

Summary
The inflammatory response to SARS/CoV‐2 (COVID‐19) infection may contribute to the risk of thromboembolic complications. α‐Defensins, antimicrobial peptides released from activated neutrophils, are anti‐fibrinolytic and prothrombotic in vitro and in mouse models. In this prospective study of 176 patients with COVID‐19 infection, we found that plasma levels of α‐defensins were elevated, tracked with disease progression/mortality or resolution and with plasma levels of interleukin‐6 (IL‐6) and D‐dimers. Immunohistochemistry revealed intense deposition of α‐defensins in lung vasculature and thrombi. IL‐6 stimulated the release of α‐defensins from neutrophils, thereby accelerating coagulation and inhibiting fibrinolysis in human blood, imitating the coagulation pattern in COVID‐19 patients. The procoagulant effect of IL‐6 was inhibited by colchicine, which blocks neutrophil degranulation. These studies describe a link between inflammation and the risk of thromboembolism, and they identify a potential new approach to mitigate this risk in patients with COVID‐19 and potentially in other inflammatory prothrombotic conditions.
Keywords: COVID‐19 infection, inflammation, interleukin‐6, thrombosis, α‐defensins
Introduction
The clinical spectrum of COVID‐19 infection ranges from asymptomatic to fatal, in part related to inflammation and vascular complications. Severely affected individuals may develop a delayed onset ‘cytokine storm’, 1 which exacerbates endothelial damage predominantly in the lung, 2 activates coagulation 3 and leads to formation of disseminated microvascular thrombi, 4 , 5 , 6 , 7 , 8 followed by pulmonary decompensation due to impaired vascular perfusion. 7 , 9 Plasma levels of D‐dimers correlate with the rate of ICU admission, the requirement for mechanical ventilation and mortality. 10 Overt thromboembolic complications (TEC) may exceed 10% of patients requiring ICU‐level care, 11 , 12 , 13 , 14 most commonly deep venous thromboses and pulmonary emboli, but in situ thrombosis within the pulmonary vasculature, 15 , 16 , 17 occlusion of central venous and dialysis catheters and extra‐corporeal membrane oxygenation (ECMO) circuits, and arterial thrombosis leading to myocardial infarction and stroke have been reported as well. 18 , 19 , 20 , 21 , 22 TEC develop in patients treated with prophylactic 14 and in some cases higher doses of heparin. 23 However, the incidence of bleeding 23 places a ceiling on the intensity of anticoagulation that is tolerated in these severely ill patients, indicating a need for alternative non‐anticoagulant forms of intervention based on a better understanding of the pathophysiology of inflammatory thrombosis. 7
One clue to this understanding comes from analysis of lungs from patients with COVID‐19, which show infiltration of neutrophils into pulmonary capillaries, generation of neutrophil extra‐cellular traps (NETs) and extravasation into alveolar spaces and inflamed parenchyma. 24 Activation of the intrinsic coagulation pathway initiated by inflammation and cell death 23 stimulates neutrophils to release antimicrobial peptides, including α‐defensins. 25 α‐Defensins accelerate fibrin polymerization, increase fiber density and incorporate within nascent fibrin clots, which impedes fibrinolysis. 25 , 26 Transgenic mice expressing human α‐defensin‐1 develop occlusive neutrophil‐rich clots resistant to heparin. 25 When α‐Def‐1 mice were given colchicine to inhibit neutrophil degranulation, plasma levels of α‐defensin‐1 fell, phenotypic reversion was seen, and heparin responsiveness was restored. 25 Based on these findings, we examined the relationship between plasma α‐defensins, activation of coagulation and clinical course in patients with COVID‐19.
Methods
Clinical criteria
All patients had a positive polymerase chain reaction (PCR) test for COVID‐19. Patients were categorized as having ‘mild’ disease when they presented with flu‐like symptoms, most often involving the respiratory tract, and fever ≤37·80C. Patients were considered to have ‘moderate’ disease when the presenting symptoms included shortness of breath, chest pain, fever >37·80C, stable respiratory and systemic symptoms, respiratory rate <25/min and oxygen saturation (pulse oximetry) >94%. ‘Severe’ disease was defined as having the presentation of moderate disease accompanied by a respiratory rate ≥25/min or oxygen saturation <94% on room air and pulmonary infiltrates on X‐ray.
Clinical trial protocol
Consecutive consenting newly diagnosed patients with ‘mild’, ‘moderate’ and ‘severe’ symptoms ≥18 years of age admitted to Hadassah Medical Center (Jerusalem, Israel) with a positive PCR test for COVID‐19 between 22 March and 20 August 2020 were enrolled. The study was approved by the Helsinki Research Ethics Commissions of Hadassah Hospital (#0204‐20). In all, 127 patients participated. Patients were classified according to disease severity as described in the preceding paragraph, including 73 patients with mild disease, 36 with moderate disease and 18 with severe disease (Fig 1). Epidemiological, demographic, clinical, treatment and outcome data were extracted from electronic medical records.
Fig 1.
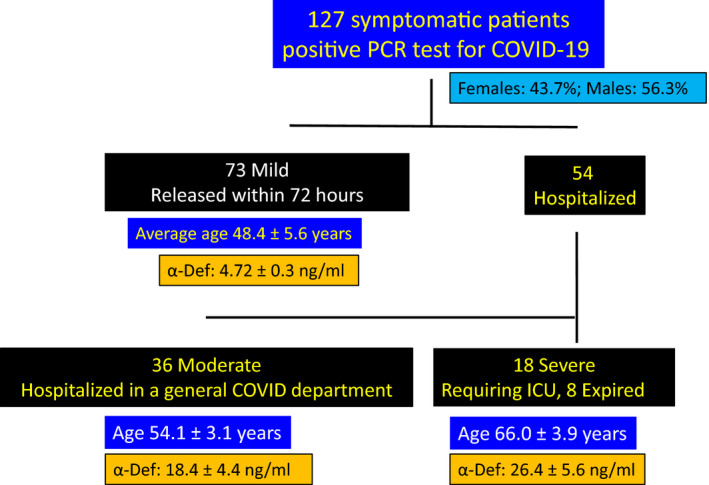
Clinical criteria of participating patients: One hundred and twenty‐seven symptomatic patients with confirmed infection with COVID‐19 were subdivided according to disease severity. This included 73 patients with mild symptoms, 36 classified as having moderate disease and 18 with severe disease. Plasma α‐defensin was measured at the time of admission. The ages of the patients in each group and plasma α‐defensin levels are shown. [Colour figure can be viewed at wileyonlinelibrary.com]
Phlebotomy
Blood was drawn from a peripheral vein in the arm into either 1·8 mg/ml ethylene diamine tetracetic acid (EDTA; final concentration) prior to measuring the complete blood count (CBC), α‐defensin and interleukin‐6 (IL‐6) or into 0·32% citrate (final concentration) to measure coagulation parameters, including thromboelastography (TEG), D‐dimers and fibrinogen. Plasma was prepared by centrifugation at 2000 × g for 10 minutes at room temperature within one hour of phlebotomy. All plasma measurements were made within 4 hours using fresh (unfrozen) samples.
Measurement of plasma components
Plasma levels of α‐defensin were measured as in Abu‐Fanne et al. 25 Samples obtained from 15 patients of comparable age admitted for elective orthopedic surgery served as controls. D‐dimers were measured using a Sysmex CS5100 automatic coagulation analyzer (Nagasaki, Japan). IL‐6 was measured using the Immulite 2000 XPi analyzer (Siemens, Los Angeles, CA, USA). No viral inactivation steps were taken prior to analysis. All results were validated by two investigators.
The effect of α‐defensin, IL‐6 and fibrinogen concentration on coagulation and fibrinolysis in COVID‐19 patients
Citrated plasma from COVID‐19 patients with moderate disease (n = 19) and healthy controls (n = 13) were compared. Plasma levels of α‐defensins and IL‐6 were measured as described in the preceding paragraph. TEG was performed using the TEG system (Haemonetics Limited, Coventry, UK). 25 TEG was performed after addition of IL‐6 (50 pg/ml), α‐defensin (64 ng/ml), and/or colchicine (10 nM) in the presence and absence of tissue plasminogen activator (tPA). Four parameters were measured: (i) reaction times (R value) from the initiation of coagulation to the beginning of clot formation; (ii) the α‐angles that reflect the contribution of platelets; (iii) the maximal amplitudes (MA, mm) that measure fibrin accretion; and (iv) Ly30% that measures the time to 30% clot lysis in the presence of 10 nM tPA. We then asked whether TEG parameters were influenced by the high concentration of fibrinogen often found in patients with COVID‐19. 27 Citrated blood samples from non‐COVID‐19 patients referred to the clinical laboratory with normal total white blood cell counts, C‐reactive protein and sedimentation rates were classified in two groups: those with plasma fibrinogen between 400 and 450 mg/dl (n = 17) and those with plasma fibrinogen between 600 and 650 mg/dl (n = 9). TEG was performed in the absence and presence of tPA (10 nM) as described above.
Histology and immunohistochemistry
Five‐µm sections cut from formalin‐fixed paraffin‐embedded lung tissues from eight autopsies of COVID‐19 patients and control tissue (normal‐appearing tissue at a distance from cancer) performed at the Hospital of the University of Pennsylvania were immunostained as described. 28 Briefly, sections were deparaffinized and steamed in 10 mM sodium citrate buffer, pH 6·0 for 10 minutes to unmask the antigen. Endogenous peroxidase was blocked with 0·3% H2O2, and the sections were immunostained with rabbit anti‐human α‐defensin antisera 29 or normal rabbit sera as negative control (1:3,000) for one hour at 37°C followed by biotinylated anti‐rabbit IgG (Vector Laboratories, Burlingame, CA, USA, 1:200 respectively) and by incubation with horseradish peroxidase (HRP)‐conjugated streptavidin (37°C for 30 min). Peroxidase was detected using the avidin–biotin complex ABC Kit (Vector Laboratories) counterstained with hematoxylin. Positive staining was visualized by the brown‐coloured [3,3‐diaminobenzidine] (DAB) reaction product. Sections were also stained by haematoxylin and eosin for histological evaluation.
Statistical analysis
Data were analyzed using the Crunch statistical software (Auckland, CA, USA). Values are presented as means ± SEM. Between‐group comparisons were performed using ANOVA or a two‐tailed Student’s t test. Pearson's correlation test was used to evaluate associations between α‐defensin and other laboratory results. Statistical significance was set at P < 0·05. The study was not powered sufficiently to draw conclusions regarding α‐defensin and premorbid conditions that impact the course of COVID‐19 (e.g. diabetes, hypertension, cardiovascular disease and gender).
Results
Patient demographics
Blood samples were collected from 127 symptomatic adults admitted to Hadassah Medical Center with a positive PCR test for COVID‐19 (Fig 1). A single sample was taken from 73 patients with mild disease who were released within 72 h to an isolation facility. Thirty‐six patients classified as having moderate disease were hospitalized in a general COVID unit, while 18 had more severe disease that required ICU care, eight of whom died of their disease (Fig 1). Patients with ‘severe’ disease were older on average (66·0 ± 3·9 years) than those with mild (48·4 ± 5·6; P < 0·02) or moderate disease (54·1 ± 3·1; P < 0·03; Fig 1). Fifteen patients admitted for elective orthopaedic surgery (63·4 ± 5·9 years) who had neutrophil counts (7·67 ± 2·27 × 103/mm3) comparable to those of COVID‐19 patients (8·11 ± 3·42 × 103/mm3) served as controls.
Correlation between plasma levels of α‐defensin and clinical course in patients with COVID‐19 infection
Plasma α‐defensin was elevated at the time of admission in all 73 COVID‐19 patients with mild disease compared to controls (4·72 ± 0·3 vs 1·53 ± 0·2 ng/ml; P < 0·02; Fig 1). Levels were higher in patients with moderate disease (18·4 ± 4·4 ng/ml; P < 0·001 vs controls; P < 0·035 vs mild) and were most elevated in those with severe disease (26·4 ± 5·6 ng/ml; P < 0·024 vs moderate; Fig 1).
Samples were collected daily from the 54 hospitalized COVID‐19 patients (Fig 1). The average durations of follow‐up and sample retrievals for patients in the moderate and severe cohorts were comparable (6·1 ± 0·8 vs 7·6 ± 0·6 days; range 1–18 days). Plasma levels of α‐defensin correlated with disease severity during hospitalization (Fig 2A). Peak values were higher in severely vs moderately affected patients (79·7 ± 8·0 vs 31·8 ± 5·7 ng/ml; P < 0·001) as were average values defined by areas under the curve throughout the hospitalization (36·9 ± 3·3 vs 13·8 ± 1·8 ng/ml respectively; P < 0·001; Fig 2A). Plasma levels of α‐defensin also rose more rapidly in the severely affected cohort (Fig 2A). Patients with severe disease were older on average than those with moderate illness (66·0 ± 3·9 vs 54·1 ± 3·1 years; P < 0·03), reflecting an association between age and mean and peak levels of α‐defensin (R = 0·36, R = 0·38; P < 0·03 respectively; Fig 2A). The predisposition to more severe clinical manifestations in the more elderly population is not due simply to an age‐dependent rise in α‐defensin levels. Plasma levels of α‐defensin were similar in healthy controls ages 18–30 years (1·4 ± 0·3 ng/ml, n = 9), ages 31–55 years (1·7 ± 0·4 ng/ml, n = 11) and 56–87 years (1·4 ± 0·2 ng/ml, n = 12). Moreover, in contrast to α‐defensin, which reflects neutrophil activation, the average total white blood cell count and neutrophil count did not rise significantly during hospitalization (7·82 ± 2·13 vs 9·18 ± 4·77 × 103/mm3; P = 0·94).
Fig 2.
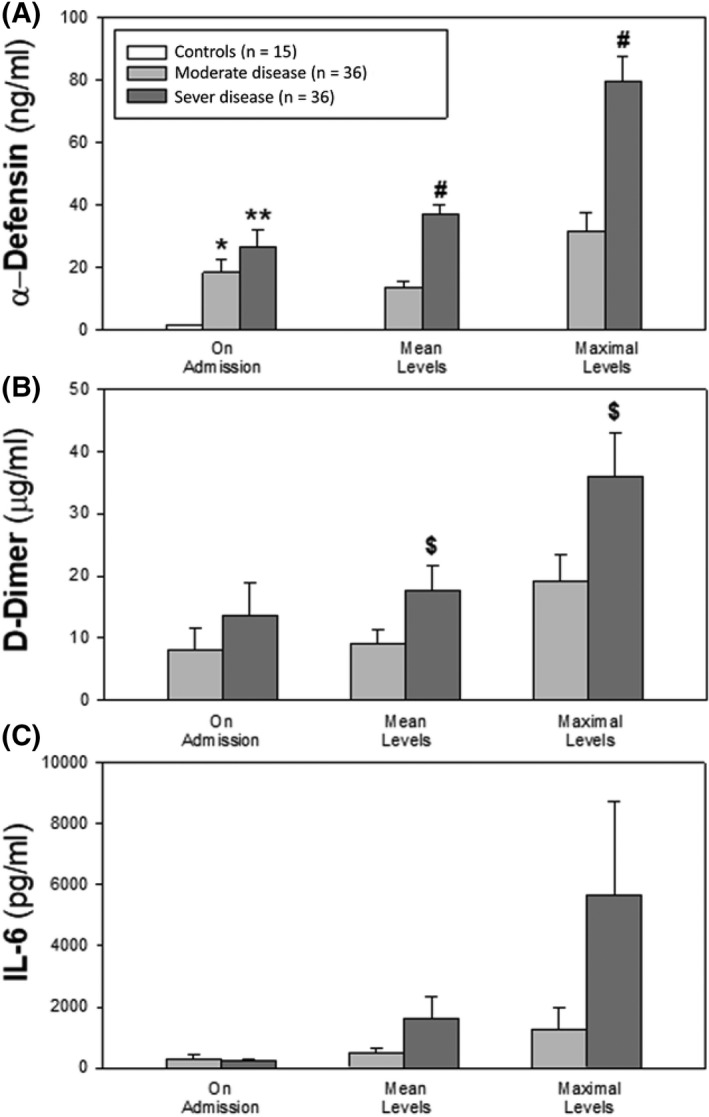
Plasma α‐defensin, D‐dimers and IL‐6 in patients with COVID‐19. Values shown are upon admission, mean levels during the hospitalization, and peak levels. (mean ± SEM; *P < 0·05 and **P ≤ 0·02 vs controls (CTR), ANOVA; #; P < 0·0001 and $; P < 0·05 vs the moderate‐disease cohort, Student's t test). Peak α‐defensin (Panel A), D‐dimer (Panel B) and IL‐6 (Panel C) levels were significantly higher than corresponding levels on admission (P = 0·01).
Deposition of α‐defensin in lung tissue
We next asked if α‐defensins are released into the lungs, a principal target organ, during COVID‐19 infection. Staining of formalin‐fixed, paraffin‐embedded sections taken at autopsy from eight patients who died with COVID‐19 infection showed intense, widespread deposition of α‐defensin. Representative sections from each autopsy were selected for staining (Fig 3). Haematoxylin and eosin‐stained sections from all specimens showed varying stages of diffuse alveolar damage (exudative, proliferative and organizing) with intra‐alveolar haemorrhage. α‐Defensin was found in neutrophils infiltrating the parenchyma. Intense heterogeneous staining for released α‐defensin in the absence of neutrophils was detected in endothelium, epithelium, vascular and airway smooth muscle cells, fibroblasts, alveolar macrophages, and in association with intravascular fibrin clots and extra‐vascular fibrin deposits within pulmonary alveoli.
Fig 3.
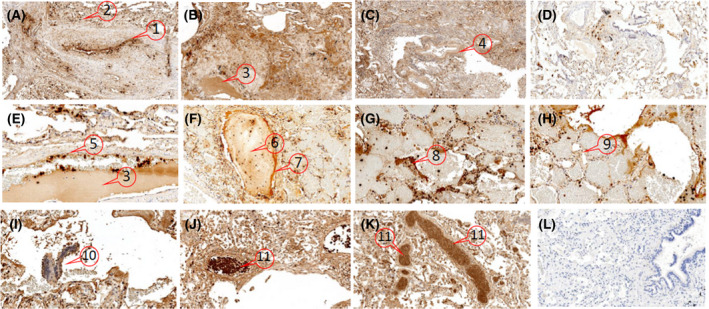
Distribution of α‐defensins in COVID‐19 lungs. α‐Defensins are distributed heterogeneously throughout all lung sections from COVID‐19 patients. Representative formalin‐fixed paraffin‐embedded lung sections from three of the eight autopsies of patients with COVID‐19 that were immunostained using rabbit anti‐HNP1‐3 sera 29 are shown (A–C, magnification 100×; E–K, magnification 200–400×). (D) Staining of ‘control’ lung tissue. (L) No antisera control, magnification 100×. The areas designated with numbers indicate: α‐defensins within neutrophils adherent to the endothelium (A‐1, E‐5) and those infiltrating the parenchyma (A‐2) and free from neutrophils in endothelial cells (A‐1, E‐5), subendothelium (A‐1), vascular smooth muscle cells (C‐4), alveolar (H‐9) and bronchial epithelium (I‐10), around and within intravascular thrombi (F‐6 and 7) and intra‐alveolar fibrin (G‐8) and within intravascular serum exudate from a fibrin clot (B‐3, E‐3). Vascular occlusion by α‐defensin‐expressing neutrophils was prominent in the lungs from several patients (J‐11, K‐11). [Colour figure can be viewed at wileyonlinelibrary.com]
Correlation between plasma levels of α‐defensin and D‐dimers during hospitalization
Based on previous findings in mice transgenic for α‐defensin‐1,26 we examined the relationship between α‐defensin and D‐dimers, a marker of active coagulation associated with disease severity and mortality. 10 Plasma levels of D‐dimers were higher in patients with severe vs mild‐to moderate COVID‐19 disease (Fig 2B), with differences reaching statistical significance for peak values (36·0 ± 7·1 vs 19·0 ± 4·1 μg/ml; P < 0·01) and mean daily values (17·6 ± 4·1 vs 9·0 ± 2·21 μg/ml respectively). Plasma α‐defensin and D‐dimers in the entire cohort correlated closely from days 3 to 7 (P < 0·01), with the most prominent association on day 5 (r = 0·74; P < 0·0001).
α‐Defensin and IL‐6 during hospitalization
We next asked if there was a correlation between α‐defensins and IL‐6, a marker of inflammation that correlates with disease severity in COVID‐19. 10 In our cohort, IL‐6 levels were markedly elevated on admission (284 ± 69 pg/ml, n = 29) compared with healthy controls (2·9 ± 0·3 pg/ml, n = 15; P < 0·05, ANOVA), and the levels increased during hospitalization (Fig 2C). Mean and peak levels were higher in those with severe vs milder disease (mean: 5 674 ± 3 061 pg/ml vs 1 288 ± 698 pg/ml; peak: 1 636 ± 69 pg/ml vs 481 ± 19 pg/ml; P < 0·01). The rise in α‐defensin correlated with increasing levels of IL‐6 (Fig 2C) on admission (two‐tailed Pearson coefficient correlation r = 0·66; P < 0·0001) and on days 4 and 5 (r = 0·48 and 0·60 respectively; P < 0·002).
α‐Defensin and clot formation ex vivo
Based on these findings, prior results demonstrating α‐defensins stimulate coagulation, 25 , 26 and the fact that IL‐6 interacts with neutrophils, 30 we examined the possibility that high levels of IL‐6 generated by lung epithelium during COVID‐19 infection 31 stimulates release of α‐defensin from neutrophils, which in turn promotes thrombotic complications, exemplified by a rise in D‐dimers. Two sets of experiments were performed to test this hypothesis. First, we examined the effect of IL‐6 on the release of α‐defensin. Second, we examined the effect of IL‐6 on coagulation.
Effect of IL‐6 and colchicine on release of α‐defensin from human neutrophils and clot formation ex vivo
Addition of pathological levels of IL‐6 (10–100 pg/ml) observed in COVID‐19 patients to isolated human neutrophils stimulated the release of α‐defensins in a concentration‐dependent manner that was inhibited by colchicine (Fig 4A). The same outcomes were seen in whole blood (data not shown). Pre‐incubation of whole blood pooled from six healthy volunteers with 50 pg/ml IL‐6 accelerated clot formation assessed by TEG (Figs 4B,C), exemplified by a decrease in R values from 6·9 ± 0·54 to 4·1 ± 0·59 min (n = 12; P < 0·03). The results were similar when purified α‐defensin (64 ng/ml) was added to whole blood (R values 4 ± 0·48 min, n = 11; P < 0·03; Fig 4C). Addition of colchicine (10 nM) to blood prior to IL‐6 inhibited shortening of the R values, but colchicine had no effect in the presence of added α‐defensin (Fig 4C). Colchicine alone (10 nM) increased the R values to 7·5 ± 0·39 min (n = 10; P < 0·03; Fig 4C) in line with our prior observations. 25
Fig 4.
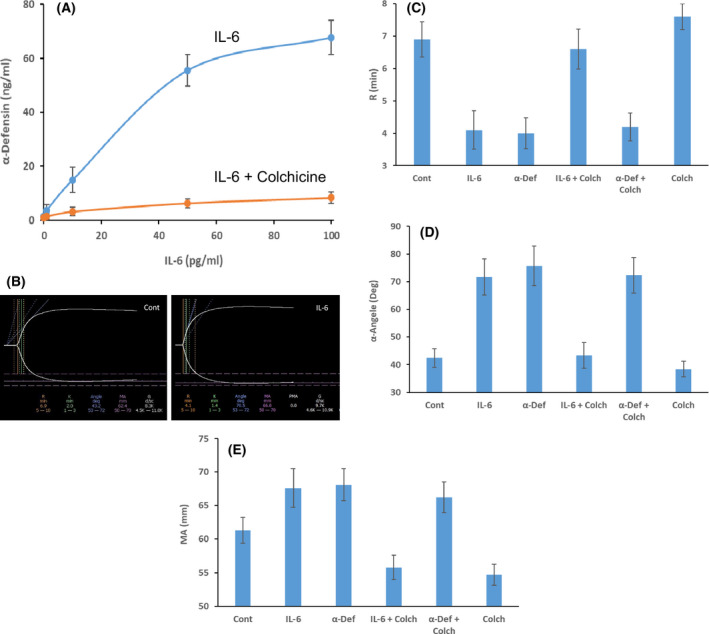
(A) Effect of IL‐6 on α‐defensins release from neutrophils. IL‐6 was added to isolated human neutrophils (10–100 pg/ml) for one hour in the presence (orange lines) or absence (blue lines) of colchicine (10 nM) with constant gentle agitation; the cells were pelleted and α‐defensins in the supernatant fluid was measured. 25 The result (mean ± SEM) of three experiments is shown. (B–E) Effect of IL‐6 on clot formation. Clot formation was assayed by thrombelastography (TEG). 25 Human blood pooled from six healthy volunteers collected in citrate was pre‐incubated with or without IL‐6 (50 pg/ml) and with or without colchicine (10 nM) for one hour as in (A). Coagulation was initiated by adding kaolin, 25 and clot formation was monitored by TEG (B), and the R values, α angles and maximal amplitude (MA) values were calculated (C–E). In other experiments, α‐defensin was added instead of IL‐6 in the presence or absence of colchicine (10 nM). The results shown are representative of three experiments. [Colour figure can be viewed at wileyonlinelibrary.com]
In addition to accelerating the rate of clot formation, IL‐6 changed the properties of the resultant clots themselves. This is exemplified by a significant increase in the α‐angles (42·3 ± 3·3 to 71·7 ± 6·5 degrees, n = 10; P < 0·03; Fig 4D) and by an increase in MAs from 61·3 ± 1·9 to 67·6 ± 2·9 mm (n = 10; P < 0·04; Fig 4E). These changes in TEG parameters related to enhanced clot strength 32 , 33 were blocked by the addition of 10 nM colchicine prior to IL‐6 (50 pg/ml) and were reproduced by the addition of α‐defensin‐1 (64 ng/ml; Figs 4D, E).
α‐Defensin, IL‐6 and fibrinolysis ex vivo
α‐Defensins also inhibit fibrinolysis in vitro 26 and in mice transgenic for human α‐defensin‐1. 25 Therefore, we asked whether neutrophils activated by IL‐6 contribute to this inhibition. Pre‐incubation of human blood from healthy volunteers with IL‐6 (50 pg/ml) in the presence of tPA (10 nM) inhibited fibrinolysis measured by a reduction in Ly30 values from 92 ± 7·1% to 65 ± 5·5% (P < 0·01; Fig 5A, B). Inhibition of fibrinolysis by IL‐6 was prevented by colchicine (10 nM; Fig 5B). The inhibitory effect of IL‐6 on Ly30 values was reproduced by adding α‐defensin (64 ng/ml; Fig 5B). In contrast to IL‐6, the inhibitory effect of α‐defensin on fibrinolysis was unaffected by the addition of colchicine (Fig 5B). Together these results indicate that activation of neutrophils by IL‐6 accelerates fibrin generation (shortened R values), increases clot parameters (increased α‐angles and MA values; Fig 4B–E) and inhibits fibrinolysis (Fig 5A, B). These findings mirror the coagulation pattern reported in COVID‐19 patients 34 and our findings in α‐defensin‐1 transgenic mice. 25
Fig 5.
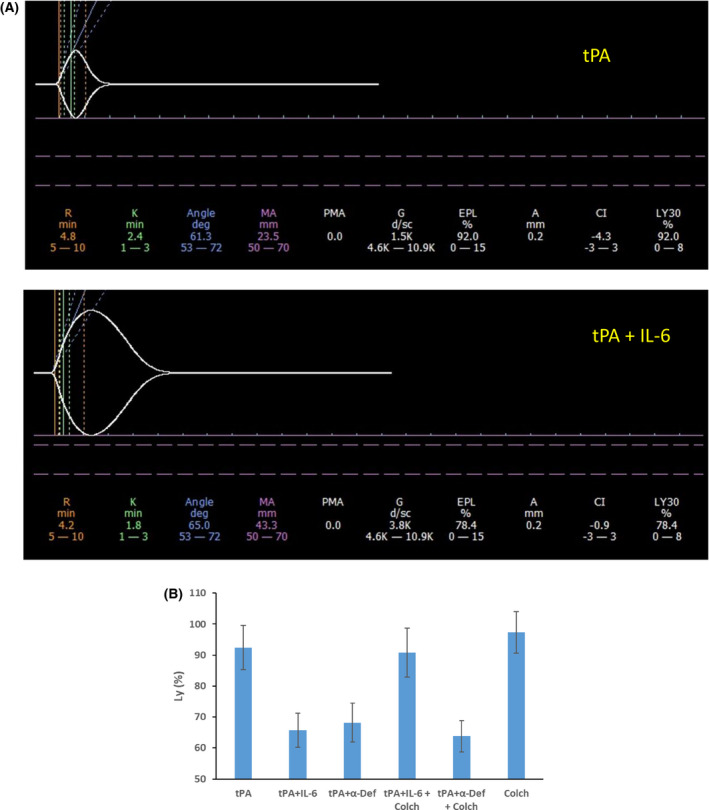
Effect of IL‐6 on fibrinolysis. (A) Clot formation and lysis were assayed by thrombelastography (TEG). 25 Human blood pooled from six healthy volunteers collected in citrate was pre‐incubated with or without IL‐6 (50 pg/ml) for one hour. Tissue plasminogen activator (tPA; 10 nM) was added to samples preincubated with IL‐6 (tPA+IL‐6) or without IL‐6 (tPA) immediately before kaolin was added to initiate coagulation. 25 The results shown are representative of three independent experiments. (B) Fibrinolysis (Ly) was calculated from experiments performed as in (A) and in triplicate in the presence of tPA alone (tPA) or tPA and IL‐6 (tPA+IL‐6). In other experiments, α‐defensin (64 ng/ml) was added instead of IL‐6 (tPA+ α‐Def). In a third set of experiments, tPA was added together with IL‐6 and colchicine (10 nM; tPA+IL‐6+Colch) or with α‐defensin and colchicine (10 nM; tPA+ α‐Def+Colch) or colchicine alone (Colch). The results (mean ± SD) were calculated from three independent experiments, each performed in triplicate. MA, maximal amplitude. [Colour figure can be viewed at wileyonlinelibrary.com]
We then examined whether the effects on the four TEG values (R, α‐angle, MA and Ly30) were due specifically to release of α‐defensin or were caused by another granule component released in response to IL‐6 and blocked by colchicine. To do so, we compared results in wild‐type (WT) mouse blood, which contains neutrophils that do not express α‐defensin, to blood from transgenic mice expressing neutrophil α‐defensin‐1. IL‐6 (50 pg/ml) shortened the R values in blood containing α‐defensin‐1‐expressing neutrophils (5·1 ± 0·5 vs 3·6 ± 0·4 min, n = 12; P < 0·05) and decreased the Ly30 values in the presence of 10 nM tPA (88·3 ± 7·2% vs 41·7 ± 4·9%, n = 9; P < 0·04), but had no effect when added to blood from WT mice (R = 7·3 ± 0·7 vs 7·1 ± 0·4 min, n = 10). The same patterns were seen in the α‐angle and MA values, which were unaffected when IL‐6 was added to blood from WT mice but increased when blood from defensin transgenic mice was studied (data not shown). Furthermore, and in contrast to results in human blood, colchicine alone had no effect on TEG parameters in blood from WT mice (R = 7·3 ± 0·7 vs 7·4 ± 0·8 min, n = 11). Together, these results indicate that IL‐6 stimulates coagulation by causing release of α‐defensin, which can be prevented by colchicine.
α‐Defensin, IL‐6, coagulation and fibrinolysis in patients with COVID‐19
We then studied the effect of α‐defensins on clot formation and fibrinolysis in blood from patients with COVID‐19. Whole blood samples from 19 patients with moderate disease and 13 healthy controls were compared. Plasma levels of IL‐6 and α‐defensins were measured and TEG performed in the presence and absence of tPA. Plasma levels of both IL‐6 and α‐defensin were elevated in samples from patients (IL‐6: 321 ± 81 pg/ml, n = 19 vs 2·1 ± 0·41 pg/ml, n = 13; P < 0·01; α‐defensins 22·3 ± 5·1 ng/ml, n = 19 vs 1·7 ± 1·1 ng/ml, n = 13; P < 0·02). The increase in IL‐6 and α‐defensins correlated with acceleration of clot formation measured by TEG, that is, R values were shorter (7·2 ± 0·62; n = 13 vs 3·9 ± 0·44 min; n = 19; P < 0·01), the α‐angles were higher (44·1 ± 3·9; n = 13 to 77·9 ± 8·6 degrees, n = 19; P < 0·001; and MA values were greater (60·4 ± 2·6; n = 13 vs 71·8 ± 3·1, n = 19; P < 0·03), consistent with reports by others. 34 To study the effect of IL‐6 and α‐defensins on fibrinolysis, tPA (10 nM) was added to whole blood as in Abu‐Fanne et al. 25 The levels of IL‐6 and α‐defensins each correlated with a reduction in the Ly30 (%) compared to controls (93·8 ± 2·1, n = 13 vs 43·8 ± 4·2, n = 19; P < 0·0001), indicating the clots were more resistant to fibrinolysis, findings in line with observations in defensin transgenic mice. 25
Fibrinogen concentration and coagulation in COVID‐19 patients
The plasma concentration of fibrinogen in COVID‐19 patients (613·4 ± 66·3 mg/dl; n = 19) was higher than in the control population (431·6 ± 47·0 mg/dl, n = 13; P < 0·03) as reported by others. 27 Although changes in fibrinogen concentration over this range (400 to 700 mg/dl) are not reported to affect the α‐angle and to have small effects on the MA, 35 we compared TEG results in samples from non‐COVID‐19 patients with plasma fibrinogen between 400 and 450 (n = 17) and 600 and 650 mg/dl (n = 9). Consistent with findings by others, 35 levels of fibrinogen in this range (422–637 mg/dl) had no significant effect on the R values (6·8 ± 0·4 vs 6·7 ± 0·9 min; P = 0·95); MA 61·8 ± 2·9 vs 63·4 ± 3·4 (P = 0·97); α‐angles 43·5 ± 5·5 vs 44·1 ± 7·2 degrees or Ly30 values after the addition of tPA (91 ± 9·5% vs 93 ± 5·3%; P = 0·92).
Discussion
In this study, plasma levels of α‐defensin tracked with disease severity on admission and with clinical improvement or deterioration thereafter. Immunohistochemical staining of lung sections from patients dying of COVID‐19 showed extra‐vasation of α‐defensin from neutrophils to intravascular thrombi and intra‐alveolar fibrin. Plasma levels of α‐defensin also correlated with previously reported elevations in plasma IL‐6 and D‐dimers. 10 IL‐6 stimulated neutrophils to release α‐defensin, which accelerated the rate of clot formation and inhibited fibrinolysis, assessed by TEG. Colchicine, which inhibits release of α‐defensin, 25 inhibited the procoagulant effect of IL‐6 in vitro and patients with COVID‐19 given colchicine developed lower plasma levels of α‐defensin and D‐dimers than those who were not treated.
One limitation of our study is that it was not powered sufficiently to draw conclusions regarding α‐defensin and premorbid conditions that impact the course of COVID‐19 (e.g. diabetes, hypertension and cardiovascular disease). A second limitation is the open‐label design of the intervention study. Larger prospective blinded studies will be needed to address both limitations. Third, additional work will be needed to study the detailed structure of clots formed in patients with COVID‐19 25 to correlate with the analyses by TEG.
Nevertheless, these data link the innate immune response in patients infected with COVID‐19 (generation of IL‐6) to inflammation (release of α‐defensin by activated neutrophils) to development of a procoagulant state, an effect that was attenuated by colchicine. This hypothesis is supported by a recent report that the levels of D‐dimers were lowed and outcome was improved in COVID‐19 patients receiving colchicine. 36 , 37 Randomized clinical trials to test this possibility are under way.
We hypothesize that our findings in patients with COVID‐19 may prove relevant to the pathogenesis of thromboembolic complications in patients with pneumonia caused by influenza and other forms of acute lung injury that are mediated at least in part by intense neutrophil activation. 38 , 39 The immunohistochemical distribution of α‐defensin we observed in the lungs of patients who died of COVID‐19 suggests that colchicine may also have beneficial effects on other neutrophil‐dependent pathogenic processes that contribute to acute lung injury, for example, those involving loss of endothelial–epithelial barrier function and injury to alveolar lining cells. 24 , 40
Author contributions
AAH conceived the study; AAH, SA, DBC, JWW, RIL, KB and KTM designed experiments; AAH, SA, DBC, SNH, KB, KTM and LAL analyzed data; SA, RA‐F, EM, NK, MH MF and CD performed and analyzed the experiments; AAH, DBC, SA and SNH wrote the paper.
Conflict of interest
None of the authors has a relevant conflict of interest.
Acknowledgements
This research was supported by grants from the Israeli Science Foundation ISF: 930/04 and 3959/19 (AH), and NIH grants DK113142 and HL123912 (DBC) and a grant from the University of Pennsylvania Institute for Translational Medicine and Therapeutics Transdisciplinary Program in Translational Medicine and Therapeutics (KB). We thank Daniel Martinez and the Pathology Core Laboratory at the Childrens Hospital of Philadelphia Research Institute for providing immunohistochemistry and digital slide scanning services.
References
- 1. Azkur AK, Akdis M, Azkur D, Sokolowska M, Veen W, Brüggen M‐C, et al. Immune response to SARS‐CoV‐2 and mechanisms of immunopathological changes in COVID‐19. Allergy. 2020;75:1564–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bryce C, Grimes Z, Pujadas E, Ahuja A, Beasley ME, Albrecht R, et al. Pathophysiology of SARS‐CoV‐2: targeting of endothelial cells renders a complex disease with thrombotic microangiopathy and aberrant immune response. The Mount Sinai COVID‐19 autopsy experience. medRxiv. Cold Spring Harbor Laboratory Press; 2020;May 22:2020.05.18.20099960. [Google Scholar]
- 3. Iba T, Levy JH, Connors JM, Warkentin TE, Thachil J, Levi M. The Unique Characteristics of COVID‐19 Coagulopathy. Crit Care. 2020;24:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barnes BJ, Adrover JM, Baxter‐Stoltzfus A, Borczuk A, Cools‐Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID‐19: Neutrophil extracellular traps. J Exp Med. 2020;217:e20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nadkarni N, Lala A, Bagiella E, Chang L, Moreno P, Pujadas E, et al. Anticoagulation, mortality, bleeding and pathology among patients hospitalized with COVID‐19. JAC. 2020;76:1815–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levi M, Thachil J, Iba T, Levy J. Coagulation abnormalities and thrombosis in patients with COVID‐19. Lancet Haematol. 2020;7:e438–e440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Connors J, Levy J. COVID‐19 and its implications for thrombosis and anticoagulation. Blood. 2020;135:2033–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fox S, Akmatbekov A, Harbert J, Li G, Brown J, Heide R. Pulmonary and cardiac pathology in African American patients with COVID‐19: an autopsy series from New Orleans. Lancet Respir Med. 2020;7:681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zaim S, Chong J, Sankaranarayanan V, Harky A. COVID‐19 and Multiorgan Response. Curr Probl Cardiol. 2020;45:100618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stefanini GG, Montorfano M, Trabattoni D, Andreini D, Ferrante G, Ancona M, et al. ST‐elevation myocardial infarction in patients With COVID‐19: clinical and angiographic outcomes. Circulation. 2020;141:2113–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Al‐Samkari H, Karp Leaf RS, Dzik WH, Carlson JCT, Fogerty AE, Waheed A, et al. COVID‐19 and coagulation: bleeding and thrombotic manifestations of SARS‐CoV‐2 infection. Blood. 2020;136:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Middeldorp S, Coppens M, van Haaps T, Foppen M, Vlaar A, Müller M, et al. Incidence of venous thromboembolism in hospitalized patients with COVID‐1. J Thromb Haemost. 2020;18:1995–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hill JB, Garcia D, Crowther M, Savage B, Peress S, Chang K, et al. Frequency of venous thromboembolism in 6513 patients with COVID‐19: a retrospective study. Blood Adv. 2020;4:5373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mueller‐Peltzer K, Krauss T, Benndorf M, Lang C, Bamberg F, Bode C, et al. Pulmonary artery thrombi are co‐located with opacifications in SARS‐CoV2 induced ARDS. Respir Med. 2020;172:106135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lax SF, Skok K, Zechner P, Kessler HH, Kaufmann N, Koelblinger C, et al. Pulmonary arterial thrombosis in COVID‐19 with fatal outcome: results from a prospective, single‐center. Clinicopathologic Case Series. Ann Intern Med. 2020;173:350–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iba K, Connors J, Levy J. The coagulopathy, endotheliopathy, and vasculitis of COVID‐19. Inflamm Res. 2020;69:1181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chun T, Judelson D, Rigberg D, Lawrence P, Cuff R, Shalhub S, et al. Managing central venous access during a health care crisis. J Vasc Surg. 2020;72:1184–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Helms J, Tacquard C, Severac F, Leonard‐Lorant I, Ohana M, Delabranche X, et al. High risk of thrombosis in patients with severe SARS‐CoV‐2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46:1089–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bemtgen X, Zotzmann V, Benk C, Rilinger J, Steiner K, Asmussen A, et al. Thrombotic circuit complications during venovenous extracorporeal membrane oxygenation in COVID 19. J Thromb Thrombolysis. 2020;11:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beyls C, Huette P, Abou‐Arab O, Berna P, Mahjoub Y. Extracorporeal membrane oxygenation for COVID‐19‐associated severe acute respiratory distress syndrome and risk of thrombosis. Br J Anaesth. 2020;125:260–e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hassanein M, Radhakrishnan Y, Sedor J, Vachharajani T, Vachharajani VT, Augustine J, et al. COVID‐19 and the kidney. Cleavland Clin J Med. 2020;87:619–31. [DOI] [PubMed] [Google Scholar]
- 23. Group CT . High risk of thrombosis in patients with severe SARS‐CoV‐2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46:1089–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. White D, MacDonald S, Bull T, Hayman M, de Monteverde‐Robb R, Sapsford D, et al. Heparin resistance in COVID‐19 patients in the intensive care unit. J Thromb Thrombolysis. 2020;50:287–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abu‐Fanne R, Stepanova V, Litvinov RI, Abdeen S, Bdeir K, Higazi M, et al. Neutrophil α‐defensins promote thrombosis in vivo by altering fibrin formation, structure, and stability. Blood. 2019;133:481–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Higazi A, Barghouti II, Abu‐Much R. Identification of an inhibitor of tissue‐type plasminogen activator‐mediated fibrinolysis in human neutrophils. A role for defensin. J Biol Chem. 1995;270:9472–7. [DOI] [PubMed] [Google Scholar]
- 27. Han H, Yang L, Liu R, Liu F, Wu K‐L, Li J, et al. Prominent changes in blood coagulation of patients with SARS‐CoV‐2 infection. Clin Chem Lab Med. 2020;58:1116–20. [DOI] [PubMed] [Google Scholar]
- 28. Hijazi N, Abu Fanne R, Abramovitch R, Yarovoi S, Higazi M, Abdeen S, et al. Endogenous plasminogen activators mediate progressive intracerebral hemorrhage after traumatic brain injury in mice. Blood. 2015;125:2558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bdeir K, Higazi A, Kulikovskaya I, Christofidou‐Solomidou M, Vinogradov S, Allen T, et al. Neutrophil alpha‐defensins cause lung injury by disrupting the capillary‐epithelial barrier. Am J Respir Cedrit Care M. 2010;181:935–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fielding CA, McLoughlin RM, McLeod L, Colmont CS, Najdovska M, Grail D, et al. IL‐6 regulates neutrophil trafficking during acuteinflammation via STAT3. J Immunol. 2008;181:2189–95. [DOI] [PubMed] [Google Scholar]
- 31. Yoshikawa T, Hill T, Li K, Peters J, Tseng K. Severe Acute Respiratory Syndrome (SARS) Coronavirus‐Induced Lung Epithelial Cytokines Exacerbate SARS Pathogenesis by Modulating Intrinsic Functions of Monocyte‐Derived Macrophages and Dendritic Cells. J of Virology. 2009;83:3039–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nystrup BN, Nis A, Windeløv AN, Annemarie B, Thomsen BA, Johansson IP. Reduced clot strength upon admission, evaluated by thrombelastography (TEG), in trauma patients is independently associated with increased 30‐day mortality. Scand J Trauma Resuscit Emerg Med. 2011;19:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jeger V, Zimmermann H, Exadaktylos A. The Role of Thrombelastography inMultiple Trauma. Emerg Med Int. 2011;ID;895674:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Panigada M, Bottino N, Tagliabue P, Grasselli G, Novembrino C, Chantarangkul V, et al. Hypercoagulability of COVID‐19 patients in intensive care unit: A report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost. 2020;18:1738–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harr JN, Moore EE, Ghasabyan A, Chin TL, Sauaia A, Banerjee A, et al. Functional fibrinogen assay indicates that fibrinogen is critical in correcting abnormal clot strength following trauma. Shock. 2013;39:45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Deftereos SG, Giannopoulos G, Vrachatis DA, Siasos GD, Giotaki SG, Gargalianos P, et al. Effect of colchicine vs standard care on cardiac and inflammatory biomarkers and clinical outcomes in patients hospitalized with coronavirus disease 2019. JAMA. 2020;3:e2013136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sandhu T, Tieng A, Chilimuri S, Franchin G. A case control study to evaluate the impact of colchicine on patients admitted to the hospital with moderate to severe COVID‐19 infection. Can J Infect Dis Med Microbiol. 2020;ID;8865954:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Poissy J, Goutay J, Caplan M, Parmentier E, Duburcq T, Lassalle F, et al. Pulmonary embolism in patients with COVID‐19 awareness of an increased prevalence. Circulation. 2020;142:184–86. [DOI] [PubMed] [Google Scholar]
- 39. Obi AT, Tignanelli CJ, Jacobs BN, Arya S, Park PK, Wakefield TW, et al. Empirical systemic anticoagulation is associated with decreased venous thromboembolism in critically ill influenza A H1N1 acute respiratory distress syndrome patients. Journal of Vascular Surgery: Venous and Lymphatic Disorders. 2020. [DOI] [PubMed] [Google Scholar]
- 40. Mitchell W. Thromboinflammation in COVID‐19 acute lung injury. Paediatr Respir Rev. 2020;35:20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]