

Abstract
The pathological aggregation of intrinsically disordered tau protein, driven by the interactions between microtubule-binding (MTB) domains, is associated with Alzheimer’s disease. The MTB domain contains either three or four repeats with sequence similarities. Compared to Amyloid β, many aspects of the misfolding and aggregation mechanisms of Tau are largely unknown. In this study, we systematically investigated the dynamics of monomer misfolding and dimerization of each MTB repeat using atomistic discrete molecular dynamics (DMD) simulations. Our results revealed all the four repeat monomers (R1–R4) were very dynamic, featuring frequent conformational conversion and lacking stable conformations. While R1, R2, and R4 monomers occasionally adopted partially helical conformations, R3 monomers frequently formed β-sheets. In dimerization simulations, R3 displayed the strongest aggregation propensity with high β-sheet contents, while R1 was the least aggregation-prone. The R2 and R4 dimers contained both helix and β-sheet structures. The β-sheets in R4 assemblies were dominant with β-hairpin conformation. In R2 and R3 dimers, inter-molecular β-sheets were mainly driven by residues around the paired helical filament (PHF) regions. Residues around the PHF6* in R2 and PHF6 in R3 had significantly higher inter-molecular contacts than other regions, suggesting that these residues play a key role in the amyloid aggregation of tau. Our results on the structural ensembles and early aggregation dynamics of each tau MTB repeat will help understand the nucleation and fibrillization of Tau.
Graphical Abstract
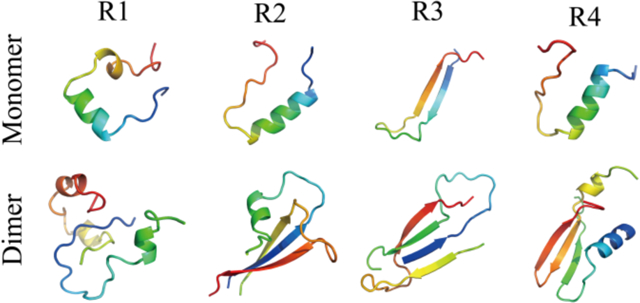
Introduction
The misfolding and pathological accumulation of intracellular tau protein and extracellular amyloid-β (Aβ) peptide are the two hallmarks of Alzheimer’s disease (AD)1–5. The microtubule-binding tau is an intrinsically disordered and highly soluble protein, which is widely abundant in the neurons of the human central nervous system2. There are six tau isoforms identified in adult human brains, consisting of a charged N-terminal region, a proline-rich region followed by either three or four imperfect microtubule-binding (i.e., MTB) repeats (i.e., 3R or 4R), and a C-terminal region6. The N-terminal and C-terminal of tau are significantly disordered7. The MTB repeat domains are more structured and play important roles in both the binding to the microtubules and the pathological aggregation into amyloid fibrils1, 8–10. Similar to other amyloid proteins11–13, patient-derived tau fibrils determined by the cryo-electron microscopy (cryo-EM) studies featured a common cross-β core structure with the β-strands aligned perpendicularly to the fibril axis. The β-sheet core regions were mainly located in the MTB repeats region14, 15. Since numerous studies suggest that small oligomer aggregates during early amyloid aggregation are more toxic mature fibrils, the characterization of monomer folding and oligomerization of the MTB repeats is important for understanding the mechanism of both functional binding to the microtubules and pathological amyloid aggregations.
Structural determination experiments, including CD, FTIR, X-ray, and NMR spectra, have demonstrated that all soluble tau isoforms can be described as random coil dominant with the lack of a stable folded structure16–20, despite some regions exhibiting a preference of helical or β-strand conformations21–23. Prior experimental reports revealed that the non-native helical intermediates in the transition from random coil were directly associated with the filament formation of tau 4R at the start of paired helical filament (PHF) formation24, 25. Each repeat has been found to contribute differently to the aggregation of tau 3R and 4R26–28. For example, the dynamic light scattering (DLS) experiment showed that R3 and R2&R3 repeats played a key important role in the aggregation patterns of 3R and 4R, respectively28, 29. The self-aggregation propensity of R1 and R4 single repeats were very weak, but necessary for amyloid aggregation of 4R26, 30. Many experiments, as well as computer simulations, found two nucleating six-residue segments, namely PHF6 (306VQIVYK311) and PHF6* (275VQIINK280), located at the beginning of the third and second repeats respectively play important roles in Tau aggregation31–34. The ion-mobility mass spectrometry experiment, together with computational simulation revealed that the fragment of residues 306–317 (i.e., Tau306–317, encompassing the PHF6 region) from R3 repeat had a much stronger aggregation propensity than residues 273–284 (Tau273–284, encompassing the PHF6* region) from R2 repeat, where the heterodimers of Tau273–284-Tau306–317 were less stable than Tau306–317 homodimers but much more stable than the homodimers of Tau273–28431. CD spectra, along with NMR experiment revealed that the monomer of R2 repeat featured helical conformation in aqueous solutions at physiological pH in the presence of copper ions27. The R3 peptide adopted a mixture conformation of random coil and β-sheet in aqueous solutions at physiological pH35. Complementary to the experimental studies, the conformation of 3R and 4R repeats and their interactions with Aβ fibrils were studied by molecular dynamics (MD) simulations36–41. All-atom replica-exchange molecular dynamics (REMD) simulations demonstrated both the 3R and 4R monomers could form dynamically helical and β-sheet conformations, where the R1 and R4 repeats displayed high helical propensity than R3 repeat36. The 3R monomer (R1–R3–R4) displayed higher β-sheet propensity than the 4R monomer due to the presence of R2 not favorable for the β-sheet formation between R1 and R339. With most prior MD studies focusing on either the aggregation of short fragments (e.g., PHF6), one of the repeats, or structures of R3 and R4 monomers, the structures and dynamics early Tau aggregation are still elusive.
Given the large system size of Tau, we propose to systemically study the monomer structures and the dimerization dynamics (i.e., the minimalist aggregates) of each of the four MTB repeats using all-atom discrete molecular dynamics (DMD) simulations42. DMD is an effective and predictive MD algorithm, which have been widely used to study amyloid aggregation of IDPs43–48. Our simulation results showed that monomers of all the four repeats were very dynamic without a stable conformation, featuring frequent conformational changes. The transient ordered structures of R1, R2, and R4 were populated with helical structures, but R3 was abundant in β-sheet conformation. In dimerization simulations, the propensities of R2 and R4 to form β-sheet aggregates were stronger than R1 but much weaker than R3. Since the stability of R1 dimer was too weak with frequent dissociation into monomers, the dimerization didn’t induce much conformational change. R2 and R4 dimers contained both helix and β-sheet structures with β-sheet as the dominant structures. The dimers were stabilized by both inter-molecular and intra-molecular β-sheets. R3 dimers were driven by inter-molecular interactions between residues around the PHF6 regions. The average number of inter-molecular contacts for the residues around PHF6* and PHF6 regions was much larger than the other regions, suggesting that these residues should play an important role in the early aggregation of tau. Overall, our simulation results uncovered the conformation and self-assembly dynamics of each tau MTB repeat, which will help better understand the aggregation of Tau.
Material and methods
Molecular systems used in simulations.
The repeat sequences used in our simulation were taken from the Uniport (P10636)49 and presented in Table 1. Both N- and C-termini were treated neutrally charged. To investigate the folding and self-assembly dynamics of each repeat, we systematically performed both one- and two-peptide simulation using all-atom DMD. For each molecular system, 40 independent trajectories were obtained starting with different coordinates and velocities. In dimerization simulations, the initial configurations with random inter-molecular distances and orientations had a minimum inter-molecular distance of at less 1.5 nm. The duration of each DMD trajectory in one- and two-peptide systems was 0.6 and 1.6 μs, respectively. The details of all the simulations were summarized in Table 2.
Table 1.
The amino acid sequences of each tau repeat used in our simulation.
| Sequence | |
|---|---|
| R1 | 244QTAPVPMPDLKNVKSKIGSTENLKHQPGGGK274 |
| R2 | 275VQIINKKLDLSNVQSKCGSKDNIKHVPGGGS305 |
| R3 | 306VQIVYKPVDLSKVTSKCGSLGNIHHKPGGGQ336 |
| R4 | 337VEVKSEKLDFKDRVQSKIGSLDNITHVPGGGN368 |
Table 2.
The details of molecule systems in our DMD simulations, including the number of simulated peptides (System), the type of repeat simulated (Peptide), the corresponding dimension of the cubic simulation box, the number of independent trajectories performed (DMD run), the length of each DMD simulations (Time), and the accumulative total simulation time (Total time).
| System | Peptide | Box (nm) | DMD run | Time (μs) | Total time (μs) |
|---|---|---|---|---|---|
| 1-peptide | R1 | 7 | 40 | 0.6 | 24 |
| R2 | 7 | 40 | 0.6 | 24 | |
| R3 | 7 | 40 | 0.6 | 24 | |
| R4 | 7 | 40 | 0.6 | 24 | |
| 2-peptide | R1 | 8.5 | 40 | 1.6 | 64 |
| R2 | 8.5 | 40 | 1.6 | 64 | |
| R3 | 8.5 | 40 | 1.6 | 64 | |
| R4 | 8.5 | 40 | 1.6 | 64 |
DMD Simulations.
All simulations were performed at 300 K using all-atom DMD algorithm with Medusa force field42. Similar to classical molecular dynamics (MD), both bonded interactions (i.e., covalent bonds, bond angles, and dihedrals) and non-bonded interactions (i.e., van der Waals, solvation, hydrogen bond, and electrostatic terms) are considered in DMD50, 51. The continuous potential functions in traditional MD are approximated by step functions in DMD42, 52. The covalent bonds and bond angles interactions (including bonds, bond angles, and dihedrals) are modeled as an infinite square well, dihedrals may feature multiple wells corresponding to the cis- or trans-conformations. The van der Waals parameters in non-bonded interactions are taken from the CHARMM force field53. Lazaridis-Karplus implicit solvation model is used for modeling the solvated energy with fully-solvated conformations as a reference state54. Hydrogen bond interactions are modeled by a reaction-like algorithms55. Screened charge-charge interactions are modeled, using Debye-Huckel approximation, by setting the Debye length approximately to 10 Å. The units of mass, time, length, and energy used in our all-atom DMD simulations with implicit water model are 1 Da, ~50 fs, 1 Å, and 1 kcal/mol, respectively. With significantly enhanced sampling efficiency, DMD is widely used to study protein folding/aggregation and protein-nanoparticle interactions both by our group and by others56–59.
Computational Analysis.
The secondary structure was calculated using the DSSP program60. The hydrogen bond was considered to be formed when the N⋯O distance was within 3.5 Å and the N–H⋯O angle was greater than 150°. A pairwise residue contact was defined when the distance between the heavy atoms of two non-sequential side-chain/main-chains was within 0.65 nm. Cluster analysis was performed using the Daura algorithm and a Ca root-mean-square deviation (RMSD) cutoff of 0.35 nm61. A two-dimensional (2D) free energy surface was constructed using −RTlnP(x, y), where P(x, y) is the probability of a conformation having a certain parameter value of x and y.
Results and discussions
Before analyzing the simulation result, the convergence of the single- and two-peptide DMD simulations for each repeat were examined by comparing the secondary structure propensity within two different time intervals (Figure S1&S2). The conformations generated within 300–450 and 450–600 ns from the single-repeat simulation and 800–1200 and 1200–1600 ns from the two-repeat simulation were used for the convergence assessments. The average secondary structure content of each repeat and the secondary structure propensity of each amino acid residue from every repeat were very similar within the two time intervals for both single- and two-peptide systems (Figure S1&S2), indicating all the molecular system simulations were reasonably converged.
Conformational ensembles of R1, R2 and R4 monomers were populated by transient, ordered structures with helical conformations, while R3 monomer was dominant with dynamically formed β-sheet.
To investigate the conformational dynamics of MTB repeats, forty independent DMD simulations were performed for the isolated monomer of each tau repeat starting from the fully extended conformation with randomized initial velocities. Each independent simulation lasted 0.6 μs. We first analyzed the conformational dynamics of each repeat by monitoring the secondary structure of each residue as a function of simulation time (e.g., representative trajectories in Fig. 1a). All the four repeats mainly adopted unstructured conformations (i.e., random coil and bend) with structured formations (e.g., helix, β-sheet, and turn) transiently observed. These ordered conformations were unstable, easily converted into the random coil and bent structures. The transient ordered structures of R1, R2, and R4 repeat were mostly helices, although less frequent β-sheets were also observed (Fig. 1a,b&d). In contrast, the dynamically-formed ordered structures of R3 repeat were predominantly β-sheets (Fig. 1c). By averaging secondary structure contents over all independent simulations (Table S1), all four isolated repeats indeed predominantly adopted unstructured conformations (>70%). The helical content for R1, R2, and R4 monomers (~9–13%) was much higher than that of R3 (<1%), while the β-sheet content of R3 (~18%) was much higher than the other repeats (~3–7%). The secondary structure propensities for each residue were also analyzed (Figure S3). There were two helices-populating regions in R1 located around residues 251–259 and 263–268 (Figure S3a). Residues 278–290 from R2 and 342–349 from R4 also displayed significant helical propensities (Figure. S3b&d). β-sheets in R3 were predominantly formed by residues 307–315 and 326–332 (Figure S3c).
Fig. 1. The conformational dynamics of Tau MTB repeat monomers.
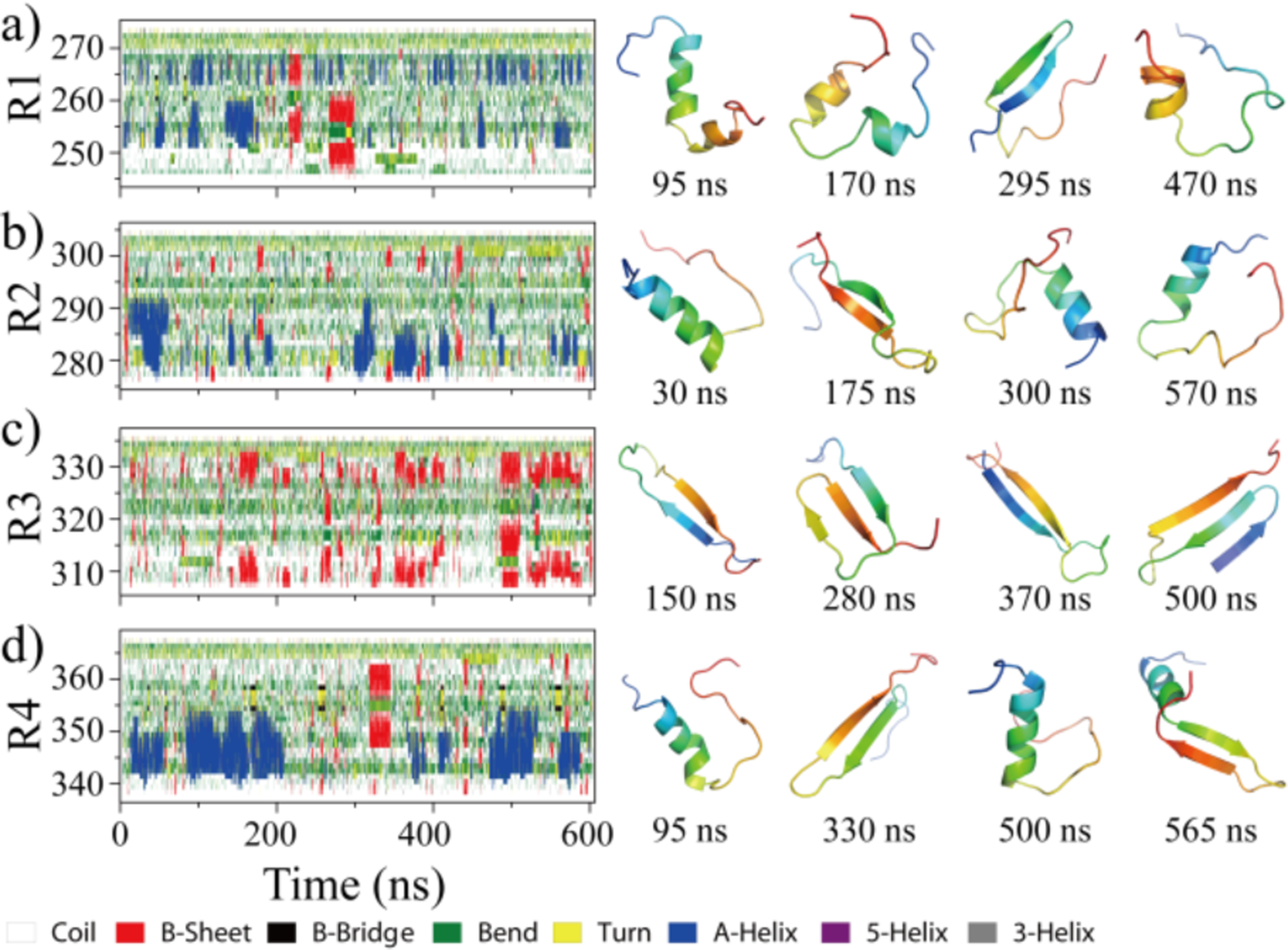
Time evolution of the secondary structure for each residue in monomer simulation is shown on the left panel from each of four tau repeats (a-d). Transient ordered conformations formed along the simulation trajectory (the time stamped blow) are presented on the right. For each tau repeat one 600ns DMD trajectory was randomly selected from 40 independent simulations.
To further characterize the structural ensemble of each repeat we calculated the potential of mean force (PMF, the effective conformational free energy landscape) as a function of the radius of gyration (Rg) and end-to-end distance (End2End) using the last 400 ns DMD trajectories from forty independent DMD simulations. As shown in Fig. 2a, the free energy basin of R1 was broad with Rg ~0.9–1.4 nm, and End2End ~1.0–4.0 nm, indicating the conformation of R1 monomer was very diverse. The most populated top 5 clusters of R1, scattered on the surface of the low free energy basin, featured helical formations. The patterns along the diagonal of the inter-residue contact frequency map (Figure S4a) further confirmed that, and the weak long-range interactions off-diagonal indicated that R1 did not form compact structures. The free energy basin of R2 (Rg ~0.9–1.2 nm and End2End ~1.0–2.5 nm) had a smaller “area” than R1 (Fig. 2b). The top 5 most populated conformations also adopted helical structures. Besides the diagonal helical contact pattern (residue 278–295), a weak β-sheet contact pattern was observed (Figure S4b), where residues 285–300 could form a β-hairpin structure with β-strand located residues 285–290 and 295–300. A similar helical region (i.e., 280–288) was also observed in another recent all-atom REMD simulation of Tau267–313 segment62. The PMF of R4 (Fig. 2d) was similar to that of R1 and R2 (Fig. 2b), with a broad free energy basin populated with various helical formations (snapshots 1, 2, 3 and 5 in Fig. 2d). The contact map of R4 had two structured contact patterns (Fig. S2c): one helical motif comprised by the N-terminal residues 342–350 (snapshots 1, 2, 3 and 5 in Fig. 2d), and one β-hairpin motif with β-strands located around residues 346–352 and 359–365 (snapshot 4 in Fig. 2d). In contrast, the PMF of R3 showed a small narrow basin with Rg ~1.1–1.3 nm and End2End ~0.7–1.5 nm (Fig. 2c). Conformational clustering analysis indicated that the top populated conformations of R3 were β-hairpins (snapshots 1–5 in Fig. 2c). The contact frequency map revealed that the β-sheets were formed by residues 307–320 and 325–333, stabilized hydrophobic and aromatic interactions (Figure S4c). For example, hydrophobic residues 308I, 309V, 313V, 315L displayed high tendencies to form contact with residue 328I. The aromatic residue 310Y also featured a strong propensity to interact with 330H. Prior all-atom full-length MTB repeat REMD simulation also demonstrated that both the 3R and 4R were predominantly adopted in random coil formation (>60%)36 and the β-sheet propensity of R3 was higher than other repeat63. The conformations of each repeat in the full-length MTB repeat (3R or 4R) might be different from pure isolated repeat due to the potential interactions between repeats may induce conformation changes. For example, prior REMD simulation study revel that the 3R monomer (R1–R3–R4) displayed higher β-sheet propensity than the 4R monomer due to the presence of R2 hindered the interactions between R1 and R336. Another recent carbon-detected NMR experiments also shown tau’s repeat region existed in extended conformation in the dispersed state and attained transient β-hairpin propensity upon liquid–liquid phase separation64. Overall, isolated tau MTB repeat monomers were intrinsically discorded lacking stable ordered conformations. R1, R2, and R4 monomers transiently formed helices, while R3 occasionally formed β-sheets.
Fig. 2. The conformational free energy landscape of Tau MTB repeat monomers.
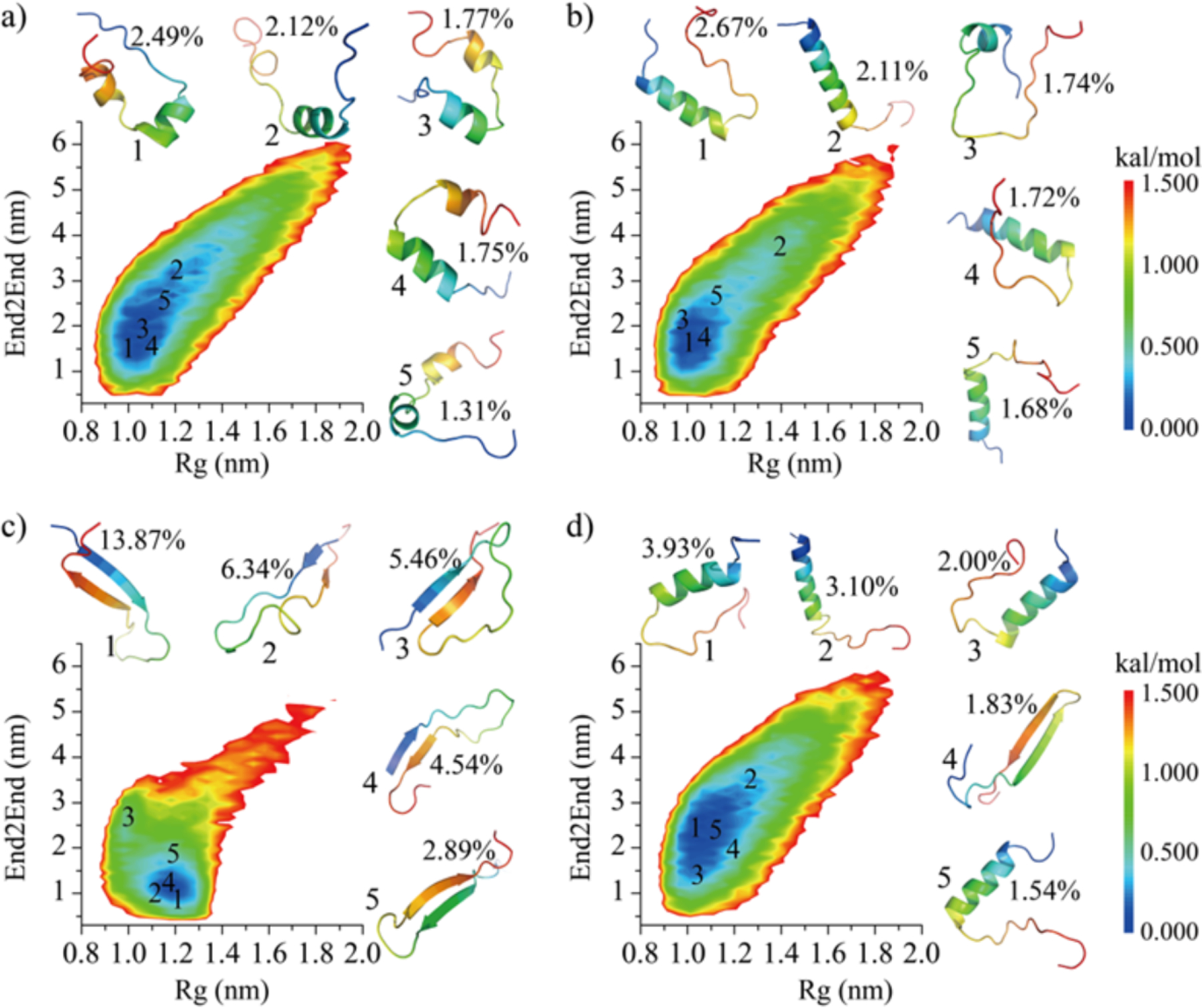
The 2D PMF as a function of radius of gyration (Rg) and end-to-end distance (End2End) for each of the four repeats (a-d). The centroid structure of the most populated top 5 clusters and their locations on the PMF are also presented as insets.
R3 displayed a significantly higher tendency to form β-sheet rich dimers than other repeats.
To investigate the contribution of each tau repeat to the full-length tau aggregation, the dimerization dynamics of each repeat was investigated. For each repeat, 40 independent simulations each lasting 1.6 μs were performed with randomized initial configurations. We first examined for each repeat the dimerization dynamics in terms of secondary structure changes, inter- and intra-chain interactions, and snapshot structures (Fig. 3). The R1 dimers were very dynamic and featured frequent conformational conversions. The probability distribution of intra-molecular hydrogen bonds and contacts for each R1 repeat observed in the two-peptide system was also similar to the one-peptide system (Figure S5a&e). The well-ordered dimer conformations with short β-sheets and helices were transient and easily converted into unstructured coils and bends (Fig. 3a). The time evolution of intra- and inter-molecular backbone hydrogen bonds, correlating to the number of structured residues, displayed constantly large fluctuation around 0–30. The averaged β-sheet contents of R2 and R4 (11%~12%, Table S1) were larger than R1(~5%), but much smaller than R3 (~35%). Compare to monomers, the intra-molecular hydrogen bonds and contacts per chain of R2 and R4 in two-peptide simulations displayed a small decrease (Figure S5) along with increased in β-sheet contents (Table S1), suggesting that the dimerization of R2 and R4 induced conformational conversion to β-sheets. Representative dimerization trajectories of R2 (Fig. 3b) and R4 (Fig. 3d) further confirmed that both R2 and R4 could assemble into metastable β-sheets rich dimer. Once the homo-dimers of R2 and R4 dissociated into monomers, random coils and bends became the dominant conformations. R3 underwent major changes in structure and dynamics upon dimerization. By forming extensive inter-molecular hydrogen bonds and contacts, both intra-chain hydrogen bonds and contacts were significantly decreased in dimers (Figure. S5c,g). The self-assembly dynamics showed that residues around PHF6 (306–311) featured the highest propensity to form inter-molecules β-sheets (Fig. 3c). The stability of β-sheet conformation in R3 dimers was significantly enhanced comparing to R3 monomers (Fig. 1c). The probability distribution of monomer and dimer in dimerization simulations (Table S2) showed that the aggregation propensity of R3 was much stronger than the other repeats. The probability distribution of the number of inter-molecular hydrogen bonds and contacts for each type of repeat in the dimeric state were further analyzed, where only these conformations two peptides were connected by at least one inter-molecular contact formed by heavy atoms were considered during the last 800 ns (Figure S6). The conformations of R3 dimer were predominant with inter-molecular contacts and hydrogen bonds, but the dimers formed by R1, R2, and R4 repeats were dominant with intra-molecular contacts and hydrogen bonds. Overall, our result was consistent with prior experimental studies showing that the self-aggregation and the quantities of aggregates at 1 h after starting the aggregation of single-repeat R3 peptide was much larger than R1, R2 and R429.
Fig. 3. The dimerization dynamics of Tau MTB repeats.
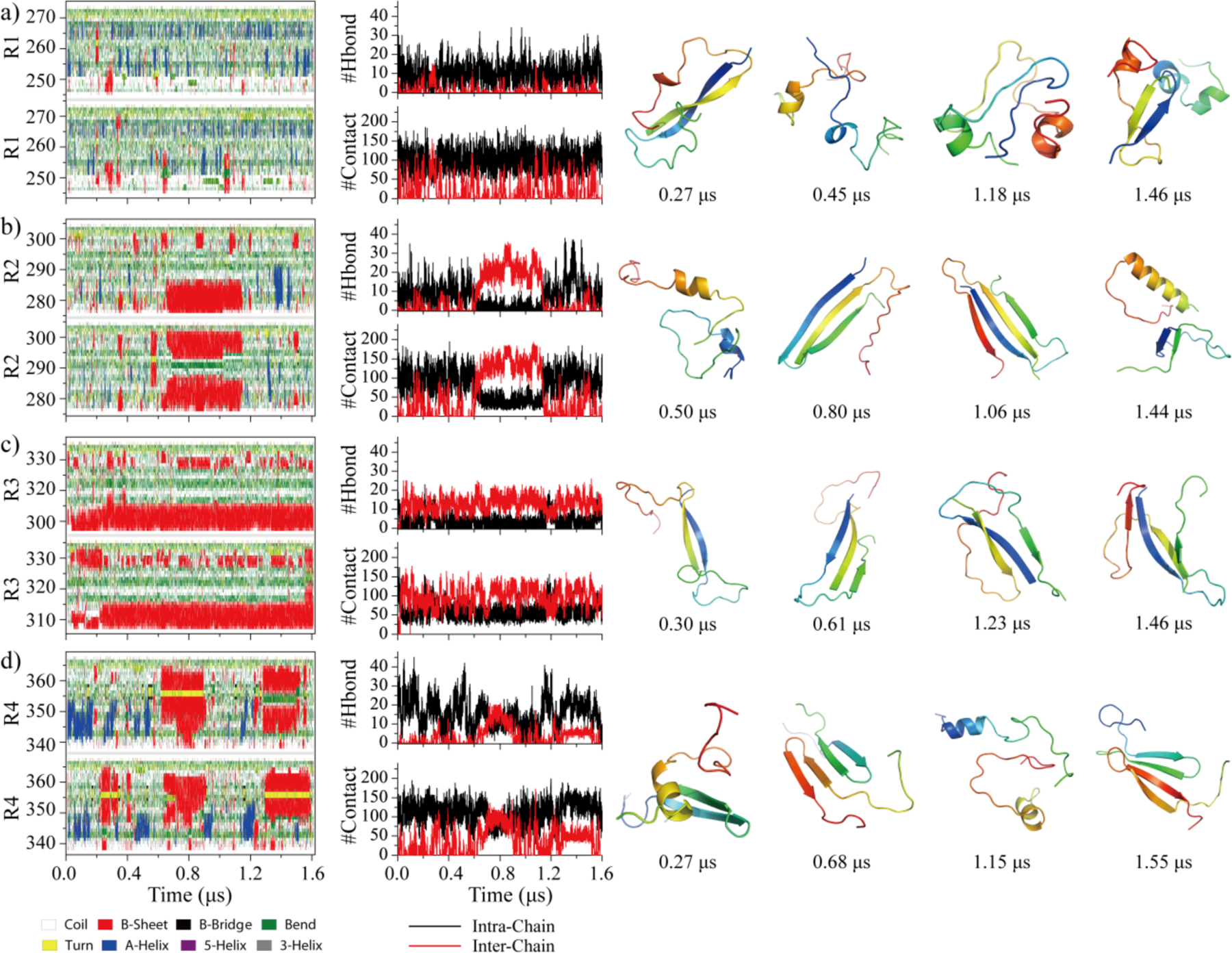
The secondary structure of each residue and the number of intra-chain and inter-chain backbone hydrogen bonds and contacts are presented as the function of simulation time. Four well-ordered dimer structures with corresponding the time stamps bellow are selected on the right.
Dimerization enhanced the β-sheet contents in R2, R3 and R4 instead of R1.
The probability of each residue adopting different secondary structures in dimerization simulations was compared with monomer simulations for each MTB repeat (Fig. 4 & Figure S7). With a weak dimerization propensity (Fig. 3), secondary structure propensity distribution of R1 in dimerization simulations was similar to that in monomer simulations (Fig. 4a & Figure. S7a). The dominant secondary structure was random coil, consistent with previous CD spectral experiments showing that the R1 mainly retained random structures in the early aggregation stage26. The partial helical structures of R1 were mainly formed by residues 251–259 and 263–268. Residues 278–290 in R2 repeat featured partial helix conformations with a probability of ~10–25% in both monomer and dimer simulations (Fig. 4b). Upon dimerization, the β-sheet propensities for residues 276–284 and 297–300 in R2 were enhanced ~10%, consistent with previous research demonstrated that residues 275–280 corresponding to the PHF6* region played a key important role in the self-assembly of tau33, 65. The mixed helices and β-sheets observed in the early R2 aggregates agreed with prior experimental CD spectral study26. Residues 307–315 and 326–332 in R3 showed strong propensities to form β-sheets in both monomer and dimer simulations (Fig. 4c). Especially in dimer simulations, the β-sheet propensities for residues around PHF6 region (residues 307–315) were increased to ~50%, suggesting an important role in the aggregation of Tau66, 67. For example, when the PHF6 fragment was deleted the amyloid aggregation of tau was significantly reduced31. Almost no helical structures were observed in R3, consistent with previous CD spectral experiments revealing that the R3 early aggregates were predominantly β-sheets26. Residues 342–349 in R4 preferred to form helixes in monomers and dimers (Fig. 4d). Upon dimerization, β-sheet propensities for residues 345–354 and 357–363 were enhanced. Prior NMR secondary chemical shifts of full-length MTB repeats, 3R and 4R, detected β-structure for 8–10 residues at the beginning of repeats R2–R4 indicating the R2, R3 and R4 had higher β-sheet structure propensity than R168. Hence, our simulation results suggested that dimerization increased the β-sheet contents in R2, R3 and R4, but not for R1.
Fig. 4. Secondary structure changes upon dimerization.
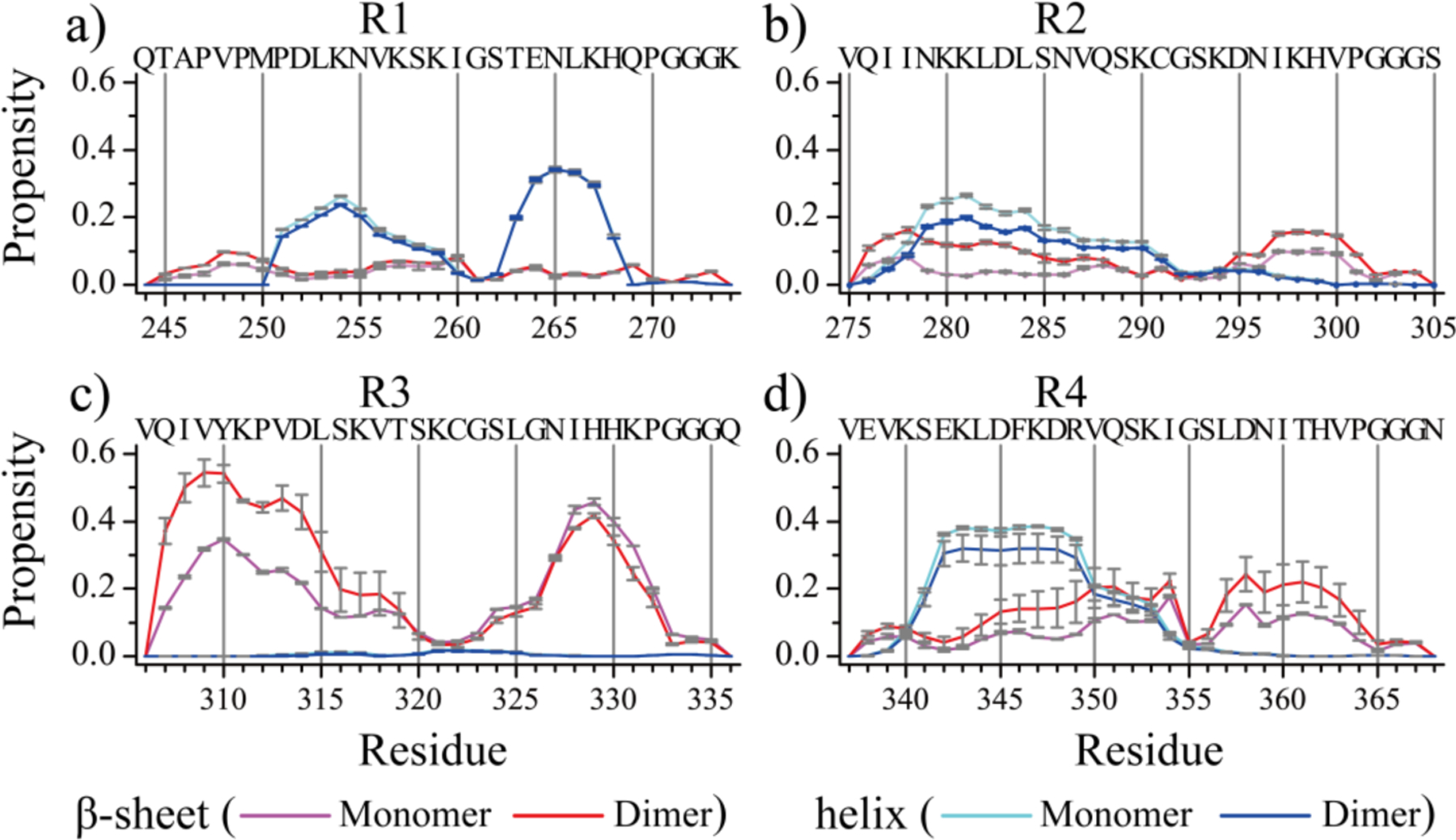
The averaged propensity of every residue adopting β-sheet and helix conformations in monomer and dimer simulations for each of the four Tau MTB repeats (a-d).
PHF6* in R2 and PHF6 in R3 played important roles for Tau MTB repeat aggregation.
To uncover the key interactions driving the aggregation of Tau MTB repeats, we computed the residue-wise contact frequency maps both inter and intra peptides in dimer simulations of each Tau repeat (Fig. 5). As expected, R1 had only weak inter-molecular contacts formed by N-terminal residues, corresponding to residues 246–260 assembled into transient parallel or anti-parallel β-sheets (e.g., snapshots Fig. 3a). The intra-chain contact frequency map was consistent with partial helical conformations (Fig. 5a). The inter-molecular contacts of R2 were mainly formed by residues 275–285 around the PHF6* region (Fig. 5b), by forming parallel or anti-parallel β-sheets with the same fragment in the other chain or interacting with residues 293–303 as parallel β-sheets (e.g., the self-assembly dynamics of R2 in Fig. 2b). R3 displayed a high aggregation propensity with most residues having larger number of inter-molecular contacts than intra-molecular contacts (Fig. 5c). The computed inter-chain contact frequency map suggested that residues around PHF6 (residues 306–315) could interact among themselves or with residues 325–334 by forming inter-molecular β-sheets. A weak intra-chain β-hairpin pattern with β-strands located around residues 310–315 and 325–330 were also observed. For instance, as shown in the dimerization trajectory in Fig. 3c, N-terminal residues around the PHF6 regions first assembled into inter-molecular β-sheet, then induced C-terminal residues converted into metastable β-sheets. In dimer simulations of R4, the inter-molecular interaction was weak, with only a faint β-sheet pattern formed by residues 343–354 observed in the contact frequency map (Fig. 5d). Therefore, the detailed analysis of inter-residue interactions revealed that PHF6* and PHF6 regions play important roles in the amyloid aggregation of Tau.
Fig. 5. The residue-wise interaction analyses.
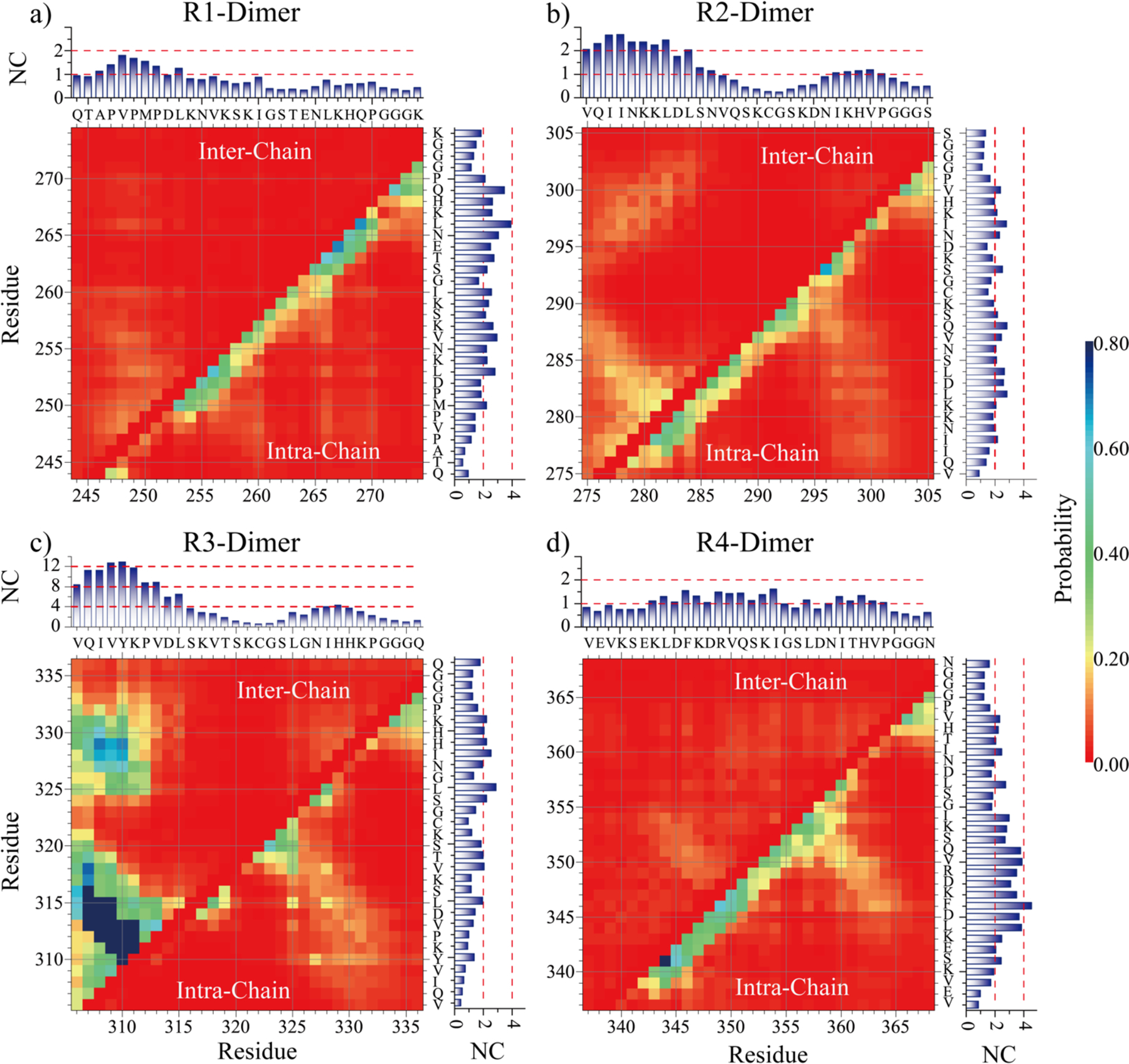
The intra-chain (lower diagonal) and inter-chain (upper diagonal) residue-wise contact frequencies for the dimer of each tau repeat. The cumulative inter- and intra-chain contact frequency of each residue with all the other residues are shown in the top panels and right panels, respectively.
Dimerization free energy landscapes of Tau MTB repeats.
To better understand the conformational dynamics of each repeat in dimerization simulations, we computed the PMF as a function of the total number of backbone hydrogen bonds (#Hbond) and the radius gyration of the assemblies (#Rg). To avoid potential biases from initial states, only the last 0.8 μs out of 1.6 μs trajectories in all forty independent simulations were used for the analysis for each repeat. The free energy basin of R1 dimerization was flat and broad with the number of backbone hydrogen bonds and Rg ranging 5–15 and 0.8–2.8 nm, respectively. Formation of inter-molecular hydrogen bonds was rare. Representative snapshots corresponding to the low energy regions labeled on the free energy surface (Fig. 6a) confirmed that the aggregation propensity of R1 was weak. Different from R1, the free energy basins of R2 dimerization featured an tail corresponding to β-sheet rich dimers with large number of hydrogen bonds and small Rg values in addition to helical monomers and dimers (Fig. 6b). The β-sheet rich dimer wasn’t stable and easily dissociated into helical monomer (Fig. 3b). Compared to R2, the β-sheet rich R4 dimers with large with large number of hydrogen bonds and small Rg values (~15–32, ~0.9–1.7 nm) became more favorable than the basin (~10–15, ~2.4–2.6 nm) corresponding isolated monomers and dimers with helixes and coils (Fig. 6d). The free energy landscape surface of R3 dimerization was completely different from R1, R2 and R4, with most of R3 aggregates featuring small Rg values (~1.2–1.5 nm) and abundant with a high number of backbone hydrogen bonds (10~15) (Fig. 6c). The averaged inter-chain hydrogen bonds were much greater than intra-chain hydrogen bonds, due to formations of stable inter-molecular β-sheets. Overall, our analysis suggested that the amyloid aggregation propensity of R2 and R4 were stronger than R1, but much weaker than R3.
Fig. 6. The dimerization free energy landscape of each tau repeat.
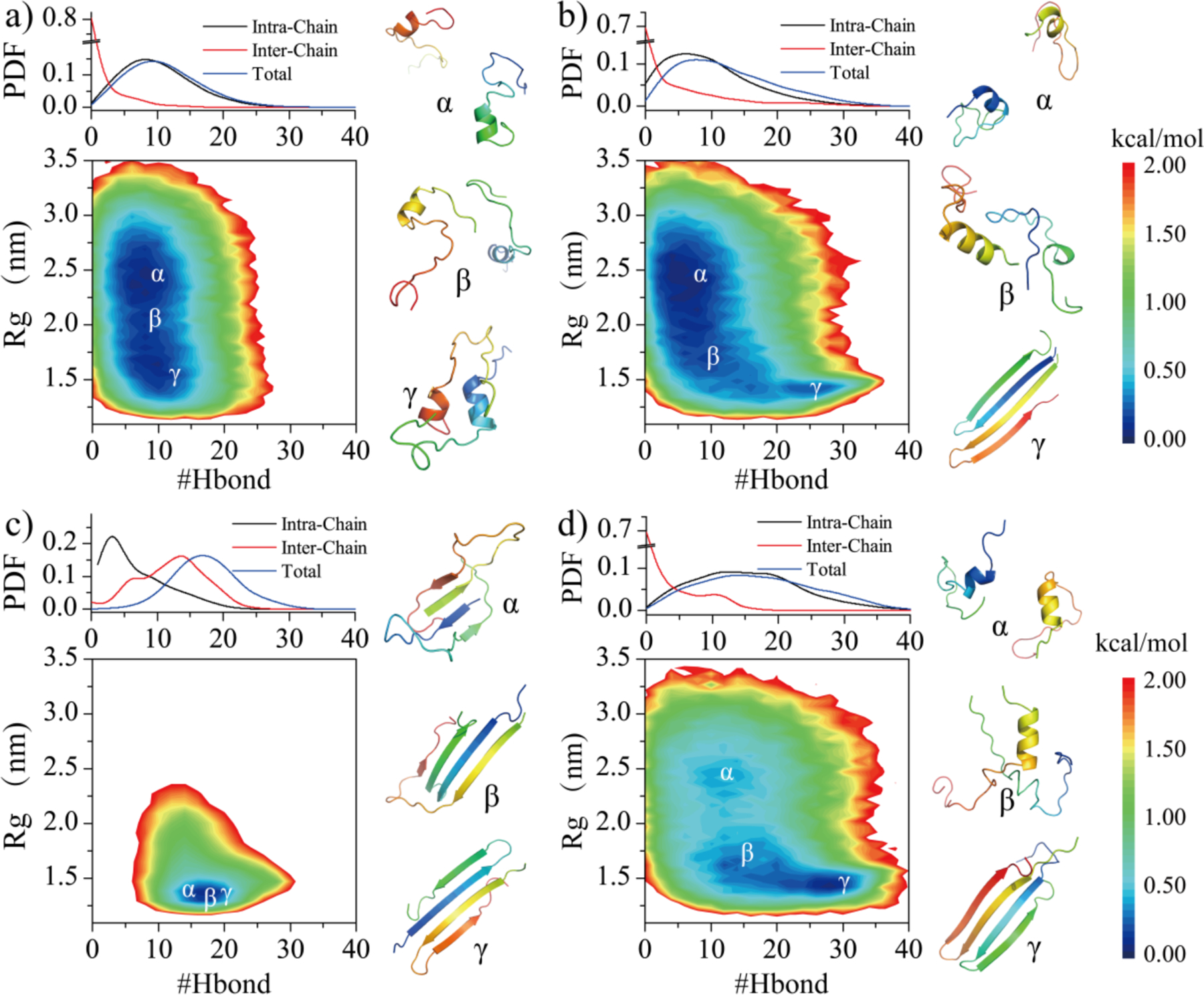
The PMF as a function of total number of backbone hydrogen bonds (#Hbond) and Rg in dimer simulations are shown for each repeat (a-d). The number of intra-chain, inter-chain, and total backbone hydrogen bonds probability distribution are shown on the top panel. Representative structures corresponding to local minima labeled in the PMFs (α, β, γ) are shown on the right.
Conclusion
We systematically investigated the conformational dynamics of monomer folding and dimerization for each of four tau MTB repeats with long timescale all-atom DMD simulations, accumulatively 24 μs for monomers and 64 μs for dimers. Our results demonstrated that all isolated single-repeat monomers are intrinsically disordered with predominantly unstructured conformations, but also displayed different secondary structure propensities in forming dynamically ordered secondary structures. The transient ordered structures of R1, R2 and R4 contained both helixes and β-sheets, but with the helical structures as the major species. The ordered structures of R3 monomers were predominantly β-sheets. The analysis of dimerization dynamics and conformational ensembles revealed that R2 and R4 had stronger aggregation propensity than R1, but much weaker than R3. The R1 dimers were unstable, lacking β-sheets, and easily dissociated into helical or unstructured monomers. The conformational ensembles and dimerization dynamics of R2 and R4 were very similar to each other, where both repeats could aggregate into metastable β-sheet rich dimers. The aggregates of R2 were abundant with inter-chain β-sheets driven by residues around the PHF6* regions, but R4 dimers were predominantly formed by β-hairpins. The R3 repeat displayed the highest amyloid aggregation propensities. Residues around PHF6 regions in R3 readily self-assembled into inter-molecular β-sheets and then induced other residues to form larger β-sheets. Both PHF6 in R3 and PHF6* in R2 formed large numbers of inter-molecular contact, and played important role in the early aggregation of tau. Our study provides a full picture of the monomer conformations and dimerization dynamics for all four MTB repeats, which will help better understand the pathology of Tau aggregation.
Supplementary Material
Acknowledgement
This work was supported in part by the National Natural Science Foundation of China under the Grant No. 11904189 (Sun), K.C.Wong Magna Fund in Ningbo University, China (Sun), NSF CBET-1553945 (Ding), and NIH R35GM119691 (Ding). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSFC, NIH and NSF.
Footnotes
Data and Software Availability.
DMD software is freely available to academic researchers at Molecules In Action (www.moleculesinaction.com). Two representative DMD trajectories for each system are available (http://dlab.clemson.edu/research/Tau-Folding/).
Conflicts of interest
There are no conflicts to declare.
Supporting Information
Supplementary Figures S1–4 and Table S1–2 (PDF).
References
- 1.Dregni AJ; Duan P; Hong M, Hydration and Dynamics of Full-Length Tau Amyloid Fibrils Investigated by Solid-State Nuclear Magnetic Resonance. Biochemistry-Us 2020, 59, 2237–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seidler PM; Boyer DR; Rodriguez JA; Sawaya MR; Cascio D; Murray K; Gonen T; Eisenberg DS, Structure-based inhibitors of tau aggregation. Nat Chem 2018, 10, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballatore C; Lee VMY; Trojanowski JQ, Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci 2007, 8, 663–672. [DOI] [PubMed] [Google Scholar]
- 4.Selkoe DJ; Hardy J, The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO molecular medicine 2016, 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nasica-Labouze J; Nguyen PH; Sterpone F; Berthoumieu O; Buchete NV; Cote S; De Simone A; Doig AJ; Faller P; Garcia A; Laio A; Li MS; Melchionna S; Mousseau N; Mu Y; Paravastu A; Pasquali S; Rosenman DJ; Strodel B; Tarus B; Viles JH; Zhang T; Wang C; Derreumaux P, Amyloid beta Protein and Alzheimer’s Disease: When Computer Simulations Complement Experimental Studies. Chemical reviews 2015, 115, 3518–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambadipudi S; Biernat J; Riedel D; Mandelkow E; Zweckstetter M, Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nature communications 2017, 8, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen D; Drombosky KW; Hou Z; Sari L; Kashmer OM; Ryder BD; Perez VA; Woodard DR; Lin MM; Diamond MI; Joachimiak LA, Tau local structure shields an amyloid-forming motif and controls aggregation propensity. Nature communications 2019, 10, 2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goode BL; Chau M; Denis PE; Feinstein SC, Structural and functional differences between 3-repeat and 4-repeat tau isoforms. Implications for normal tau function and the onset of neurodegenetative disease. The Journal of biological chemistry 2000, 275, 38182–9. [DOI] [PubMed] [Google Scholar]
- 9.Levy SF; Leboeuf AC; Massie MR; Jordan MA; Wilson L; Feinstein SC, Three- and four-repeat tau regulate the dynamic instability of two distinct microtubule subpopulations in qualitatively different manners. Implications for neurodegeneration. The Journal of biological chemistry 2005, 280, 13520–8. [DOI] [PubMed] [Google Scholar]
- 10.Xu L; Zheng J; Margittai M; Nussinov R; Ma B, How Does Hyperphopsphorylation Promote Tau Aggregation and Modulate Filament Structure and Stability? ACS chemical neuroscience 2016, 7, 565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liberta F; Loerch S; Rennegarbe M; Schierhorn A; Westermark P; Westermark GT; Hazenberg BPC; Grigorieff N; Fandrich M; Schmidt M, Cryo-EM fibril structures from systemic AA amyloidosis reveal the species complementarity of pathological amyloids. Nature communications 2019, 10, 1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao Q; Boyer DR; Sawaya MR; Ge P; Eisenberg DS, Cryo-EM structure and inhibitor design of human IAPP (amylin) fibrils. Nature structural & molecular biology 2020, 27, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kollmer M; Close W; Funk L; Rasmussen J; Bsoul A; Schierhorn A; Schmidt M; Sigurdson CJ; Jucker M; Fandrich M, Cryo-EM structure and polymorphism of Abeta amyloid fibrils purified from Alzheimer’s brain tissue. Nature communications 2019, 10, 4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fitzpatrick AWP; Falcon B; He S; Murzin AG; Murshudov G; Garringer HJ; Crowther RA; Ghetti B; Goedert M; Scheres SHW, Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falcon B; Zhang W; Murzin AG; Murshudov G; Garringer HJ; Vidal R; Crowther RA; Ghetti B; Scheres SHW; Goedert M, Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cleveland DW; Hwo SY; Kirschner MW, Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. Journal of molecular biology 1977, 116, 227–47. [DOI] [PubMed] [Google Scholar]
- 17.Schweers O; Schonbrunn-Hanebeck E; Marx A; Mandelkow E, Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. The Journal of biological chemistry 1994, 269, 24290–7. [PubMed] [Google Scholar]
- 18.Perez M; Valpuesta JM; Medina M; Montejo de Garcini E; Avila J, Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction. Journal of neurochemistry 1996, 67, 1183–90. [DOI] [PubMed] [Google Scholar]
- 19.Barghorn S; Davies P; Mandelkow E, Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry-Us 2004, 43, 1694–703. [DOI] [PubMed] [Google Scholar]
- 20.Mylonas E; Hascher A; Bernado P; Blackledge M; Mandelkow E; Svergun DI, Domain conformation of tau protein studied by solution small-angle X-ray scattering. Biochemistry-Us 2008, 47, 10345–53. [DOI] [PubMed] [Google Scholar]
- 21.Eliezer D; Barre P; Kobaslija M; Chan D; Li X; Heend L, Residual structure in the repeat domain of tau: echoes of microtubule binding and paired helical filament formation. Biochemistry-Us 2005, 44, 1026–36. [DOI] [PubMed] [Google Scholar]
- 22.Gamblin TC, Potential structure/function relationships of predicted secondary structural elements of tau. Biochimica et biophysica acta 2005, 1739, 140–9. [DOI] [PubMed] [Google Scholar]
- 23.Sadqi M; Hernandez F; Pan U; Perez M; Schaeberle MD; Avila J; Munoz V, Alpha-helix structure in Alzheimer’s disease aggregates of tau-protein. Biochemistry-Us 2002, 41, 7150–5. [DOI] [PubMed] [Google Scholar]
- 24.Hiraoka S; Yao TM; Minoura K; Tomoo K; Sumida M; Taniguchi T; Ishida T, Conformational transition state is responsible for assembly of microtubule-binding domain of tau protein. Biochemical and biophysical research communications 2004, 315, 659–63. [DOI] [PubMed] [Google Scholar]
- 25.Ozenne V; Schneider R; Yao M; Huang JR; Salmon L; Zweckstetter M; Jensen MR; Blackledge M, Mapping the potential energy landscape of intrinsically disordered proteins at amino acid resolution. Journal of the American Chemical Society 2012, 134, 15138–48. [DOI] [PubMed] [Google Scholar]
- 26.Mizushima F; Minoura K; Tomoo K; Sumida M; Taniguchi T; Ishida T, Fluorescence-coupled CD conformational monitoring of filament formation of tau microtubule-binding repeat domain. Biochemical and biophysical research communications 2006, 343, 712–8. [DOI] [PubMed] [Google Scholar]
- 27.Ma Q; Li Y; Du J; Liu H; Kanazawa K; Nemoto T; Nakanishi H; Zhao Y, Copper binding properties of a tau peptide associated with Alzheimer’s disease studied by CD, NMR, and MALDI-TOF MS. Peptides 2006, 27, 841–9. [DOI] [PubMed] [Google Scholar]
- 28.Sugino E; Nishiura C; Minoura K; In Y; Sumida M; Taniguchi T; Tomoo K; Ishida T, Three-/four-repeat-dependent aggregation profile of tau microtubule-binding domain clarified by dynamic light scattering analysis. Biochemical and biophysical research communications 2009, 385, 236–40. [DOI] [PubMed] [Google Scholar]
- 29.Okuyama K; Nishiura C; Mizushima F; Minoura K; Sumida M; Taniguchi T; Tomoo K; Ishida T, Linkage-dependent contribution of repeat peptides to self-aggregation of three- or four-repeat microtubule-binding domains in tau protein. The FEBS journal 2008, 275, 1529–39. [DOI] [PubMed] [Google Scholar]
- 30.Tomoo K; Yao TM; Minoura K; Hiraoka S; Sumida M; Taniguchi T; Ishida T, Possible role of each repeat structure of the microtubule-binding domain of the tau protein in in vitro aggregation. Journal of biochemistry 2005, 138, 413–23. [DOI] [PubMed] [Google Scholar]
- 31.Ganguly P; Do TD; Larini L; LaPointe NE; Sercel AJ; Shade MF; Feinstein SC; Bowers MT; Shea JE, Tau assembly: the dominant role of PHF6 (VQIVYK) in microtubule binding region repeat R3. The journal of physical chemistry. B 2015, 119, 4582–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu H; Zhong H; Liu X; Zhou S; Tan S; Liu H; Yao X, Disclosing the Mechanism of Spontaneous Aggregation and Template-Induced Misfolding of the Key Hexapeptide (PHF6) of Tau Protein Based on Molecular Dynamics Simulation. ACS chemical neuroscience 2019, 10, 4810–4823. [DOI] [PubMed] [Google Scholar]
- 33.Smit FX; Luiken JA; Bolhuis PG, Primary Fibril Nucleation of Aggregation Prone Tau Fragments PHF6 and PHF6. The journal of physical chemistry. B 2017, 121, 3250–3261. [DOI] [PubMed] [Google Scholar]
- 34.Arya S; Ganguly P; Arsiccio A; Claud SL; Trapp B; Schonfeld GE; Liu X; Lazar Cantrell K; Shea JE; Bowers MT, Terminal Capping of an Amyloidogenic Tau Fragment Modulates Its Fibrillation Propensity. The journal of physical chemistry. B 2020, 124, 8772–8783. [DOI] [PubMed] [Google Scholar]
- 35.Ma QF; Li YM; Du JT; Kanazawa K; Nemoto T; Nakanishi H; Zhao YF, Binding of copper (II) ion to an Alzheimer’s tau peptide as revealed by MALDI-TOF MS, CD, and NMR. Biopolymers 2005, 79, 74–85. [DOI] [PubMed] [Google Scholar]
- 36.Luo Y; Ma B; Nussinov R; Wei G, Structural Insight into Tau Protein’s Paradox of Intrinsically Disordered Behavior, Self-Acetylation Activity, and Aggregation. The journal of physical chemistry letters 2014, 5, 3026–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X; Dong X; Wei G; Margittai M; Nussinov R; Ma B, The distinct structural preferences of tau protein repeat domains. Chemical communications 2018, 54, 5700–5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weismiller HA; Murphy R; Wei G; Ma B; Nussinov R; Margittai M, Structural disorder in four-repeat Tau fibrils reveals a new mechanism for barriers to cross-seeding of Tau isoforms. The Journal of biological chemistry 2018, 293, 17336–17348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo Y; Dinkel P; Yu X; Margittai M; Zheng J; Nussinov R; Wei G; Ma B, Molecular insights into the reversible formation of tau protein fibrils. Chemical communications 2013, 49, 3582–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller Y; Ma B; Nussinov R, Synergistic interactions between repeats in tau protein and Abeta amyloids may be responsible for accelerated aggregation via polymorphic states. Biochemistry-Us 2011, 50, 5172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Derreumaux P; Man VH; Wang J; Nguyen PH, Tau R3–R4 Domain Dimer of the Wild Type and Phosphorylated Ser356 Sequences. I. In Solution by Atomistic Simulations. The journal of physical chemistry. B 2020, 124, 2975–2983. [DOI] [PubMed] [Google Scholar]
- 42.Ding F; Tsao D; Nie H; Dokholyan NV, Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure 2008, 16, 1010–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu C; Beck MV; Griffith JD; Deshmukh M; Dokholyan NV, Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America 2018, 115, 4661–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun Y; Huang J; Duan X; Ding F, Direct Observation of beta-Barrel Intermediates in the Self-Assembly of Toxic SOD128–38 and Absence in Nontoxic Glycine Mutants. Journal of chemical information and modeling 2021, 61, 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y; Gao Y; Hill SE; Huard DJE; Tomlin MO; Lieberman RL; Paravastu AK; Hall CK, Simulations and Experiments Delineate Amyloid Fibrilization by Peptides Derived from Glaucoma-Associated Myocilin. The journal of physical chemistry. B 2018, 122, 5845–5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y; Bunce SJ; Radford SE; Wilson AJ; Auer S; Hall CK, Thermodynamic phase diagram of amyloid-beta (16–22) peptide. Proceedings of the National Academy of Sciences of the United States of America 2019, 116, 2091–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Voelker MJ; Barz B; Urbanc B, Fully Atomistic Abeta40 and Abeta42 Oligomers in Water: Observation of Porelike Conformations. Journal of chemical theory and computation 2017, 13, 4567–4583. [DOI] [PubMed] [Google Scholar]
- 48.Sun Y; Ding F, Thermo- and pH-responsive fibrillization of squid suckerin A1H1 peptide. Nanoscale 2020, 12, 6307–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshida H; Goedert M, Phosphorylation of microtubule-associated protein tau by AMPK-related kinases. Journal of neurochemistry 2012, 120, 165–76. [DOI] [PubMed] [Google Scholar]
- 50.Proctor EA; Dokholyan NV, Applications of Discrete Molecular Dynamics in biology and medicine. Current opinion in structural biology 2016, 37, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shirvanyants D; Ding F; Tsao D; Ramachandran S; Dokholyan NV, Discrete molecular dynamics: an efficient and versatile simulation method for fine protein characterization. The journal of physical chemistry. B 2012, 116, 8375–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun Y; Ding F, alphaB-Crystallin Chaperone Inhibits Abeta Aggregation by Capping the beta-Sheet-Rich Oligomers and Fibrils. The journal of physical chemistry. B 2020, 124, 10138–10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M, Charmm - a Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comput Chem 1983, 4, 187–217. [Google Scholar]
- 54.Lazaridis T; Karplus M, Effective energy functions for protein structure prediction. Current opinion in structural biology 2000, 10, 139–145. [DOI] [PubMed] [Google Scholar]
- 55.Ding F; Borreguero JM; Buldyrey SV; Stanley HE; Dokholyan NV, Mechanism for the alpha-helix to beta-hairpin transition. Proteins 2003, 53, 220–8. [DOI] [PubMed] [Google Scholar]
- 56.Faridi A; Sun YX; Mortimer M; Aranha RR; Nandakumar A; Li YH; Javed I; Kakinen A; Fan QQ; Purcell AW; Davis TP; Ding F; Faridi P; Ke PC, Graphene quantum dots rescue protein dysregulation of pancreatic beta-cells exposed to human islet amyloid polypeptide. Nano Res 2019, 12, 2827–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Srinivasan E; Rajasekaran R, Molecular binding response of naringin and naringenin to H46R mutant SOD1 protein in combating protein aggregation using density functional theory and discrete molecular dynamics. Progress in biophysics and molecular biology 2019, 145, 40–51. [DOI] [PubMed] [Google Scholar]
- 58.Brodie NI; Popov KI; Petrotchenko EV; Dokholyan NV; Borchers CH, Solving protein structures using short-distance cross-linking constraints as a guide for discrete molecular dynamics simulations. Science advances 2017, 3, e1700479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun YX; Ding F, Thermo- and pH-responsive fibrillization of squid suckerin A1H1 peptide. Nanoscale 2020, 12, 6307–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kabsch W; Sander C, Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–637. [DOI] [PubMed] [Google Scholar]
- 61.Daura X; Gademann K; Jaun B; Seebach D; van Gunsteren WF; Mark AE, Peptide folding: When simulation meets experiment. Angew Chem Int Edit 1999, 38, 236–240. [Google Scholar]
- 62.Chatterjee S; Salimi A; Lee JY, Intrinsic Origin of Tau Protein Aggregation: Effects of Histidine Tautomerism on Tau267–312 Monomer. ACS chemical neuroscience 2020, 11, 3814–3822. [DOI] [PubMed] [Google Scholar]
- 63.Dong X; Bera S; Qiao Q; Tang Y; Lao Z; Luo Y; Gazit E; Wei G, Liquid-Liquid Phase Separation of Tau Protein Is Encoded at the Monomeric Level. The journal of physical chemistry letters 2021, 12, 2576–2586. [DOI] [PubMed] [Google Scholar]
- 64.Ambadipudi S; Reddy JG; Biernat J; Mandelkow E; Zweckstetter M, Residue-specific identification of phase separation hot spots of Alzheimer’s-related protein tau. Chemical science 2019, 10, 6503–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sahara N; Maeda S; Murayama M; Suzuki T; Dohmae N; Yen SH; Takashima A, Assembly of two distinct dimers and higher-order oligomers from full-length tau. The European journal of neuroscience 2007, 25, 3020–9. [DOI] [PubMed] [Google Scholar]
- 66.von Bergen M; Friedhoff P; Biernat J; Heberle J; Mandelkow EM; Mandelkow E, Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proceedings of the National Academy of Sciences of the United States of America 2000, 97, 5129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goux WJ; Kopplin L; Nguyen AD; Leak K; Rutkofsky M; Shanmuganandam VD; Sharma D; Inouye H; Kirschner DA, The formation of straight and twisted filaments from short tau peptides. The Journal of biological chemistry 2004, 279, 26868–75. [DOI] [PubMed] [Google Scholar]
- 68.Mukrasch MD; Biernat J; von Bergen M; Griesinger C; Mandelkow E; Zweckstetter M, Sites of tau important for aggregation populate {beta}-structure and bind to microtubules and polyanions. The Journal of biological chemistry 2005, 280, 24978–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.