proportion of chronic HBV infection, the rate of HCC among Lynch syndrome kindreds was 8%.30
This study has several strengths, including the prospective design, the enrollment of diverse consecutive patients with HCC seen at a medical center, and the use of CLIA-certified genetic testing, which enabled us to recontact patients, implement germline-based precision treatment changes, and recommend cascade testing for family members when clinically indicated. To our knowledge, this is the first study to systematically perform germline genetic testing in patients with HCC.
This study had several limitations. Recruitment for the prospective cohort was from a single tertiary care medical center and may not be generalizable to other centers. The sample set was biased toward patients who had undergone liver transplantation, which favors earlier-stage HCC. The gnomAD data set was used as the comparison group, whereas the ideal comparison would be with a matched control population from our institution. Nominal P values are shown for the sake of hypothesis generation, but a larger, dedicated study will be needed to replicate the results for FANCA and BRIP1. Finally, the retrospective cohort comprises patients referred for testing, resulting in selection bias.
In conclusion, universal testing of patients with HCC to identify germline variants should be considered, consistent with the INTERCEPT study supporting universal testing of all patients with cancer.9 At a minimum, a thorough family history should be obtained for all patients and they should be referred for testing if they meet established criteria for hereditary breast or ovarian cancer or Lynch syndrome. Future investigations may better define the subgroups of patients who would maximally benefit from genetic testing.
Appendix
TABLE A1.
Cancer Predisposition Multigene Panel Used in the Prospective Cohort
K. Rajender Reddy
Consulting or Advisory Role: Mallinckrodt, Pfizer
Research Funding: Mallinckrodt, Intercept Pharmaceuticals, BMSi, Exact Sciences, Target RWE, Gilead Sciences, Merck
Jewel Samadder
Consulting or Advisory Role: Janssen Research & Development, Cancer Prevention Pharmaceuticals, Recursion Pharmaceuticals
Sarah M. Nielsen
Employment: Invitae
Stock and Other Ownership Interests: Invitae
Travel, Accommodations, Expenses: Invitae
Kathryn E. Hatchell
Employment: Invitae
Stock and Other Ownership Interests: Invitae
Edward D. Esplin
Employment: Invitae
Stock and Other Ownership Interests: Invitae
Bryson W. Katona
Travel, Accommodations, Expenses: Janssen
Maarouf A. Hoteit
Consulting or Advisory Role: Eisai, HepQuant
Kirk J. Wangensteen
Research Funding: Calico LLC
No other potential conflicts of interest were reported.
SUPPORT
Supported by the National Institutes of Health (R03DK123543 and P30DK50306 Pilot grant to K.J.W.).
DATA SHARING STATEMENT
A data sharing statement provided by the authors is available with this article at DOI https://doi.org/10.1200/PO.21.00079.
AUTHOR CONTRIBUTIONS
Conception and design: Anil K. Rustgi, Maarouf A. Hoteit, Katherine L. Nathanson, Kirk J. Wangensteen
Financial support: Kim M. Olthoff, K. Rajender Reddy, Kirk J. Wangensteen
Administrative support: Anya Mezina, Neil Philips, Zoe Bogus, Kirk J. Wangensteen
Provision of study materials or patients: K. Rajender Reddy, Sarah M. Nielsen, Kathryn E. Hatchell, Edward D. Esplin, Maarouf A. Hoteit
Collection and assembly of data: Anya Mezina, Neil Philips, Zoe Bogus, Noam Erez, Ruoming Fan, Sarah M. Nielsen, Kathryn E. Hatchell, Edward D. Esplin, Kirk J. Wangensteen
Data analysis and interpretation: Anya Mezina, Rui Xiao, N. Jewel Samadder, Sarah M. Nielsen, Edward D. Esplin, Bryson W. Katona, Katherine L. Nathanson, Kirk J. Wangensteen
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by the authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
K. Rajender Reddy
Consulting or Advisory Role: Mallinckrodt, Pfizer
Research Funding: Mallinckrodt, Intercept Pharmaceuticals, BMSi, Exact Sciences, Target RWE, Gilead Sciences, Merck
Jewel Samadder
Consulting or Advisory Role: Janssen Research & Development, Cancer Prevention Pharmaceuticals, Recursion Pharmaceuticals
Sarah M. Nielsen
Employment: Invitae
Stock and Other Ownership Interests: Invitae
Travel, Accommodations, Expenses: Invitae
Kathryn E. Hatchell
Employment: Invitae
Stock and Other Ownership Interests: Invitae
Edward D. Esplin
Employment: Invitae
Stock and Other Ownership Interests: Invitae
Bryson W. Katona
Travel, Accommodations, Expenses: Janssen
Maarouf A. Hoteit
Consulting or Advisory Role: Eisai, HepQuant
Kirk J. Wangensteen
Research Funding: Calico LLC
No other potential conflicts of interest were reported.
REFERENCES
- 1.Yang JD, Hainaut P, Gores GJ, et al. : A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol 16:589-604, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wangensteen KJ, Chang KM: Multiple roles for hepatitis B and C viruses and the host in the development of hepatocellular carcinoma. Hepatology 73:1-11, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forner A, Reig M, Bruix J: Hepatocellular carcinoma. Lancet 391:1301-1314, 2018 [DOI] [PubMed] [Google Scholar]
- 4.Yu MW, Chang HC, Liaw YF, et al. : Familial risk of hepatocellular carcinoma among chronic hepatitis B carriers and their relatives. J Natl Cancer Inst 92:1159-1164, 2000 [DOI] [PubMed] [Google Scholar]
- 5.Hassan MM, Spitz MR, Thomas MB, et al. : The association of family history of liver cancer with hepatocellular carcinoma: A case-control study in the United States. J Hepatol 50:334-341, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turati F, Edefonti V, Talamini R, et al. : Family history of liver cancer and hepatocellular carcinoma. Hepatology 55:1416-1425, 2012 [DOI] [PubMed] [Google Scholar]
- 7.Huang KL, Mashl RJ, Wu Y, et al. : Pathogenic germline variants in 10,389 adult cancers. Cell 173:355-370.e14, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marrero JA, Kulik LM, Sirlin CB, et al. : Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American Association for the Study of Liver Diseases. Hepatology 68:723-750, 2018 [DOI] [PubMed] [Google Scholar]
- 9.Samadder NJ, Riegert-Johnson D, Boardman L, et al. : Comparison of universal genetic testing vs guideline-directed targeted testing for patients with hereditary cancer syndrome. JAMA Oncol 7:230-237, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nykamp K, Anderson M, Powers M, et al. : Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med 19:1105-1117, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karczewski KJ, Francioli LC, Tiao G, et al. : The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434-443, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu C, Hart SN, Polley EC, et al. : Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA 319:2401-2409, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalia SS, Adelman K, Bale SJ, et al. : Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet Med 19:249-255, 2017 [DOI] [PubMed] [Google Scholar]
- 14.Sun W, Anderson B, Redman J, et al. : CFTR 5T variant has a low penetrance in females that is partially attributable to its haplotype. Genet Med 8:339-345, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Hamosh A, FitzSimmons SC, Macek M Jr, et al. : Comparison of the clinical manifestations of cystic fibrosis in black and white patients. J Pediatr 132:255-259, 1998 [DOI] [PubMed] [Google Scholar]
- 16.Hayano T, Matsui H, Nakaoka H, et al. : Germline variants of prostate cancer in Japanese families. PLoS One 11:e0164233, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicolosi P, Ledet E, Yang S, et al. : Prevalence of germline variants in prostate cancer and implications for current genetic testing guidelines. JAMA Oncol 5:523-528, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beltran H, Eng K, Mosquera JM, et al. : Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol 1:466-474, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramus SJ, Song H, Dicks E, et al. : Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J Natl Cancer Inst 107:djv214, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rafnar T, Gudbjartsson DF, Sulem P, et al. : Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet 43:1104-1107, 2011 [DOI] [PubMed] [Google Scholar]
- 21.Cybulski C, Gorski B, Huzarski T, et al. : CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet 75:1131-1135, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jenkins MA, Croitoru ME, Monga N, et al. : Risk of colorectal cancer in monoallelic and biallelic carriers of MYH mutations: A population-based case-family study. Cancer Epidemiol Biomarkers Prev 15:312-314, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Cleary SP, Cotterchio M, Jenkins MA, et al. : Germline MutY human homologue mutations and colorectal cancer: A multisite case-control study. Gastroenterology 136:1251-1260, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Win AK, Dowty JG, Cleary SP, et al. : Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology 146:1208-1211. e1-5, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertolotto C, Lesueur F, Giuliano S, et al. : A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 480:94-98, 2011 [DOI] [PubMed] [Google Scholar]
- 26,.Qin Y, Yao L, King EE, et al. : Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet 42:229-233, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao L, Schiavi F, Cascon A, et al. : Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA 304:2611-2619, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Damiola F, Pertesi M, Oliver J, et al. : Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: Results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res 16:R58, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heikkinen K, Rapakko K, Karppinen SM, et al. RAD50 and NBS1 are breast cancer susceptibility genes associated with genomic instability. Carcinogenesis 27:1593-1599, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laycock-van Spyk S, Thomas N, Cooper DN, et al. : Neurofibromatosis type 1-associated tumours: Their somatic mutational spectrum and pathogenesis. Hum Genomics 5:623-690, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adam R, Spier I, Zhao B, et al. : Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. Am J Hum Genet 99:337-351, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCabe N, Turner NC, Lord CJ, et al. : Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 66:8109-8115, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Muth A, Crona J, Gimm O, et al. : Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J Intern Med 285:187-204, 2019 [DOI] [PubMed] [Google Scholar]
- 34.Giardiello FM, Petersen GM, Brensinger JD, et al. : Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 39:867-869, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nalepa G, Clapp DW: Fanconi anaemia and cancer: An intricate relationship. Nat Rev Cancer 18:168-185, 2018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
A data sharing statement provided by the authors is available with this article at DOI https://doi.org/10.1200/PO.21.00079.