

Keywords: fibrosis, ketone ester, ketosis, NAFLD, NASH
Abstract
Nutritional ketosis as a therapeutic tool has been extended to the treatment of metabolic diseases, including obesity, type 2 diabetes, and nonalcoholic fatty liver disease (NAFLD). The purpose of this study was to determine whether dietary administration of the ketone ester (KE) R,S-1,3-butanediol diacetoacetate (BD-AcAc2) attenuates markers of hepatic stellate cell (HSC) activation and hepatic fibrosis in the context of high-fat diet (HFD)-induced obesity. Six-week-old male C57BL/6J mice were placed on a 10-wk ad libitum HFD (45% fat, 32% carbohydrates, 23% proteins). Mice were then randomized to one of three groups (n = 10 per group) for an additional 12 wk: 1) control (CON), continuous HFD; 2) pair-fed (PF) to KE, and 3) KE (HFD + 30% energy from BD-AcAc2, KE). KE feeding significantly reduced histological steatosis, inflammation, and total NAFLD activity score versus CON, beyond improvements observed for calorie restriction alone (PF). Dietary KE supplementation also reduced the protein content and gene expression of profibrotic markers (α-SMA, COL1A1, PDGF-β, MMP9) versus CON (P < 0.05), beyond reductions observed for PF versus CON. Furthermore, KE feeding increased hepatic markers of anti-inflammatory M2 macrophages (CD163) and also reduced proinflammatory markers [tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and cellular communication network factor 1 (CCN1)] versus CON and PF (P ≤ 0.05), in the absence of changes in markers of total hepatic macrophage content (F4/80 and CD68; P > 0.05). These data highlight that the dietary ketone ester BD-AcAc2 ameliorates histological NAFLD and inflammation and reduces profibrotic and proinflammatory markers. Future studies to further explore potential mechanisms are warranted.
NEW & NOTEWORTHY To our knowledge, this is the first study focusing on hepatic outcomes in response to dietary ketone ester feeding in male mice with HFD-induced NAFLD. Novel findings include that dietary ketone ester feeding ameliorates NAFLD outcomes via reductions in histological steatosis and inflammation. These improvements were beyond those observed for caloric restriction alone. Furthermore, dietary ketone ester feeding was associated with greater reductions in markers of hepatic fibrogenesis and inflammation compared with control and calorie-restricted mice.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a progressive disease of the liver, comprising a spectrum of liver injury ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), fibrosis, and end-stage cirrhosis and hepatocellular carcinoma. In the general population, the prevalence of NAFLD ranges from 10% to 30% and as high as 80%–100% in individuals with obesity (1, 2). Progression of NAFLD to NASH and more advanced fibrosis and cirrhosis is the most rapidly increasing indication for liver transplantation in the United States (3) and is considered an independent risk factor for cardiovascular, liver‐related, and all‐cause mortality (4, 5).
Hepatic fibrosis is a complex process orchestrated by a multitude of cells and signaling pathways culminating in the activation of hepatic stellate cells (6). Activation, proliferation, and migration of hepatic stellate cells result in increasing amounts of type I and III collagen and fibronectin deposited in the extracellular matrix, contributing to hepatic fibrosis and a worsening NAFLD/NASH phenotype (6, 7). Currently, there are no Food and Drug Administration (FDA)‐approved pharmacological treatments for NAFLD/NASH and associated fibrosis. Indeed, the prevalence of NAFLD and the multiple factors that contribute to its onset highlight the need to develop novel and therapeutic interventions to prevent the progression of disease (8).
The recent application of nutritional ketosis as a therapeutic tool has extended beyond its origin as a treatment for refractory epileptic seizures to the treatment of metabolic diseases such as obesity, type 2 diabetes, and NAFLD (9, 10). The ketone bodies acetoacetate (AcAc), β‐hydroxybutyrate (β-HB), and acetone are small molecules derived from increased mitochondrial flux of hepatic fatty acid oxidation-derived acetyl-CoA, a process amplified during prolonged fasting, starvation, very low carbohydrate intake, and glycogen‐depleting exercise (11–13). It has long been established that ketone bodies act as an alternative substrate for fuel, particularly in the brain, and exert anticatabolic effects in the liver, adipose, heart, and skeletal muscle (12, 14–16). Moreover, hepatic macrophage metabolism of endogenous AcAc is known to attenuate high-fat diet (HFD)-induced hepatic fibrogenesis (16). The growing body of evidence for nutritional ketosis has prompted interest in the ability to increase circulating ketones in the absence of total energy or carbohydrate restriction using exogenous ketone esters.
Exogenous ketone supplements can be ingested in the form of ketone salts (e.g., sodium‐potassium β‐HB) or ketone esters (KE) (both monoester and diester forms). The R,S-1,3-butanediol acetoacetate diester (BD-AcAc2) has been extensively studied (17). Dietary BD-AcAc2 produces modest increases in circulating ketone concentrations ranging from 0.5 to 1.0 mM and reductions in adiposity and body weight, with limited understanding of the contributing mechanisms (18–21). In addition, little is known regarding the possible effects of BD-AcAc2 on liver outcomes in diet-induced obese mice. We hypothesized that dietary KE feeding with BD-AcAc2 would beneficially attenuate NAFLD outcomes compared with calorie restriction alone in male mice with diet-induced obesity.
MATERIALS AND METHODS
Animals and Diets
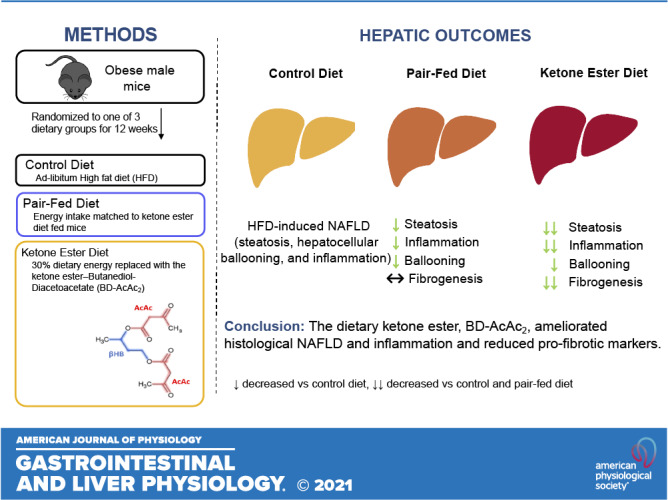
A portion of the animal characteristics have been previously published elsewhere (22). Five-week-old male C57BL/6J mice (n = 30) were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were allowed to acclimate for 7 days upon arrival at the University of Alabama at Birmingham animal facility. Mice were single housed and maintained in a standard 12:12 light-dark cycle (lights on, 6 AM) room at 22°C–23°C. At 6 wk of age, all mice were given ad libitum access to a high-fat diet (HFD) containing 45% kcal from fat (Cat. No. 104212; Dyets Inc., Bethlehem, PA; Table 1) and remained on the diet for 10 wk. Mice were then randomized to one of three groups (n = 10 per group): 1) control group (CON, remained on HFD), 2) pair-fed (PF; HFD with energy intake matched to the mean intake of the KE group from the previous day and provided in a single allotment at ∼10 AM each morning), and 3) KE (HFD + 30% carbohydrate energy replaced by KE; Table 1). Mice remained in these conditions for an additional 12 wk. BD-AcAc2 was purchased from SavInd (Urbana, IL) and shipped to Dyets Inc. for production. The KE group was acclimated to the diet with a mixture of the powdered KE group diet and CON diet (50:50) during the first week of the experimental phase. All analyses were performed with 10 animals per group, except for the PF group, which had nine after removal of one animal that was not eating during week 7 of the experimental phase of the study, for reasons unknown. The University of Alabama at Birmingham Institutional Animal Care and Use Committee approved the investigation, and all conditions conformed to the care procedures used by the University of Alabama at Birmingham (UAB) Animal Resources Program.
Table 1.
High-fat control and ketone ester diet
| Ingredient | Amount, kcal/g | CON, g/kg | KE, g/kg | CON, kcal/g | KE, kcal/g |
|---|---|---|---|---|---|
| Casein | 3.6 | 200.0 | 200.0 | 716.00 | 716.00 |
| Cornstarch | 3.6 | 72.8 | 0.0 | 262.08 | 0.00 |
| Dyetrose | 3.8 | 100.0 | 0.0 | 380.00 | 0.00 |
| Sucrose | 4.0 | 162.8 | 27.5 | 651.20 | 110.00 |
| Cellulose | 0.0 | 50.0 | 50.0 | 0.00 | 0.00 |
| Soybean oil | 9.0 | 25.0 | 25.0 | 225.00 | 225.00 |
| t-Butylhydroquinone | 0.0 | 0.005 | 0.005 | 0.00 | 0.00 |
| Lard | 9.0 | 177.5 | 177.5 | 1,597.50 | 16.00 |
| Salt mix 21008 | 1.6 | 10.0 | 10.0 | 16.00 | 16.00 |
| Dicalcium phosphate | 0.0 | 13.0 | 13.0 | 0.00 | 0.00 |
| Calcium carbonate | 0.0 | 5.5 | 5.5 | 0.00 | 0.00 |
| Potassium citrate H2O | 0.0 | 16.5 | 16.6 | 0.00 | 0.00 |
| Vitamin mix 300050 | 3.9 | 10.0 | 10.0 | 39.20 | 39.20 |
| l-cystine | 4.0 | 3.0 | 3.0 | 12.00 | 12.00 |
| Choline bitrate | 0.0 | 2.0 | 2.0 | 0.00 | 0.00 |
| Peanut flavor | 4.0 | 10.0 | 10.0 | 40.00 | 40.00 |
| Ketone ester | 4.7 | 0.0 | 252.0 | 0.00 | 1,184.45 |
| Total | - | 858.11 | 802.02 | 3,938.98 | 3,940.15 |
Diets are from Dyets Inc. Data in this table have been previously reported (22). CON, control; KE, ketone ester.
Body Composition
Body composition was measured by quantitative magnetic resonance (EchoMRI 3-in-1 version 2013; Echo Medical Systems, Houston, TX).
Blood β-Hydroxybutyrate Measurements
Whole blood D-β-HB concentrations were measured with a Nova Vet ketone meter (Nova Biomedical, Waltham, MA) from a tail nick. Animals had food withheld for 4 h and refed for 4 h before testing at the start of the dark cycle. This measure was performed during week 6 of the experimental phase of the study. AcAc concentrations are not measurable by the ketone meter and are not available.
Tissue Collection and Histology
Before being euthanized, mice were fasted for 6 h. Immediately following decapitation, livers were quickly excised, snap frozen in liquid nitrogen, and stored at −80°C. Snap-frozen liver tissue was thawed in 10% formalin for 24 h, then embedded in paraffin, serially sliced, and stained with hematoxylin and eosin (H&E) to examine liver morphology by IDEXX RADIL (Columbia, MO). In addition, slides were stained with Picro-Sirius Red to examine collagen deposition/fibrosis. Liver specimens were graded and scored by an individual blinded to the study groups for changes in NAFLD activity score (NAS; comprising fat accumulation, inflammation, and hepatocellular ballooning) according to the Brunt scale (23).
Western Blot Analysis
Western blot analysis was completed in liver homogenates, as described previously (24). Briefly, Triton X-100 cell lysates were prepared, and Laemmli gel loading buffer (No. 161–0737; Bio-Rad Laboratories, Hercules, CA) was added to the lysate (3 µg protein/uL) and boiled at 100°C for 10 min. Following gel electrophoresis and transfer, the membrane was incubated overnight in blocking solution [5% dry milk in Tris-buffered saline (TBS), 0.1% Tween 20 buffer] followed by overnight incubation in the primary antibody. Membranes were washed with TBS-Tween 20 buffer and placed in a horseradish peroxidase-conjugated secondary antibody (Nos. 7074 and 7076; Cell Signaling Technology, Danvers, MA). Blots (n = 8–10 per group) were analyzed via densiometric analysis (Image Lab 4.0). Amido-black staining was used to control for differences in protein loading. The total protein staining for each lane, quantified by laser densitometry, was used to correct for any differences in protein loading or transfer of all band densities. The primary antibodies used are as follows: α-smooth muscle actin (αSMA; No. 5694, Abcam, MA), collagen 1 A1 (COL1A1) (No. 8784, Santa Cruz Biotechnologies, Dallas, TX), cluster of differentiation 68 (CD68; No. 17832, Santa Cruz Biotechnology), F4/80 (No. 52664, Santa Cruz Biotechnology), interleukin-1β (IL-1β; No. 52012, Santa Cruz Biotechnology), CD11b (No. 110–89474; Novus Biologicals; Centennial, CO), CD163 (No. 58965, Santa Cruz Biotechnology), tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL; No.7877, Santa Cruz Biotechnology), cellular communication network factor 1 (CCN1; No. 24448, Abcam), matrix metalloproteinase 2 (MMP2; No. 97779, Abcam), MMP9 (No. AB9016, EMD Millipore, Billerica, MA), and platelet‐derived growth factor β (PDGFβ; No. 3169, Cell Signaling Technology).
mRNA Expression Analysis
RNA was extracted from liver tissue using a commercially available kit (No. 74104, Germantown, MD), and a cDNA library was synthesized (Promega, Madison, WI). RNA and cDNA purity and quality were assessed with a Nanodrop spectrometer. Quantitative real-time PCR (qPCR) was conducted using SYBR Green reagents (No. 172–5121, Bio-Rad Laboratories) and primer pairs (Sigma, St. Louis, MO). Data are represented relative to cyclophilin B (Ppib) using the 2−ΔΔCT method. See Table 2 for primer sequences.
Table 2.
Primer sequences
| Gene Name | Forward (5′–3′) | Reverse (3′–5′) |
|---|---|---|
| αSma | AAACAGGAATACGACGAAG | CAGGAATGATTTGGAAAGGA |
| Col1a1 | ACGCCATCAAGGTCTACTGC | ACGCCATCAAGGTCTACTGC |
| cd68 | GTGTCTGATCTTGCTAGGACC | GTGCTTTCTGTGGCTGTAG |
| Il-1b | TCACAGCAGCACATCAACAA | TGTCCTCATCCTGGAAGGTC |
| Tnfα | AGGCACTCCCCCAAAAGATG | CTTGGTGGTTTGCTACCACG |
| Mcp1 | AGCTGTAGTTTTTTGTCACCAAGC | GTGCTGAAGACCTTAGGGCA |
| Cd163 | TCCTTCTGGAATAGATTGGG | TCCTTCTGGAATAGATTGGG |
| Arg1 | GCAACCTGTGTCCTTTCTCCTGAA | TCGCAAGCCAATGTACACGATGTC |
| Mrc1 | GTTCACCTGGAGTGATGGTTCTC | AGGACATGCCAGGGTCACCTTT |
| Ppib | TGGAGATGAATCTGTAGGAC | CAAATCCTTTCTCTCCTGTAG |
Statistical Analysis
Differences among groups (CON, PF, and KE) were analyzed by one-way ANOVA. A significant main effect (P ≤ 0.05) was followed up with Fisher’s least significant difference post hoc comparisons to explore significant differences among groups. Significance was set a priori at P ≤ 0.05 (two-tailed). Data are expressed as means ± SE. All statistical analyses were conducted with GraphPad Prism version 8.0 (San Diego, CA).
RESULTS
Animal Characteristics
Select animal characteristics have been reported previously (22). Ten weeks of HFD feeding increased body weight of the entire cohort from 20.8 ± 0.2 g to 42.9 ± 0.8 g (means ±SD) (22). At the end of the experimental phase, 12 wk of KE supplementation significantly lowered body weight, compared with CON (P ≤ 0.05; Table 3). Despite matching energy provisions, final body weight of the PF group was 23% higher than the KE group (Table 3). Fat mass was 74% lower for KE versus CON (P ≤ 0.05) and 54% lower for KE versus PF (P ≤ 0.05; Table 3). Both KE and PF groups displayed similar reductions in lean body mass versus CON, with the KE group displaying the greatest reduction (P ≤ 0.05; Table 3). Circulating D-β-HB concentrations were elevated in the KE group (0.51 ± 0.12 mM) compared with CON (0.32 ± 0.06) and PF (0.32 ± 0.10 mM; P ≤ 0.05 for all comparisons; Table 3) groups. Compared with the CON group, both PF and KE groups displayed a significantly lower liver weight at the end of the study (50% and 58% lower, respectively; P ≤ 0.05), whereas there was no significant difference in liver weight for KE versus PF (P > 0.05; Table 3).
Table 3.
Animal characteristics
| Variable | CON | PF | KE |
|---|---|---|---|
| Food intake, kcal/day | 11.2 ± 0.2a | 8.2 ± 1.6b | 8.2 ± 1.6b |
| Final body mass, g | 46.2 ± 0.9a | 34.5 ± 1.6b | 26.2 ± 0.9c |
| Final fat mass, g | 20.0 ± 0.7a | 11.3 ± 1.5b | 5.1 ± 0.4c |
| Final lean mass, g | 25.0 ± 0.5a | 21.9 ± 0.4b | 19.8 ± 0.5c |
| Liver mass, g | 2.4 ± 0.2a | 1.0 ± 0.1b | 1.2 ± 0.1b |
| Liver:body mass ratio, % | 5.6 ± 0.4a | 2.5 ± 0.2b | 2.9 ± 0.2b |
| Serum β-HB, mM | 0.3 ± 0.02a | 0.3 ± 0.03a | 0.5 ± 0.04b |
Serum β‐hydroxybutyrate (β-HB) concentrations were measured in whole blood from a tail nick following a 4-h food withholding/4-h refeeding period at the start of the dark cycle (6 PM). Values are means ± SE. P ≤ 0.05, different letters indicate statistically significant differences. CON, control; KE, ketone ester; PF, pair-fed.
KE Supplementation Attenuates NAFLD Activity Scores
Overall, total NAFLD activity scores were significantly lower for KE versus CON and PF (P ≤ 0.05 for all comparisons; Fig. 1, A and B). In addition, KE supplementation significantly attenuated histological steatosis, inflammation, and ballooning scores versus CON and steatosis and inflammation versus PF (P ≤ 0.05; Fig. 1, A and B).
Figure 1.

Effect of a dietary ketone ester on liver phenotype: histological steatosis and inflammation ×10 magnification (A) and nonalcoholic fatty liver disease (NAFLD) activity score (B). Black arrows indicate hepatic inflammation. White arrows indicate hepatocyte ballooning. Black lines in A represent scale bar. CON, control; KE, ketone ester; PF, pair-fed. Values are means ± SE (n = 8–10). P ≤ 0.05, different letters indicate statistically significant differences.
KE Supplementation Attenuates Markers of HSC Activation and Fibrogenesis
Increased histological fibrosis and markers of fibrogenesis and hepatic stellate cell activation are hallmarks of NAFLD/NASH progression. No differences were observed in histological fibrosis following 22 wk of HFD feeding (representative images in Fig. 2A). αSma and Col1A1 mRNA expression, markers of collagen deposition, were significantly lower in the KE and PF groups versus the CON group (P ≤ 0.05, Fig. 2C). KE supplementation significantly lowered α-SMA hepatic protein content compared with CON and PF groups (P ≤ 0.05, Fig. 2D), whereas no differences in COL1A1 protein content were observed among groups (P > 0.05, Fig. 2D). In addition, signatures of lower fibrogenesis and hepatic stellate cell activation were observed in livers of mice in the KE group versus the CON group, including PDGFβ and MMP9 protein content (P ≤ 0.05, Fig. 2E), with no significant differences observed for PF versus CON or KE versus PF (P > 0.05 for both comparisons, Fig. 2E).
Figure 2.

Effect of a dietary ketone ester on fibrosis and activation of hepatic stellate cells (HSCs) and fibrogenesis: representative Picro-Sirius Red (PSR) staining (A), hepatic markers of fibrosis mRNA expression (B), hepatic markers of fibrosis protein content (C), markers of hepatic stellate cell activation and fibrogenesis protein content (D), and representative Western blots (E). Black lines in A represent scale bar.CON, control; KE, ketone ester; PF, pair-fed. Values are means ± SE (n = 8–10). P ≤ 0.05, different letters indicate statistically significant differences.
Effect of KE Supplementation on Markers of Hepatic Inflammation
Activation of inflammatory pathways is linked to NAFLD/NASH progression. KE metabolism and signaling has been shown to have beneficial effects on inflammatory pathways in several tissues (9). Hence, we sought to explore the effect of a dietary KE on markers of hepatic inflammation. KE and PF feeding significantly reduced mRNA expression of Cd68, a marker of macrophage content, versus CON (P ≤ 0.05, Fig. 3A). Despite this, there were no significant differences in CD68 and F4/80 protein content for the CON group versus KE groups (P > 0.05, Fig. 3B). There was no effect of diet on the hepatic M1 inflammatory markers Il-1β and Tnfα mRNA expression (P > 0.05), whereas KE and PF significantly reduced Mcp1 mRNA expression versus CON (P ≤ 0.05, Fig. 3A). KE supplementation significantly increased CD11b protein content versus CON (P ≤ 0.05, Fig. 3B), whereas there was no significant effect of the KE on IL-1β protein content (P > 0.05, Fig. 3B). Given that caloric restriction is generally associated with reductions in inflammation, reasons as to why protein content of F4/80, IL-1β, and CD11b was significantly elevated for PF versus CON are unclear (P ≤ 0.05, Fig. 3B). We also explored measures of anti-inflammatory M2 macrophage markers. KE feeding significantly increased Cd163 mRNA expression versus CON and CD163 protein content compared with PF and CON (P ≤ 0.05, Fig. 3, A and B). Hepatic mRNA expression of the M2 markers Arg1 and Mrc1 did not differ among groups (P > 0.05, Fig. 3A). KE and PF also lowered inflammatory marker TRAIL protein content compared with CON, with significantly greater differences compared with PF (P ≤ 0.05, Fig. 3, C and E). In addition, KE significantly attenuated CCN1 protein content compared with PF and CON (P ≤ 0.05, Fig. 3, D and E).
Figure 3.

Effect of a dietary ketone ester on markers of inflammation: hepatic inflammatory markers mRNA expression (A), hepatic inflammatory markers protein content (B), TRAIL protein content (C), CCN1 protein content (D), and representative Western blots (E). CCN1, Cellular Communication Network Factor 1; CON, control; KE, ketone ester; PF, pair-fed; TRAIL, tumor necrosis factor (TNF)-related apoptosis-inducing ligand. Values are means ± SE (n = 8–10). P ≤ 0.05, different letters indicate statistically significant differences.
DISCUSSION
Achieving nutritional ketosis in the form of an exogenous KE may serve as a potential therapeutic avenue to mitigate metabolic disease. In particular, exogenous KE feeding has been shown to exert beneficial effects on adiposity and glucose metabolism and brown adipose tissue (22, 25–27). However, no studies have explored the impact of an exogenous dietary KE on liver outcomes in the setting of NAFLD. In this study, we demonstrate the efficacy of the KE BD-AcAc2 (30% by kcal) in attenuating NAFLD progression in obese mice receiving a high-fat, low-carbohydrate diet. Importantly, these data highlight that dietary KE supplementation ameliorates HFD-induced hepatic steatosis and inflammation, as well as attenuating profibrotic and proinflammatory markers in the liver beyond a pair-fed control group of mice receiving similar energy provisions.
As we have previously reported, replacing carbohydrate energy with BD-AcAc2 in the setting of a HFD reduced mean weekly energy intake by 26% versus CON (22). Furthermore, KE feeding was associated with a greater reduction in body weight (23%) compared with PF mice that received matched-energy provisions. Additional weight loss may have resulted from overall increases in energy expenditure, alterations in the partitioning of nutrients, loss of energy, or lower energy availability of the KE (22). Because ∼25% of BD-AcAc2 is metabolized to the nonphysiological S(l)-β-HB enantiomer, which is oxidized more slowly and to a lesser extent than the R enantiomer (28, 29), the difference in body weight observed between the KE and PF groups, in part, may be attributable to lower energy availability of the KE as an energy substrate. Furthermore, digestive efficiency of the KE may have also played a role. KE feeding has been shown to increase fecal production and fecal energy content compared with HFD-fed control mice (unpublished observations). Indeed, these factors may have impacted hepatic outcomes in this study and should be considered in future studies.
At present, weight loss via caloric restriction is the only approved and most effective therapeutic tool for the histological regression of NAFLD/NASH in both humans and animal models (30–35). In the current study, KE- and calorie-restricted pair-feeding significantly ameliorated NAS outcomes compared with HFD-fed controls, and these data are in line with existing literature demonstrating the use of caloric restriction to mitigate NAFLD (31, 34). Interestingly, KE feeding almost completely abrogated NAS compared with HFD-fed control and pair-fed groups (NAS = 0.4 ± 0.3, vs. 5.9 ± 0.5 and 3.5 ± 0.7, respectively). Furthermore, to our knowledge, we are the first to demonstrate an added benefit of an exogenous KE to reduce histological hepatic steatosis and inflammation going beyond benefits observed with caloric restriction alone (pair-fed group).
Activation of hepatic stellate cells is the main driver of liver fibrogenesis (36, 37). Paracrine signals from cellular injury, proinflammatory immune cell activation and infiltration, and systemic metabolic dysregulation can directly or indirectly induce stellate cell activation. Indeed, dietary KE feeding significantly reduced markers of hepatic stellate cell activation and transcriptional regulators of fibrogenesis (PDGFβ, MMP9, α-SMA, and COL1A1) versus obese control mice, and these improvements were beyond those observed with pair-feeding alone. Supporting this, macrophage mitochondrial oxidation of the ketone AcAc has been shown to modulate macrophage metabolic plasticity and attenuate liver fibrosis (16). In particular, macrophage AcAc metabolism was shown to decrease hepatic stellate cell activation, matrix deposition, and fibrogenesis. The authors also demonstrated that this effect was lost with the knockout of succinyl-coenzyme A-oxoacid transferase (SCOT), the terminal enzyme required for the oxidation of AcAc. Overall, these data demonstrate for the first time, to our knowledge, that exogenous ketone feeding with BD-AcAc2 protects against hepatic fibrogenic activation in obese male mice. However, future studies are required to elucidate the roles of mitochondrial ketone metabolism and/or signaling among the varying cell types in the liver (hepatocyte, Kupffer cell, stellate cell) and the role of AcAc versus β-HB on different immune and fibrosis effectors in NAFLD/NASH amelioration.
The hallmark of NAFLD progression to NASH is hepatocellular inflammation, mediated by a shift in the balance between activation of proinflammatory M1 macrophages and a reduction of anti-inflammatory M2 macrophages (38). Here, we demonstrate that KE supplementation increased M2 macrophage marker CD163, in the absence of changes in markers of total hepatic macrophage protein content (CD68, F4/80). CD163 is a scavenger receptor expressed exclusively on cells of monocyte/macrophage lineage (39) and plays a key role in anti-inflammatory and tissue regeneration processes (40) through downregulation in nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) proinflammatory processes (41). Although these data suggest that BD-AcAc2 may serve as a modulator of anti-inflammatory pathways, it should be noted that whether CD163 is pro- or anti-inflammatory in NAFLD is not conclusive (42), and thus, further research is required to elucidate its exact role.
CCN1 is a matrix protein that regulates cell death and immune cell adhesion (43) that is strongly associated with the pathogenesis of NASH in rodents and humans. (44, 45). BD-AcAc2 significantly lowered CCN1 protein content compared with HFD controls and pair-feeding. However, increased hepatic CCN1 expression has been shown to play a protective role against hepatic fibrosis and stellate cell activation (46, 47). Thus, further studies are required to delineate the mechanisms by which BD-AcAc2 attenuates CCN1 expression and to better understand its complex role in the pathogenesis of NAFLD. In a similar fashion, the ligand protein TRAIL, which plays a pivotal role in accentuating hepatic steatosis, inflammation, and apoptosis in NAFLD/NASH pathogenesis in human and murine models (48–51), was significantly decreased in mice fed a KE-supplemented diet. CCN1 signaling has been shown to enable the cytotoxicity of the inflammatory cytokine TNF-α and enhance the apoptotic activity of TRAIL through integrin-mediated signaling pathways (52–54). Nutritional ketosis achieved using BD-AcAc2 may interrupt the proinflammatory and proapoptotic signaling cascades of CCN1 and TRAIL by an unknown mechanism to ultimately attenuate NAFLD/NASH.
Surprisingly, KE feeding alone had no effect on classical M1 markers of inflammation, including Il-1b, Tnf-α, and Mpc1, well-established proinflammatory mediators implicated in the progression of NASH and fibrosis development (55, 56). Although, CD11b, a regulator of monocyte recruitment during an inflammatory response (57), was significantly elevated with KE feeding compared with obese control mice. β-HB has been shown to attenuate IL-1β and TNFα transcription, likely via inhibition of NFkB signaling in various tissues, including liver and cultured hepatocytes (58–61). The blood ketone concentrations achieved in this study (0.5 ± 0.04 mM) may explain some of the differing results observed in these well-established markers, with previous studies often reporting effects at concentrations > 5 mM (9). Indeed, further experiments at varying cumulative ketone body concentrations are required to provide greater clarity.
Diets high in simple sugars such as sucrose increase hepatic de novo lipogenesis (62). Hence, simply reducing sucrose content of the KE diet may have contributed to some of the improvements in liver outcomes observed. Future studies should match energy provisions of simple carbohydrates to address this potential confounding factor. Furthermore, KE produced significantly greater weight loss compared with PF animals receiving similar energy provisions. In addition, greater improvements in histological NAFLD outcomes occurred with the KE diet compared with pair-feeding, despite similar improvements in insulin and glucose tolerance for both groups compared with CON animals [measures previously reported by our group (22)], suggesting that the improvements in liver outcomes induced by KE feeding beyond pair-feeding are likely not related to improvements in insulin resistance solely. Indeed, future studies are needed to examine the extent to which the KE exerts direct effects on specific cell types in liver versus the expected improvements in hepatic health produced by weight loss alone.
Conclusions
In summary, we demonstrate for the first time, to our knowledge, that the dietary KE BD-AcAc2 abrogated histological and transcriptional markers of NAFLD/NASH in obese mice on a high-fat diet via a reduction in histological hepatic steatosis, lobular inflammation, and hepatocellular ballooning, beyond improvements observed for caloric restriction alone. These benefits histologically observed with KE feeding were accompanied by reductions in molecular markers of inflammation, hepatic stellate cell activation, and fibrogenesis. With no FDA‐approved pharmacological therapies for NAFLD/NASH currently available, dietary KE administration may provide an economic, safe, and practical treatment for prevention and treatment of NAFLD/NASH.
GRANTS
The project was supported by the University of Alabama (UAB) Nutrition Obesity Research Center (NORC) Pilot and Feasibility Award P30DK056336 (to E. P. Plaisance) from the National Institute of Diabetes and Digestive and Kidney Diseases and a Named New Investigator Award P30DK056336; the National Heart and Lung Institute Predoctoral Training Award T32HL105349 (to R. A. H. Davis); the UAB Postdoctoral Training Program in Obesity-Related Research T32 Postdoctoral Award T32DK062710 (to S. E. Deemer); and in part by the Veterans Affairs-Merit Grant I01BX003271 (to R. S. Rector). This work was supported with resources and the use of facilities at the Harry S Truman Memorial Veterans Hospital in Columbia, Missouri.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.P.M., R.A.H.D., S.E.D., B.M.R., E.P.P. and R.S.R. conceived and designed research; M.P.M., R.A.H.D., S.E.D., B.M.R., E.P.P. and R.S.R. performed experiments; M.P.M. and R.S.R. analyzed data; M.P.M., R.P.C., E.P.P. and R.S.R. interpreted results of experiments; M.P.M. and R.S.R. prepared figures; M.P.M., E.P.P. and R.S.R. drafted manuscript; M.P.M., R.P.C., R.A.H.D., S.E.D., B.M.R., E.P.P. and R.S.R. edited and revised manuscript; M.P.M., R.P.C., R.A.H.D., S.E.D., B.M.R., E.P.P. and R.S.R. approved final version of manuscript.
REFERENCES
- 1.Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, Harrison SA, Brunt EM, Sanyal AJ. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67: 328–357, 2018. doi: 10.1002/hep.29367. [DOI] [PubMed] [Google Scholar]
- 2.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 34: 274–285, 2011. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 3.Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, Ahmed A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 148: 547–555, 2015. doi: 10.1053/j.gastro.2014.11.039. [DOI] [PubMed] [Google Scholar]
- 4.Stepanova M, Rafiq N, Makhlouf H, Agrawal R, Kaur I, Younoszai Z, McCullough A, Goodman Z, Younossi ZM. Predictors of all-cause mortality and liver-related mortality in patients with non-alcoholic fatty liver disease (NAFLD). Dig Dis Sci 58: 3017–3023, 2013. doi: 10.1007/s10620-013-2743-5. [DOI] [PubMed] [Google Scholar]
- 5.Targher G, Marra F, Marchesini G. Increased risk of cardiovascular disease in non-alcoholic fatty liver disease: causal effect or epiphenomenon? Diabetologia 51: 1947–1953, 2008. doi: 10.1007/s00125-008-1135-4. [DOI] [PubMed] [Google Scholar]
- 6.Gandhi CR. Hepatic stellate cell activation and pro-fibrogenic signals. J Hepatol 67: 1104–1105, 2017. doi: 10.1016/j.jhep.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marcher A-B, Bendixen SM, Terkelsen MK, Hohmann SS, Hansen MH, Larsen BD, Mandrup S, Dimke H, Detlefsen S, Ravnskjaer K. Transcriptional regulation of hepatic stellate cell activation in NASH. Sci Rep 9: 2324–2324, 2019. doi: 10.1038/s41598-019-39112-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leoni S, Tovoli F, Napoli L, Serio I, Ferri S, Bolondi L. Current guidelines for the management of non-alcoholic fatty liver disease: a systematic review with comparative analysis. World J Gastroenterol 24: 3361–3373, 2018. doi: 10.3748/wjg.v24.i30.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puchalska P, Crawford PA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab 25: 262–284, 2017. doi: 10.1016/j.cmet.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schugar RC, Crawford PA. Low-carbohydrate ketogenic diets, glucose homeostasis, and nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care 15: 374–380, 2012. doi: 10.1097/MCO.0b013e3283547157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balasse EO, Féry F. Ketone body production and disposal: effects of fasting, diabetes, and exercise. Diabetes Metab Rev 5: 247–270, 1989. doi: 10.1002/dmr.5610050304. [DOI] [PubMed] [Google Scholar]
- 12.Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev 60: 143–187, 1980. doi: 10.1152/physrev.1980.60.1.143. [DOI] [PubMed] [Google Scholar]
- 13.Yancy WS, Olsen MK, Guyton JR, Bakst RP, Westman EC. A low-carbohydrate, ketogenic diet versus a low-fat diet to treat obesity and hyperlipidemia: a randomized, controlled trial. Ann Intern Med 140: 769–777, 2004. doi: 10.7326/0003-4819-140-10-200405180-00006. [DOI] [PubMed] [Google Scholar]
- 14.Cahill JG. Starvation in man. Clin Endocrinol Metab 5: 397–415, 1976. doi: 10.1016/S0300-595X(76)80028-X. [DOI] [PubMed] [Google Scholar]
- 15.Owen OE, Felig P, Morgan AP, Wahren J, Cahill GF. Liver and kidney metabolism during prolonged starvation. J Clin Invest 48: 574–583, 1969. doi: 10.1172/JCI106016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puchalska P, Martin SE, Huang X, Lengfeld JE, Daniel B, Graham MJ, Han X, Nagy L, Patti GJ, Crawford PA. Hepatocyte-macrophage acetoacetate shuttle protects against tissue fibrosis. Cell Metab 29: 383–398.e387, 2019. doi: 10.1016/j.cmet.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunengraber H. Potential of ketone body esters for parenteral and oral nutrition. Nutrition 13: 233–235, 1997. doi: 10.1016/s0899-9007(96)00409-1. [DOI] [PubMed] [Google Scholar]
- 18.Ari C, Kovács Z, Juhasz G, Murdun C, Goldhagen CR, Koutnik AP, Poff AM, Kesl SL, D’Agostino DP. Exogenous ketone supplements reduce anxiety-related behavior in Sprague-Dawley and Wistar Albino Glaxo/Rijswijk rats. Front Mol Neurosci 9: 137, 2017. doi: 10.3389/fnagi.2017.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carpenter RG, Grossman S. Plasma fat metabolites and hunger. Physiol Behav 30: 57–63, 1983. doi: 10.1016/0031-9384(83)90038-0. [DOI] [PubMed] [Google Scholar]
- 20.Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa-Veloso K, Ho M, Roberts A, Robertson J, Vanitallie TB, Veech RL. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol 63: 401–408, 2012. doi: 10.1016/j.yrtph.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poff A, Ari C, Arnold P, Seyfried T, D'agostino D. Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. Int J Cancer 135: 1711–1720, 2014. doi: 10.1002/ijc.28809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis RAH, Deemer SE, Bergeron JM, Little JT, Warren JL, Fisher G, Smith DL , Jr, Fontaine KR, Dickinson SL, Allison DB, Plaisance EP. Dietary R, S-1,3-butanediol diacetoacetate reduces body weight and adiposity in obese mice fed a high-fat diet. FASEB J 33: 2409–2421, 2019. doi: 10.1096/fj.201800821RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 94: 2467–2474, 1999. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 24.Moore MP, Cunningham RP, Kelty TJ, Boccardi LR, Nguyen NY, Booth FW, Rector RS. Ketogenic diet in combination with voluntary exercise impacts markers of hepatic metabolism and oxidative stress in male and female Wistar rats. Appl Physiol Nutr Metab 45: 35–44, 2020. doi: 10.1139/apnm-2019-0042. [DOI] [PubMed] [Google Scholar]
- 25.Ari C, Murdun C, Koutnik AP, Goldhagen CR, Rogers C, Park C, Bharwani S, Diamond DM, Kindy MS, D'Agostino DP, Kovács Z. Exogenous ketones lower blood glucose level in rested and exercised rodent models. Nutrients 11: 2330, 2019. doi: 10.3390/nu11102330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deemer SE, Davis RAH, Gower BA, Koutnik AP, Poff AM, Dickinson SL, Allison DB, D'Agostino DP, Plaisance EP. Concentration-dependent effects of a dietary ketone ester on components of energy balance in mice. Front Nutr 6: 56, 2019. doi: 10.3389/fnut.2019.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srivastava S, Kashiwaya Y, King MT, Baxa U, Tam J, Niu G, Chen X, Clarke K, Veech RL. Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB J 26: 2351–2362, 2012. doi: 10.1096/fj.11-200410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stubbs BJ, Cox PJ, Evans RD, Santer P, Miller JJ, Faull OK, Magor-Elliott S, Hiyama S, Stirling M, Clarke K. On the metabolism of exogenous ketones in humans. Front Physiol 8: 848, 2017. doi: 10.3389/fphys.2017.00848, 10.3389/fpls.2017.00848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Webber RJ, Edmond J. Utilization of L (+)-3-hydroxybutyrate, D (-)-3-hydroxybutyrate, acetoacetate, and glucose for respiration and lipid synthesis in the 18-day-old rat. J Biol Chem 252: 5222–5226, 1977. doi: 10.1016/S0021-9258(19)63335-1. [DOI] [PubMed] [Google Scholar]
- 30.Elias MC, Parise ER, de Carvalho L, Szejnfeld D, Netto JP. Effect of 6-month nutritional intervention on non-alcoholic fatty liver disease. Nutrition 26: 1094–1099, 2010. doi: 10.1016/j.nut.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 31.Larson-Meyer DE, Newcomer BR, Heilbronn LK, Volaufova J, Smith SR, Alfonso AJ, Lefevre M, Rood JC, Williamson DA, Ravussin E, Pennington CT; Pennington CALERIE Team. Effect of 6-month calorie restriction and exercise on serum and liver lipids and markers of liver function. Obesity (Silver Spring) 16: 1355–1362, 2008. doi: 10.1038/oby.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linden MA, Fletcher JA, Meers GM, Thyfault JP, Laughlin MH, Rector RS. A return to ad libitum feeding following caloric restriction promotes hepatic steatosis in hyperphagic OLETF rats. Am J Physiol Gastrointest Liver Physiol 311: G387–G395, 2016. doi: 10.1152/ajpgi.00089.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linden MA, Lopez KT, Fletcher JA, Morris EM, Meers GM, Siddique S, Laughlin MH, Sowers JR, Thyfault JP, Ibdah JA, Rector RS. Combining metformin therapy with caloric restriction for the management of type 2 diabetes and nonalcoholic fatty liver disease in obese rats. Appl Physiol Nutr Metab 40: 1038–1047, 2015. doi: 10.1139/apnm-2015-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rector RS, Uptergrove GM, Morris EM, Borengasser SJ, Laughlin MH, Booth FW, Thyfault JP, Ibdah JA. Daily exercise vs. caloric restriction for prevention of nonalcoholic fatty liver disease in the OLETF rat model. Am J Physiol Gastrointest Liver Physiol 300: G874–G883, 2011. doi: 10.1152/ajpgi.00510.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thoma C, Day CP, Trenell MI. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: a systematic review. J Hepatol 56: 255–266, 2012. doi: 10.1016/j.jhep.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 36.Czochra P, Klopcic B, Meyer E, Herkel J, Garcia-Lazaro JF, Thieringer F, Schirmacher P, Biesterfeld S, Galle PR, Lohse AW, Kanzler S. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J Hepatol 45: 419–428, 2006. doi: 10.1016/j.jhep.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Giraudi PJ, Becerra VJB, Marin V, Chavez-Tapia NC, Tiribelli C, Rosso N. The importance of the interaction between hepatocyte and hepatic stellate cells in fibrogenesis induced by fatty accumulation. Exp Mol Pathol 98: 85–92, 2015. doi: 10.1016/j.yexmp.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 38.Alisi A, Carpino G, Oliveira FL, Panera N, Nobili V, Gaudio E. The role of tissue macrophage-mediated inflammation on NAFLD pathogenesis and its clinical implications. Mediators Inflamm 2017: 8162421, 2017. doi: 10.1155/2017/8162421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pulford K, Micklem K, McCarthy S, Cordell J, Jones M, Mason DY. A monocyte/macrophage antigen recognized by the four antibodies GHI/61, Ber-MAC3, Ki-M8 and SM4. Immunology 75: 588–595, 1992. [PMC free article] [PubMed] [Google Scholar]
- 40.Marchetti V, Yanes O, Aguilar E, Wang M, Friedlander D, Moreno S, Storm K, Zhan M, Naccache S, Nemerow G, Siuzdak G, Friedlander M. Differential macrophage polarization promotes tissue remodeling and repair in a model of ischemic retinopathy. Sci Rep 1: 76–76, 2011. doi: 10.1038/srep00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akahori H, Karmali V, Polavarapu R, Lyle AN, Weiss D, Shin E, Husain A, Naqvi N, Van Dam R, Habib A, Choi CU, King AL, Pachura K, Taylor WR, Lefer DJ, Finn AV. CD163 interacts with TWEAK to regulate tissue regeneration after ischaemic injury. Nat Commun 6: 7792, 2015. doi: 10.1038/ncomms8792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, Schuppan D, Grønbæk H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 16: 145–159, 2019. doi: 10.1038/s41575-018-0082-x. [DOI] [PubMed] [Google Scholar]
- 43.Chen C-C, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol 41: 771–783, 2009. doi: 10.1016/j.biocel.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bian Z, Peng Y, You Z, Wang Q, Miao Q, Liu Y, Han X, Qiu D, Li Z, Ma X. CCN1 expression in hepatocytes contributes to macrophage infiltration in nonalcoholic fatty liver disease in mice. J Lipid Res 54: 44–54, 2013. doi: 10.1194/jlr.M026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ju L, Sun Y, Xue H, Chen L, Gu C, Shao J, Lu R, Luo X, Wei J, Ma X, Bian Z. CCN1 promotes hepatic steadies and inflammation in non-alcoholic steatohepatitis. Sci Rep 10: 1–13, 2020. doi: 10.1038/s41598-020-60138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borkham-Kamphorst E, Steffen BT, Van de Leur E, Haas U, Tihaa L, Friedman SL, Weiskirchen R. CCN1/CYR61 overexpression in hepatic stellate cells induces ER stress-related apoptosis. Cell Signal 28: 34–42, 2016. doi: 10.1016/j.cellsig.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 47.Kim K-H, Chen C-C, Monzon RI, Lau LF. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol 33: 2078–2090, 2013. doi: 10.1128/MCB.00049-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cartland SP, Harith HH, Genner SW, Dang L, Cogger VC, Vellozzi M, Di Bartolo BA, Thomas SR, Adams LA, Kavurma MM. Non-alcoholic fatty liver disease, vascular inflammation and insulin resistance are exacerbated by TRAIL deletion in mice. Sci Rep 7: 1–12, 2017. doi: 10.1038/s41598-017-01721-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirsova P, Ibrahim SH, Bronk SF, Yagita H, Gores GJ. Vismodegib suppresses TRAIL-mediated liver injury in a mouse model of nonalcoholic steatohepatitis. PLoS One 8: e70599-e70599, 2013. doi: 10.1371/journal.pone.0070599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hirsova P, Weng P, Salim W, Bronk SF, Griffith TS, Ibrahim SH, Gores GJ. TRAIL deletion prevents liver, but not adipose tissue, inflammation during murine diet-induced obesity. Hepatol Commun 1: 648–662, 2017. doi: 10.1002/hep4.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Volkmann X, Fischer U, Bahr MJ, Ott M, Lehner F, Macfarlane M, Cohen GM, Manns MP, Schulze-Osthoff K, Bantel H. Increased hepatotoxicity of tumor necrosis factor-related apoptosis-inducing ligand in diseased human liver. Hepatology 46: 1498–1508, 2007. doi: 10.1002/hep.21846. [DOI] [PubMed] [Google Scholar]
- 52.Bai T, Chen CC, Lau LF. Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages. J Immunol 184: 3223–3232, 2010. doi: 10.4049/jimmunol.0902792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen C-C, Young JL, Monzon RI, Chen N, Todorović V, Lau LF. Cytotoxicity of TNFalpha is regulated by integrin-mediated matrix signaling. EMBO J 26: 1257–1267, 2007. doi: 10.1038/sj.emboj.7601596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franzen CA, Chen CC, Todorović V, Juric V, Monzon RI, Lau LF. Matrix protein CCN1 is critical for prostate carcinoma cell proliferation and TRAIL-induced apoptosis. Mol Cancer Res 7: 1045–1055, 2009. doi: 10.1158/1541-7786.MCR-09-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Braunersreuther V, Viviani GL, Mach F, Montecucco F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J Gastroenterol 18: 727–735, 2012. doi: 10.3748/wjg.v18.i8.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palacios RS, Roderfeld M, Hemmann S, Rath T, Atanasova S, Tschuschner A, Gressner OA, Weiskirchen R, Graf J, Roeb E. Activation of hepatic stellate cells is associated with cytokine expression in thioacetamide-induced hepatic fibrosis in mice. Lab Invest 88: 1192–1203, 2008. doi: 10.1038/labinvest.2008.91. [DOI] [PubMed] [Google Scholar]
- 57.Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol 27: 339–362, 2009. doi: 10.1146/annurev.immunol.021908.132554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bae HR, Kim DH, Park MH, Lee B, Kim MJ, Lee EK, Chung KW, Kim SM, Im DS, Chung HY. β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 7: 66444–66454, 2016. doi: 10.18632/oncotarget.12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fu S-P, Li S-N, Wang J-F, Li Y, Xie S-S, Xue W-J, Liu H-M, Huang B-X, Lv Q-K, Lei L-C, Liu G-W, Wang W, Liu J-X. BHBA suppresses LPS-induced inflammation in BV-2 cells by inhibiting NF-κB activation. Mediators Inflamm 2014: 983401–983401, 2014. doi: 10.1155/2014/983401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamanashi T, Iwata M, Kamiya N, Tsunetomi K, Kajitani N, Wada N, Iitsuka T, Yamauchi T, Miura A, Pu S, Shirayama Y, Watanabe K, Duman RS, Kaneko K. Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci Rep 7: 7677–7677, 2017. doi: 10.1038/s41598-017-08055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Youm Y-H, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D'Agostino D, Planavsky N, Lupfer C, Kanneganti TD, Kang S, Horvath TL, Fahmy TM, Crawford PA, Biragyn A, Alnemri E, Dixit VD. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 21: 263–269, 2015. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moore JB, Gunn PJ, Fielding BA. The role of dietary sugars and de novo lipogenesis in non-alcoholic fatty liver disease. Nutrients 6: 5679–5703, 2014. doi: 10.3390/nu6125679. [DOI] [PMC free article] [PubMed] [Google Scholar]