

Abstract
Inhaled gene therapy poses a unique potential of curing chronic lung diseases, which are currently managed primarily by symptomatic treatments. However, it has been challenging to achieve therapeutically relevant gene transfer efficacy in the lung due to the presence of numerous biological delivery barriers. Here, we introduce a simple approach that overcomes both extracellular and cellular barriers to enhance gene transfer efficacy in the lung in vivo. We endowed tetra(piperazino)fullerene epoxide (TPFE)‐based nanoparticles with non‐adhesive surface polyethylene glycol (PEG) coatings, thereby enabling the nanoparticles to cross the airway mucus gel layer and avoid phagocytic uptake by alveolar macrophages. In parallel, we utilized a hypotonic vehicle to facilitate endocytic uptake of the PEGylated nanoparticles by lung parenchymal cells via the osmotically driven regulatory volume decrease (RVD) mechanism. We demonstrate that this two‐pronged delivery strategy provides safe, wide‐spread and high‐level transgene expression in the lungs of both healthy mice and mice with chronic lung diseases characterized by reinforced delivery barriers.
Keywords: fullerene, gene delivery, hypotonic vehicles, inhalation, RVD effect
Tetra(piperazino)fullerene (TPFE)‐based nanoparticles (NPs), surface‐coated with polyethylene glycol (PEG), were engineered. The PEGylation allows the NPs to avoid interactions with mucin glycoproteins and phagocytic uptake by macrophages. After the NPs access the target lung cells, a hypotonic vehicle solution enhances their endocytosis via the osmotically driven regulatory volume effect, thereby providing highly efficient transgene expression.
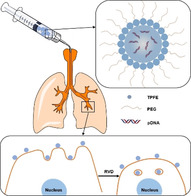
Current standards‐of‐care for chronic lung diseases, such as cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD) and asthma, manage disease symptoms at best,[1] except for a few daily administered oral tablets available to genetically defined subpopulations of CF patients.[2] Discovery of numerous genetic targets or therapeutic nucleic acids has promoted gene therapy as a broadly applicable therapeutic strategy to potentially cure patients with these notoriously refractory lung diseases.[3] In particular, localized gene therapy via an inhaled route provides a unique opportunity to exert therapeutic impacts directly on the lung, while mitigating the risk of systemic adverse effects by minimizing extra‐pulmonary gene transfer.[4] However, both airways and alveolar sacs, two primary compartments of the lung, harbor challenging biological barriers that hamper efficient delivery of inhaled gene therapy to target lung parenchymal cells.[5] Specifically, lung airways are covered with a highly adhesive and nano‐porous mucus gel layer that readily traps inhaled foreign matters and rapidly removes them from the lung via physiological mucus clearance mechanisms.[6] On the other hand, an army of alveolar macrophages populated throughout the alveolar sacs (12–35 cells per alveolar sac[7]) are specialized in engulfing and subsequently degrading foreign particulate matters.[8]
A cationic carbon nanocluster, TPFE, is an attractive gene transfer agent that provides robust DNA compaction, endocytic cellular uptake, protection against endosomal nuclease and cytoplasmic DNA release.[9] Further, individual fullerene molecule can be cleared from human body due to its small particle diameter of 1 nm (i.e., smaller than the size cutoff of glomerular filtration).[10] However, TPFE‐based gene delivery nanoparticles were previously shown to mediate highly efficient transgene expression in the liver and spleen, but not in the lung, following systemic administration.[11] Although the administration via an inhaled route might change this reality, TPFE per se is unlikely to do so due to its positively charged and hydrophobic nature that makes it prone to mucus entrapment and/or macrophage uptake.[12] Thus, we here implemented a simple strategy of endowing TPFE‐based gene delivery nanoparticles with non‐adhesive surface PEG coatings by chemically conjugating PEG polymer chains to the piperazine component of TPFE. Of note, we and others have demonstrated that nanoparticles densely coated with PEG are capable of efficiently penetrating the airway mucus,[13] and resisting macrophage uptake,[12] thereby increasing the probability of particle access to target lung cells.
Despite the aforementioned benefits, PEGylation might interfere with nanoparticle interactions with, and subsequent uptake by, target cells (i.e., PEG dilemma).[14] We thus employed an additional strategy of modulating vehicle osmolality to enhance endocytosis of nanoparticles, the process called RVD.[15] The RVD is initiated by hypotonically driven cell swelling that stimulates fusion of intracellular vesicles with plasma membrane to prevent cell lysis.[16] Subsequent activation of P2 receptors triggers the osmotically‐driven release of intracellular ions and water, followed by internalization of plasma membrane to homeostatically reform the lost vesicles.[17] During this process, small particulate matters that successfully reach the vicinity of target cell membrane would be endocytosed. We thus have investigated here whether a hypotonic vehicle facilitates the uptake of PEGylated TPFE (TPFE‐PEG)‐based gene delivery nanoparticles by lung epithelial cells in vivo via the RVD mechanism.
We first synthesized TPFE using a previously established method.[18] Briefly, C60 was reacted with 1‐tert‐butyloxycarbonyl piperazine to yield 1‐tert‐butyloxycarbonyl‐protected TPFE (Boc‐TPFE), followed by deprotection to obtain TPFE (Figure S1). We confirmed successful synthesis of Boc‐TPFE and TPFE using nuclear magnetic resonance (1H‐NMR) spectroscopy and mass spectroscopy (Figure S2–S5). Subsequently, 5 kDa PEG polymers were chemically conjugated to TPFE via covalent coupling of N‐hydroxysuccinimide of PEG and secondary amine of piperazine to yield PEGylated TPFE (TPFE‐PEG). 1H‐NMR analysis revealed the PEG to TPFE molar ratio of TPFE‐PEG to be 2.96 (Figure 1 A).
Figure 1.
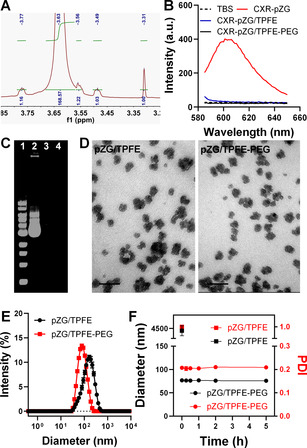
Physicochemical properties of TPFE‐based gene delivery nanoparticles. A) 1H‐NMR spectrum of TPFE‐PEG. B) Fluorescence spectra of CXR‐labeled pAAV‐ZsGreen1 plasmid (CXR‐pZG) and CXR‐pZG‐loaded TPFE (CXR‐pZG/TPFE) and TPFE‐PEG (CXR‐pZG/TPFE‐PEG). Fluorescence quenching indicates successful compaction of CXR‐pZG. C) Electrophoretic migration assay demonstrating efficient compaction of pZG by nanoparticles: DNA ladder (1), naked pZG (2), pZG/TPFE (3) and pZG/TPFE‐PEG (4). D) Representative TEM images (scale bars=100 nm) and E) hydrodynamic diameter profiles of pZG/TPFE and pZG/TPFE‐PEG (n=3). F) Colloidal stability of pZG/TPFE (solid squares) and pZG/TPFE‐PEG (solid circles) in BALF at 37 °C over time (n=3), based on the changes in hydrodynamic diameter (black) and PDI (red). Hydrodynamic diameters and PDI values of pZG/TPFE and pZG/TPFE‐PEG were measured by dynamic light scattering (DLS).
We next went on to prepare DNA‐loaded nanoparticles by condensing plasmid DNA using either TPFE only or a blend of TPFE and TPFE‐PEG at a molar ratio of 5:1 (N/P ratio is 20; Figure S6). To confirm successful DNA compaction, we employed two different experimental methods. Leveraging the unique fluorescence quenching property of C60 of TPFE, we monitored fluorescence spectra of carboxy‐X‐rhodamine (CXR)‐labeled plasmid DNA encoding fluorescent Zsgreen protein (pZG hereafter) condensed by either TPFE or the blend. We found that the fluorescence of CXR‐pZG was completely quenched by TPFE or the blend (Figure 1 B), indicating successful compaction of CXR‐pZG. We then conducted the more conventional electrophoretic migration assay to validate this observation. The DNA band was invisible when (unlabeled) pZG was condensed by TPFE or the blend, an indicative of full compaction, whereas naked pZG (lane 2) migrated away from the well (Figure 1 C). Collectively, the findings here underscore that inclusion of PEG does not compromise the ability of TPFE to form stable nanocomplexes with plasmid DNA.
We next conducted fundamental physicochemical characterization of these TPFE‐based gene delivery nanoparticles carrying either pZG or plasmid DNA encoding luciferase (pd1GL3‐RL; pGL3 hereafter). Transmission electron microscopy and dynamic light scattering revealed that both non‐PEGylated (plasmid DNA/TPFE hereafter) and PEGylated (plasmid DNA/TPFE‐PEG hereafter) nanoparticles exhibited similar cluster‐shaped morphology and hydrodynamic diameters ranging from 73 to 90 nm based on z‐average (number mean: 32–59 nm), respectively (Figure 1 D,E, Figure S7–8). However, while non‐PEGylated nanoparticles, including pZG/TPFE and pGL3/TPFE, exhibited highly positive ζ‐potential values (>25 mV), PEGylated nanoparticles, including pZG/TPFE‐PEG and pGL3/TPFE‐PEG, possessed near‐neutral surface charges (<3.5 mV) (Table S1), suggesting that the otherwise positively charged particle surfaces were efficiently shielded by PEG. It is critical to retain desired particle properties in physiologically relevant conditions for in vivo application,[5a] and thus we assessed colloidal stability of pZG/TPFE and pZG/TPFE‐PEG in mouse bronchoalveolar lavage fluid (BALF). The size and polydispersity index (PDI) of pZG/TPFE increased instantaneously upon incubation in BALF, whereas pZG/TPFE‐PEG exhibited excellent colloidal stability, as evidenced by unperturbed size and PDI at least up to 5 hours (Figure 1 F).
The next rational step was to investigate whether our PEGylation strategy endowed TPFE‐based gene delivery nanoparticles with abilities to resist adhesive interactions with mucus and/or phagocytosis by alveolar macrophages. To do this, we incubated pZG/TPFE and pZG/TPFE‐PEG in a mucin solution and monitored the changes in their particle sizes. Of note, we prepared the mucin solution by dissolving porcine gastric mucin in ultrapure water to assess exclusively the impact of mucin (without being affected by electrolytes in an ionic solution). We found that the mucin solution mediated rapid (within 15 minutes) and significant aggregation of pZG/TPFE. However, hydrodynamic diameters of pZG/TPFE‐PEG were unchanged upon the incubation at least up to an hour (Figure 2 A), suggesting that the surface PEG coatings efficiently prevented adhesive interactions with mucin. In parallel, we compared uptake of pZG/TPFE and pZG/TPFE‐PEG by mouse alveolar macrophages in vitro where we found that our PEGylation strategy markedly reduced phagocytosis of TPFE‐based gene delivery nanoparticles; quantitatively, macrophage uptake was ≈80 % and ≈20 % for pZG/TPFE and pZG/TPFE‐PEG, respectively (Figure 2 B).
Figure 2.
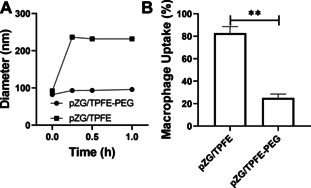
The ability of TPFE‐based gene delivery nanoparticles to overcome biological delivery barriers in the lung. A) Colloidal stability of pZG/TPFE (solid squares) and pZG/TPFE‐PEG (solid circles) in a mucin solution at 37 °C over time (n=3), which indicates the ability of nanoparticles to resist adhesive interactions with mucin. Hydrodynamic diameters and PDI values of pZG/TPFE and pZG/TPFE‐PEG were measured by DLS. B) In vitro uptake of pZG/TPFE and pZG/TPFE‐PEG by mouse alveolar macrophages (MH‐S) after a 2‐hour incubation (n=5). Error bars represent SEM. **p<0.01.
Encouraged by our in vitro observation, we investigated the ability of TPFE‐based gene delivery nanoparticles to mediate reporter transgene expression in the lungs of wild‐type C57BL/6 mice following intratracheal administration. Here, we engineered nanoparticles to carry pZG or pGL3 to evaluate distribution or overall level of transgene expression, respectively, and used ultrapure water as a vehicle to potentially exploit the hypotonically driven RVD effect. As shown by representative confocal images of lung tissue sections, pZG/TPFE‐PEG mediated widespread and uniform transgene expression throughout airways and alveolar sacs (>65 % coverage), unlike naked pZG and pZG/TPFE that exhibited negligible fluorescence (<2 % coverage) (Figures 3 A,B). In parallel, lung homogenate‐based luciferase assay revealed that pGL3/TPFE‐PEG provided approximately two orders of magnitude greater luciferase activity compared to naked pGL3 and pGL3/TPFE in the lungs of C57/BL/6 mice (Figure 3 C). The findings here agree with our previous observations that dense surface PEG coatings markedly enhance gene transfer efficacy of polymer‐ and peptide‐based gene delivery nanoparticles following localized administration.[12, 19] Of note, we confirmed that meaningful luciferase activity was not detected in mononuclear phagocyte systems, including liver and spleen, and kidney (Figure S9), suggesting that the plasmid payload (i.e., pGL) was unlikely translocated to extra‐pulmonary organs.
Figure 3.
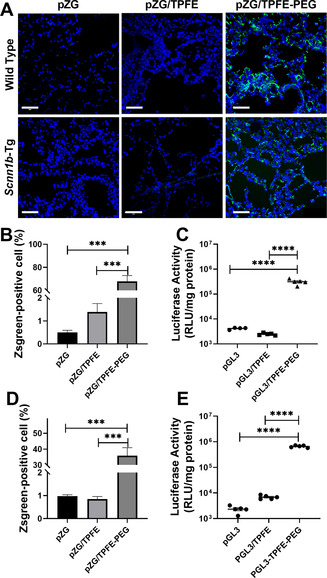
In vivo reporter transgene expression mediated by TPFE‐based delivery nanoparticles following intratracheal administration. A) Representative confocal images demonstrating distribution of transgene expression mediated by naked pZG (Left), pZG/TPFE (Middle) and pZG/TPFE‐PEG (Right) in the lungs of wild‐type C57BL/6 (Upper) and Scnn1b‐Tg mice (Lower). Scale bar=100 μm. Image‐based analysis demonstrating the percentages of cells with reporter transgene expression in the lungs of B) wild‐type C57BL/6 and D) Scnn1b‐Tg mice (n=3). Overall level of transgene expression mediated by naked pd1GL3‐RL (pGL3), pGL3/TPFE and pGL3/TPFE‐PEG in the lungs of C) wild‐type C57BL/6 and E) Scnn1b‐Tg mice, determined by homogenate‐based luciferase assay (n=5). *p<0.05, **p<0.01, ***p<0.005 and **** p<0.001.
To establish clinical relevance, we next repeated these studies using a transgenic mouse model of chronic lung diseases, specifically the mice overexpressing epithelial sodium channel (ENaC) β subunit, backgrounded on C57BL/6 (Scnn1b‐Tg mice). Scnn1b‐Tg mice exhibit key pathologies of CF and COPD, including mucus accumulation/stasis and chronic lung inflammation.[20] These pathological features essentially reinforce biological delivery barriers to inhaled gene transfer to lung epithelial cells in both airways and airspace.[5a] Nevertheless, pZG/TPFE‐PEG provided remarkably widespread transgene expression (≈35 % coverage) in the lungs of Scnn1b‐Tg mice (Figure 3 A,D), although the coverage was reduced compared to what we observed in in healthy mouse lungs (Figure 3 B). We also found that pGL3/TPFE‐PEG retained the ability to mediate high‐level overall transgene expression in the lungs of Scnn1b‐Tg mice regardless of the more challenging delivery barriers (Figure 3 E). Overall, our in vitro and in vivo studies collectively demonstrated that effective surface shielding by PEG renders TPFE‐based gene delivery nanoparticles highly efficacious in transfecting parenchymal cells in the lungs of both healthy and diseased mice, most likely due to the ability to breach the aforementioned delivery barriers.
We next sought to validate our hypothesis that a hypotonic vehicle could enhance gene transfer efficacy in the lung by facilitating endocytosis of gene delivery nanoparticles via the RVD effect. We first confirmed that physicochemical properties (i.e., hydrodynamic diameter, PDI and ζ‐potential) of PEGylated nanoparticles, including pZG/TPFE‐PEG and pGL3/TPFE‐PEG, were virtually identical when suspended in vehicles of varying osmolality (Table S2). The finding excludes the possibility that gene transfer efficacy might be affected by the alteration of particle characteristics in vehicles with varying ionic strengths. We then compared reporter transgene expression mediated by pZG/TPFE‐PEG intratracheally administered in a hypotonic (i.e., ultrapure water), a mildly hypotonic (0.45 % saline) or an isotonic (0.9 % saline) vehicle. As shown by representative confocal images, reporter fluorescence signal attenuated with increases in vehicle osmolality both in airways and airspace (Figure 4 A). We also repeated the experiment with pGL3/TPFE‐PEG to quantify the overall level of transgene expression and observed a similar trend where the luciferase activity was greatest when a hypotonic vehicle was used, followed by mildly hypotonic and isotonic vehicles (Figure 4 B). Quantitatively, pGL3/TPFE‐PEG administered in ultrapure water provided an order of magnitude greater transgene expression compared to the identical nanoparticles administered in 0.9 % saline. To investigate whether the enhanced transgene expression was indeed driven by the RVD effect, we dosed mice with pGL3/TPFE‐PEG suspended in water containing a P2 antagonist (i.e., suramin, 100 nM), a potent inhibitor of the RVD effect.[15, 16] We found that the luciferase activity was significantly reduced to the level observed with pGL3/TPFE‐PEG administered in 0.9 % saline when the RVD was inhibited by suramin (Figure 4 B). In contrast, luciferase activity was virtually identical when pGL3/TPFE‐PEG was administered in 0.9 % saline irrespective of suramin treatment (Figure 4 B). We also confirmed the RVD‐mediated enhancement of transgene expression in vitro using human lung epithelial cell lines (Figure S10–11). The findings here underscore that the hypotonically driven RVD effect played a significant role on mediating efficient transgene expression by pGL3/TPFE‐PEG following intratracheal administration.
Figure 4.
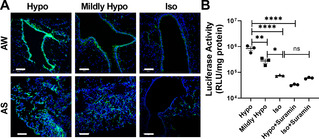
The effect of vehicle osmolality on TPFE‐PEG‐mediated transgene expression following intratracheal administration. A) Representative confocal images demonstrating distribution of transgene expression mediated by pZG/TPFE‐PEG administered in water‐ or saline‐based vehicle solutions with varying osmolality, including hypotonic (Hypo, water), mildly hypotonic (Mildly Hypo, 0.45 % saline) and isotonic (Iso, 0.9 % saline) vehicles in the airway (AW) and alveolar sacs (AS) of wild‐type C57BL/6 mice. The cell nuclei are stained with DAPI (blue). Scale bars=100 μm. B) Overall level of transgene expression mediated by pGL3/TPFE‐PEG administered in hypotonic (±100 nM suramin), mildly hypotonic and isotonic vehicles (±100 nM suramin) in the lungs of wild‐type C57BL/6 mice (n=3). Error bars represent SEM. *p<0.05, **p<0.01 and ****p<0.001.
We finally conduced comprehensive in vitro and in vivo safety assessments. We first confirmed in vitro that cell viability was unperturbed by pGL3/TPFE‐PEG at a broad plasmid DNA concentrations (Figure S12). We then evaluated local and systemic safety profiles of mice intratracheally received pGL3/TPFE‐PEG (in ultrapure water). Tissue sections of primary organs harvested from the treated mice, including lung, heart, liver, spleen and kidney, stained with hematoxylin and eosin did not exhibit any sign of acute inflammation or tissue damage, similar to the observation with untreated mice (Figure S13). We also confirmed that T helper type 1 and 2 cytokine levels and immune cell counts in BALF were not altered by the treatment (Figure S14–15). In addition, blood biochemistry parameters quantified from untreated control and treated mice were virtually identical and fell in normal ranges (Table S3). The findings here suggest that our pulmonary gene delivery approach presents excellent local and systemic biocompatibility and safety.
In conclusion, we here demonstrate that a marriage of PEGylated TPFE‐based gene delivery nanoparticles and osmotically driven RVD effect synergistically enhances gene transfer efficacy in the lung. We conducted a panel of in vitro and in vivo studies to confirm that the enhancement was attributed to the ability to overcome numerous biological barriers present in the lung. Specifically, our PEGylation strategy endows TPFE‐based gene delivery nanoparticles with abilities to overcome key extracellular barriers, even in a pathological condition characterized by the reinforced barrier properties. We also discovered that a hypotonic vehicle facilitated endocytic uptake of the PEGylated nanoparticles by lung cells via the RVD effect, presumably providing a means to overcome the PEG dilemma if any. The elegantly simple combinatorial delivery strategy introduced here may pave a way for clinical development of pulmonary gene therapy.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the National Institutes of Health (R01HL136617 and P30EY001765) and the Cystic Fibrosis Foundation (SUK18I0).
D. Chen, S. Liu, D. Chen, J. Liu, J. Wu, H. Wang, Y. Su, G. Kwak, X. Zuo, D. Rao, H. Cui, C. Shu, J. S. Suk, Angew. Chem. Int. Ed. 2021, 60, 15225.
Contributor Information
Prof. Chunying Shu, Email: shucy@iccas.ac.cn.
Prof. Jung Soo Suk, Email: jsuk@jhmi.edu.
References
- 1.Bereza B. G., Nielsen A. T., Valgardsson S., Hemels M. E., Einarson T. R., Int. J. Chronic Obstruct. Pulm. Dis. 2015, 10, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a.Hoffman L. R., Ramsey B. W., Chest 2013, 143, 207–213; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b.Rafeeq M. M., Murad H. A. S., J. Transl. Med. 2017, 15, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a.Kim J., Jeon S., Kang S. J., Kim K.-R., Thai H. B. D., Lee S., Kim S., Lee Y.-S., J. Controlled Release 2020, 322, 108–121; [DOI] [PubMed] [Google Scholar]
- 3b.Johnson L. G., Olsen J. C., Sarkadi B., Moore K. L., Swanstrom R., Boucher R. C., Nat. Genet. 1992, 2, 21–25; [DOI] [PubMed] [Google Scholar]
- 3c.Da Silva A. L., Martini S. V., Abreu S. C., Samary C. d. S., Diaz B. L., Fernezlian S., De Sá V. K., Capelozzi V. L., Boylan N. J., Goya R. G., J. Controlled Release 2014, 180, 125–133; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d.Mohamed A., Kunda N. K., Ross K., Hutcheon G. A., Saleem I. Y., Eur. J. Pharm. Biopharm. 2019, 136, 1–8. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a.Wan T., Ping Y., Adv. Drug Delivery Rev. 2021, 168, 196–216; [DOI] [PubMed] [Google Scholar]
- 4b.Chow M. T., Chang R. Y. K., Chan H.-K., Adv. Drug Delivery Rev. 2021, 168, 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a.Chen D., Liu J., Wu J., Suk J. S., Expert Opin. Drug Delivery 2020, 10.1080/17425247.2021.1854222; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b.Ferrari S., Geddes D. M., Alton E. W., Adv. Drug Delivery Rev. 2002, 54, 1373–1393; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c.Kim N., Duncan G. A., Hanes J., Suk J. S., J. Controlled Release 2016, 240, 465–488; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d.Duncan G. A., Jung J., Hanes J., Suk J. S., Mol. Ther. 2016, 24, 2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a.Button B., Cai L.-H., Ehre C., Kesimer M., Hill D. B., Sheehan J. K., Boucher R. C., Rubinstein M., Science 2012, 337, 937–941; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b.Cone R. A., Adv. Drug Delivery Rev. 2009, 61, 75–85. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a.Bowden D. H., Environ. Health Perspect. 1984, 55, 327–341; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b.Ochs M., Nyengaard J. R., Jung A., Knudsen L., Voigt M., Wahlers T., Richter J., Gundersen H. J. G., Am. J. Respir. Crit. Care Med. 2004, 169, 120–124. [DOI] [PubMed] [Google Scholar]
- 8.Gomes dos Reis L., Svolos M., Hartwig B., Windhab N., Young P. M., Traini D., Expert Opin. Drug Delivery 2017, 14, 319–330. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a.Nakamura E., Isobe H., Chem. Rec. 2010, 10, 260–270; [DOI] [PubMed] [Google Scholar]
- 9b.Isobe H., Nakanishi W., Tomita N., Jinno S., Okayama H., Nakamura E., Chem. Asian J. 2006, 1, 167–175; [DOI] [PubMed] [Google Scholar]
- 9c.Isobe H., Sugiyama S., Fukui K. i., Iwasawa Y., Nakamura E., Angew. Chem. Int. Ed. 2001, 40, 3364–3367; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 3468–3471. [Google Scholar]
- 10.
- 10a.Bolskar R. D., in Encyclopedia of Nanotechnology (Ed.: Bhushan B.), Springer Netherlands, Dordrecht, 2016, pp. 1267–1281; [Google Scholar]
- 10b.Minami K., Okamoto K., Kent D., Harano K., Noiri E., Nakamura E., Sci. Rep. 2014, 4, 4916–4922; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c.Peng Y., Yang D., Lu W., Hu X., Hong H., Cai T., Acta Biomater. 2017, 61, 193–203. [DOI] [PubMed] [Google Scholar]
- 11.Maeda-Mamiya R., Noiri E., Isobe H., Nakanishi W., Okamoto K., Doi K., Sugaya T., Izumi T., Homma T., Nakamura E., Proc. Natl. Acad. Sci. USA 2010, 107, 5339–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a.Suk J. S., Kim A. J., Trehan K., Schneider C. S., Cebotaru L., Woodward O. M., Boylan N. J., Boyle M. P., Lai S. K., Guggino W. B., J. Controlled Release 2014, 178, 8–17; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b.Mastorakos P., Da Silva A. L., Chisholm J., Song E., Choi W. K., Boyle M. P., Morales M. M., Hanes J., Suk J. S., Proc. Natl. Acad. Sci. USA 2015, 112, 8720–8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a.Tang B. C., Dawson M., Lai S. K., Wang Y.-Y., Suk J. S., Yang M., Zeitlin P., Boyle M. P., Fu J., Hanes J., Proc. Natl. Acad. Sci. USA 2009, 106, 19268–19273; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b.Schneider C. S., Xu Q., Boylan N. J., Chisholm J., Tang B. C., Schuster B. S., Henning A., Ensign L. M., Lee E., Adstamongkonkul P., Sci. Adv. 2017, 3, 1601556-1601566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a.Hatakeyama H., Akita H., Harashima H., Biol. Pharm. Bull. 2013, 36, 892–899; [DOI] [PubMed] [Google Scholar]
- 14b.Hatakeyama H., Akita H., Harashima H., Adv. Drug Delivery Rev. 2011, 63, 152–160. [DOI] [PubMed] [Google Scholar]
- 15.van der Wijk T., Tomassen S. F., Houtsmuller A. B., de Jonge H. R., Tilly B. C., J. Biol. Chem. 2003, 278, 40020–40025. [DOI] [PubMed] [Google Scholar]
- 16.Okada Y., Maeno E., Shimizu T., Dezaki K., Wang J., Morishima S., J. Physiol. 2001, 532, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemoine J., Farley R., Huang L., Gene Ther. 2005, 12, 1275–1282. [DOI] [PubMed] [Google Scholar]
- 18.Isobe H., Tomita N., Nakamura E., Org. Lett. 2000, 2, 3663–3665. [DOI] [PubMed] [Google Scholar]
- 19.Osman G., Rodriguez J., Chan S. Y., Chisholm J., Duncan G., Kim N., Tatler A. L., Shakesheff K. M., Hanes J., Suk J. S., J. Controlled Release 2018, 285, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mall M., Grubb B. R., Harkema J. R., O'Neal W. K., Boucher R. C., Nat. Med. 2004, 10, 487–493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary