

Abstract
Hydrogen atom abstraction (HAT) from C(sp3)–H bonds of naturally abundant alkanes for alkyl radical generation represents a promising yet underexplored strategy in the alkylation reaction designs since involving stoichiometric oxidants, excessive alkane loading, and limited scope are common drawbacks. Here we report a photo-induced and chemical oxidant-free cross-dehydrogenative coupling (CDC) between alkanes and heteroarenes using catalytic chloride and cobalt catalyst. Couplings of strong C(sp3)–H bond-containing substrates and complex heteroarenes, have been achieved with satisfactory yields. This dual catalytic platform features the in situ engendered chlorine radical for alkyl radical generation and exploits the cobaloxime catalyst to enable the hydrogen evolution for catalytic turnover. The practical value of this protocol was demonstrated by the gram-scale synthesis of alkylated heteroarene with merely 3 equiv. alkane loading.
Subject terms: Homogeneous catalysis, Synthetic chemistry methodology
Hydrogen atom abstraction from C(sp3)–H bonds of naturally abundant alkanes for alkyl radical generation represents a promising yet underexplored strategy in the alkylation reaction designs. Here the authors show a photo-induced and chemical oxidant-free cross-dehydrogenative coupling between alkanes and heteroarenes using catalytic chloride and cobalt catalyst.
Introduction
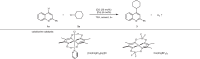
Alkyl radical (R·), one of the most fundamental intermediates in organic synthesis, constitutes important approaches toward rapid molecular construction. Its generation from functionalised precursors such as aliphatic carboxylic acids, boronic reagents, halides, and others have been well-established and broadly applied as efficient paradigms in routine synthesis1–12. Of equal importance, direct C(sp3) radical generation from the readily available non-functionalised alkanes represents a more straightforward and sustainable method13. However, due to the high bond dissociation energies (BDEs)14,15, current strategies involving the homolysis of strong C(sp3)–H bonds mostly rely on the hydrogen atom transfer (HAT) with electrophilic heteroatom radicals, for instance, bromo-16–18, nitrogen-15,19, or oxygen-centred radicals19–22 (Fig. 1a). In this context, robust redox catalysts or strongly oxidising reagents are required, whereas precise control over the site selectivity among ubiquitous C(sp3)–H bonds in a molecule with broad substrate scope imposes grand challenges.
Fig. 1. From inspiration to reaction design.

a Bond dissociation energies (BDEs) of aliphatic C–H bonds and some protonated heteroatom species. b The representative chlorine radical generation method in HAT reactions. c The designed dehydrogenative Minisci alkylation enabled by catalytic chlorine radical generation. d The proposed dehydrogenative Minisci alkylation mechanism; SET single-electron transfer, LMCT ligand-to-metal charge transfer, SH2 bimolecular homolytic substitution, FG functional group.
Chlorine radical (Cl·) is an efficient HAT agent that could cleave various C(sp3)–H bonds. Using chloride (Cl−) in Cl· generation brings several benefits in organic syntheses because it is innocuous and abundant in diverse salt forms. Nevertheless, the unfavourable chloride-to-chlorine oxidation ( = 1.36 V vs NHE)23 and untamed reactivity of Cl· compared with other halide analogues17,18,24–26 make chlorine radical-promoted alkylation rarely explored. In this endeavour, few strategies have been disclosed for the efficient usage of Cl·, including (a) the direct single-electron transfer (SET) from Cl− to photocatalyst under photothermal conditions27–30; (b) the ligand-to-metal charge transfer (LMCT), which has been employed for the coupling of alkanes and organohalides by metallophotoredox catalysis31–36; (c) the photolysis of in situ generated Cl2 via electrooxidation of HCl37; (d) the bimolecular homolytic substitution (SH2) between chloroborate and an oxy radical for the alkane borylation38. These pioneering examples demonstrate the potential of Cl− to realise the HAT process via Cl· intermediate (Fig. 1b).
The Minisci alkylation involves the coupling between heteroarenes and nucleophilic alkyl radicals. In views of the diversity of heteroarenes and their countless applications in material science, agrochemicals, and the pharmaceutical industry, the Minisci alkylation plays a pivotal role in synthetic chemistry. Since the key of the Minisci alkylation is the alkyl radical generation, it prompts us to consider the possibility of merging the chlorine radical-mediated alkyl radical generation manifold with the Minisci alkylation, which could allow the heteroarene diversification simply through the coupling with widely available C(sp3)-H feedstocks39. Furthermore, an H2-evolving Minisci alkylation is more desirable for its highest step- and atom-economy40,41. Owing to the strong aliphatic C–H bonds and the net oxidative nature, excessive alkane (normally as solvent) and oxidant loadings, high temperature, or precious catalysts are not uncommon. Therefore, a catalytic cross-dehydrogenative Minisci alkylation without stoichiometric chemical oxidants is long-sought-after39. Our group has a long-term interest in the arene functionalisation, which was facilitated by the excited aromatics under catalyst-free conditions42–47. Recently, we documented a simple and clean Minisci alkylation reaction via formal dehydrative coupling of heteroarenes with alcohols. Taking advantage of the superior redox properties of the excited heteroarenes, metallophotoredox catalysts could be strategically avoided48.
Based on these literature precedents49,50, we envisioned that heteroarene itself could be an intrinsic photosensitiser, allowing efficient Cl− oxidation in the Minisci reaction. In this work, a dehydrogenative Minisci alkylation using catalytic Cl− under photochemical conditions is presented (Fig. 1c). Owing to the strong hydrogen atom affinity of Cl·, a wide range of inactivated and activated C(sp3)–H bonds could be arylated with good functional group tolerance and substrate diversity, and notably, with the strategic introduction of the cobaloxime catalyst, we formulate a chemical oxidant-free heteroarene alkylation protocol by releasing H2.
Results
Reaction design and optimisation
The proposed dehydrogenative alkylation pathway is depicted in Fig. 1d. It was hypothesised that the excited heteroarenes I could oxidise the Cl− under irradiation to generate the Cl· for the aliphatic C–H abstraction from alkane 2. The addition of the so-formed alkyl radical to another equivalent of heteroarene 1-H+ followed by hydrogen atom removal would give the desired alkylated heteroarene 3-H+. Meanwhile, to efficiently quench the generated radical intermediate II, a readily accessible cobaloxime catalyst [Co(dmgH)2(py)]Cl was introduced to the system51–55 not only to prevent the over-reduction of intermediate II but also to serve as a terminal oxidant for the rearomatisation of alkylated intermediate III though H2 evolution.
In our initial evaluation, 2-phenylquinoline (1a) and cyclohexane (2a) were chosen as model substrates with trifluoroacetic acid (TFA) and a catalytic amount of Bu4NCl and cobaloxime [Co(dmgH)2(py)]Cl in CHCl3. Delightfully, the desired coupling product 3 could be obtained in good conversion and product yield. After considerable efforts, the optimal reaction conditions yielded 80% of 3 when 20 mol% Bu4NCl, 5 mol% [Co(dmgH)2(py)]Cl, and 3 equiv. of TFA were used in CHCl3 under photo-irradiation for 20 h (Table 1, entry 1). During the optimisations, three key reaction components including Bu4NCl, [Co(dmgH)2(py)]Cl, and CHCl348 were identified, all of which could potentially serve as the Cl· sources. The reaction could proceed smoothly in the presence of any of these chlorides; otherwise, no reaction occurred (entries 2-8). Importantly, the cobaloxime catalyst could improve the reaction productivity by minimising the over-reduction of quinoline to tetrahydroquinoline or other off-target decomposition reactivities. A significant solvent effect was observed in this transformation. In the chlorinated solvent CHCl3, heteroarene 1a was alkylated efficiently without side reaction detected; however, when other solvents were used, poor selectivity and side product formations were often observed (e.g. in entry 4, 10% tetrahydroquinoline was obtained). Additionally, we found that the reaction could be partially suppressed by oxygen (entry 9), and no reaction occurred without light (entry 10).
Table 1.
Key results in reaction optimisations.
 | |||||
|---|---|---|---|---|---|
| Entrya | [Cl−] | [Co] | Solvent | Conversion (%) | Yield (%) |
| 1 | Bu4NCl | [Co(dmgH)2(py)]Cl | CHCl3 | 84 | 80 |
| 2 | – | [Co(dmgH)2(py)]Cl | CHCl3 | 56 | 56 |
| 3 | Bu4NCl | – | CHCl3 | 83 | 65 |
| 4 | Bu4NCl | [Co(dmgH)2(py)]Cl | MeCN | 100 | 70 |
| 5 | – | [Co(dmgH)2(py)]Cl | MeCN | 64 | 46 |
| 6 | Bu4NCl | Co(dmgBF2)2 | MeCN | 78 | 69 |
| 7 | – | Co(dmgBF2)2 | CHCl3 | 60 | 58 |
| 8 | – | Co(dmgBF2)2 | MeCN | 3 | 0 |
| 9b | Bu4NCl | [Co(dmgH)2(py)]Cl | CHCl3 | 48 | 41 |
| 10c | Bu4NCl | [Co(dmgH)2(py)]Cl | CHCl3 | 1 | 0 |
dmg dimethylglyoxime, TFA trifluoroacetic acid.
aReaction conditions: 1a (0.2 mmol), 2a (0.6 mL), [Cl−] (20 mol%), [Co] (5 mol%), and TFA (0.6 mmol) in solvent (1.5 mL) under light irradiated at 20–25 °C for 20 h under N2. Yields were determined by 1H NMR using CH2Br2 as the internal standard.
bThe reaction was conducted under air.
cThe reaction was run in dark.
Substrate scope
After obtaining the optimal reaction conditions, we approached the substrate scope to different C(sp3)–H species using 2-phenylquinoline (1a) as the coupling partner (Fig. 2). Simple cyclic alkanes containing five to twelve carbons afforded the corresponding alkylated heteroarenes 3 to 7 in moderate to good yields, and so did bridged alkanes norbornane and adamantane (8 and 9). Carbonyl compound cyclopentanone was functionalised at the β-position and afforded the desired product 11. Benzylic C–H abstractions of methylbenzene derivatives also provided benzylated heteroarenes (11–14)56. It was not surprising that ethylated quinoline 15 was produced as the major product with diethyl ether (Et2O) as the alkane source48; and the similar C–O cleavage was observed with 1,2-dimethoxyethane (DME) to give the deethoxylated products 16; gratifyingly, the non-cleaved ethereal compounds 17 and 18 could be obtained by decreasing the acid amount to 1.2 to 2 equiv37. However, the C–O cleavage was still inevitable with tetrahydrofuran (THF) and methanol (19–22), possibly due to the relatively high ring strain and oxidation potential of the alcoholic oxygen atom. Reactions with tetrahydropyran (THP) and its analogues proceeded smoothly and gave good yields of the products 23–26. α-C(sp3)–H functionalisation of amine derivatives, for example, amides, sulfonamide and phosphoramide, were all successful (27–33). Interestingly, the HAT of the N-methyl group of N,N-dimethylformamide (DMF) is more favourable than the formyl one and the alkylated heteroarene 29 was obtained as the major product. Few alkane substrates, for instance, cyclododecane, adamantane and cyclopentane were inert at ambient temperature, and a slight temperature increase to 55–60 °C was helpful for their transformations (7, 9, and 10). The unique regioselectivity of Cl· could be displayed from some substrates. For example, the HAT on adamantane is slightly favourable on the tertiary C–H bond in comparison to the secondary one (9), yet 2-methyltetrahydrofuran (MeTHF) only afforded the secondary carbon-functionalised product (20); C4 functionalisation of the N-methylpyrrolidinone (NMP) primarily occurred at the secondary C4 position rather the primary C5 one (31). Other than alkyl substrates, the amidation of heteroarene is also viable with formamide, which furnished the amido product 34. Noticeably, the potential application of this reaction was briefly demonstrated by a challenging gram-scale reaction of heteroarene 1a with 3 equiv. of unactivated alkane 3e, and delightfully the desired product 7 could be obtained in 54% isolated yield.
Fig. 2. Substrate scope of alkane.

Reaction conditions: 1a (0.2 mmol), 2 (0.6 mL for liquid, or 10 equiv. for solid), Bu4NCl (0.04 mmol), [Co(dmgH)2(py)]Cl (0.01 mmol), and TFA (0.6 mmol) in CHCl3 (1.5 mL) under light irradiated at 20–25 °C for 20 h under N2, unless otherwise specified, and the yields were isolated ones. Reaction conditions for the gram-scale synthesis: 1a (4.88 mmol), 2e (14.6 mmol), Bu4NCl (0.49 mmol), [Co(dmgH)2(py)]Cl (0.025 mmol), and TFA (14.6 mmol) in CHCl3 (8 mL) under light irradiated at 55–60 °C for 72 h under N2. aThe reaction was run for 36 h. b1.2 equiv. of TFA was used. c2 equiv. of TFA was used. dThe reaction was heated to 55–60 °C. eA mixture of congeners, C1:C2 = 1:0.9. fA mixture of trans isomers. gA mixture of congeners, C4:C5 = 1:0.26; dmg dimethylglyoxime, TFA trifluoroacetic acid, THF tetrahydrofuran, MeTHF 2-methyltetrahydrofuran, DME 1,2-dimethoxyethane.
Further substrate and functional group tolerance were examined by coupling various heterocycles 1 with cyclohexane (2a). Satisfyingly, a broad variety of functional groups such as halides, cyano, acetyl, ester, amino, nitro, and sulfonamide are compatible with our method, as shown in Fig. 3. Quinoline, pyridine, and isoquinoline moieties afforded the corresponding alkylated products with up to 75% yield (35–56); other heterocycles like pyrimidine (57 and 58), pyrazine (59), quinoxalinone (60), and purine (61–63) were also proven to be feasible (substrates that are incompatible or with low reactivity in the reaction are listed in Supplementary Information Fig. 3). Complex heterocycles bearing different aliphatic C(sp3)–H bonds were examined, including alanine and menthol-derived pyridines (64 and 65), and nicotine (66) were successfully turned into the desired products with moderate yields. Alkylation of hydrocinchonine, a common chiral ligand candidate in synthetic chemistry, gave an appreciable yield of the product (67) with its hydroxy group preserved. Fasudil, a potent Rho-kinase inhibitor and vasodilator, also provided the desired product (68) after protecting its amino group.
Fig. 3. Substrate scope of heteroarenes.

Reaction conditions: 1 (0.2 mmol), 2a (0.6 mL), Bu4NCl (0.04 mmol), [Co(dmgH)2(py)]Cl (0.01 mmol), and TFA (0.6 mmol) in CHCl3 (1.5 mL) under light irradiated at 20–25 °C for 20 h under N2, unless otherwise specified, and the yields were isolated ones. aThe reaction was run for 36 h. b4 equiv. of TFA was used. c5 equiv. of TFA was used. dThe reaction was heated to 55–60 °C. eSeparable mixture, mono:di = 1.4:1. fSeparable mixture, mono:di = 3.9:1. gSeparable mixture, mono:di = 5.7:1; dmg dimethylglyoxime, TFA trifluoroacetic acid.
Mechanistic investigations
To gain insight into this transformation, a series of experiments were conducted (See Supplementary Information for details). We found that radical quenchers, 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) and 3,5-di-tert-4-butylhydroxytoluene (BHT), significantly suppressed the product formation. Noticeably, a radical adduct 69 was detected by GC-MS in the case of TEMPO, which indicated an alkyl radical-involved mechanistic scenario (Fig. 4a). The involvement of R· was also evidenced by a radical trapping experiment by subjecting the alkene 70 to our alkane arylation reaction with heteroarene 1a and Bu4NCl (Fig. 4b), in which a cycloalkylated product 71 was isolated. All these results supported the presence of R· in the plausible mechanism. To probe more details of the chlorine species, a diallyl sulfonamide 72 was submitted to the alkylation reaction of 1a with a stoichiometric amount of Bu4NCl. As expected, a cyclochlorinated compound 73 was isolated, suggesting the formation of Cl· in our case (Fig. 4c). According to some literature37,57, chlorine (Cl2) might not be an active intermediate in our system as the characteristic alkenyl dichlorination products were not observed. On the contrary, no cyclochlorination occurred when the same diallyl compound 72 was irradiated with Bu4NCl and cobaloxime catalyst in the absence of heteroarene and chlorinated solvent, indicating that the LMCT-induced Cl· generation by the cobaloxime catalyst might not be operative in this reaction. Also, replacing cyclohexane (2a) with cyclohexyl chloride (74) gave no desired product, indicating that 74 is not an active intermediate in generating the cyclohexyl radical (Fig. 4d). The light on-and-off experiment showed that continuous irradiation was essential for the product formation (Fig. 4e), and the low reaction quantum yield (<1) does not support the radical chain process (see Supplementary Information). Besides, both parallel and competing kinetic isotope effects were examined, giving kH/kD = 1.64 and 1.56, respectively. This suggested that the alkyl C–H cleavage might not be the rate-determining step (Fig. 4f)8. Moreover, the H2 evolution was confirmed by the GC-TCD analysis (Fig. 4g). Lastly, from the fluorescence quenching experiments, strong interaction between excited heteroarene 1a and Cl− was observed only in the presence of an acid, emphasising the indispensable role of TFA in protonating the heteroarene for Cl− oxidation (Fig. 4h, see Supplementary Information for details). Taken together, these experiments provide support for the surmised radical generation (R· and Cl·) and H2 evolution reaction pathways. More detailed mechanistic studies are still ongoing in our lab. Finally, as a logical extension based on the current mechanistic framework, we believed that this coupling reaction could be realised under visible light irradiation with the catalytic introduction of a more conjugated heteroarene. While this dehydrogenative coupling reaction was proven unsuccessful with most of the commercially available photosensitizers, 43% of the alkylated heteroarene 3 could be furnished with 5 mol% of 2,4-diphenylquinoline as the photocatalyst (See supplementary Information Table 3)50. This encouraging result could enlighten more visible light-promoted Cl· generation strategies for coupling reactions.
Fig. 4. Mechanistic studies.

See Supplementary Information for more detailed reaction conditions and descriptions, including a radical quenching, b R· trapping, c Cl· trapping, d intermediate investigation, e light on/off experiment, f kinetic isotope effect, g H2 detection, and h fluorescence quenching. Ar = 2-phenylquin-4-yl, Cy, cyclohexyl. TEMPO 2,2,6,6-tetramethylpiperidine 1-oxyl, BHT 3,5-di-tert-4-butylhydroxytoluene; dmg dimethylglyoxime, TFA trifluoroacetic acid.
Discussion
We have developed an approach toward photo-induced dehydrogenative Minisci alkylation, which couples a wide range of heterocycles with strong aliphatic C–H bonds, while the merger of catalytic Cl· generation and hydrogen evolution grants simple and chemical oxidant-free reaction conditions. Mechanistic studies were revealed to support the direct Cl· generation via the SET between Cl− and excited heteroarene. As a powerful platform, complex molecules bearing diverse C(sp3)–H patterns and functional groups were feasible for alkylations, and a large-scale synthesis was demonstrated with low alkane loading. We believed that this work would advance the excited arene chemistry and inspire future cross-dehydrogenative coupling designs.
Methods
General experimental procedure for the cross-dehydrogenative coupling of alkanes and heteroarenes
To a 10 mL pyrex microwave vial equipped with a Teflon-coated magnetic stirring bar was added heteroarene 1 (0.2 mmol) and [Co(dmgH)2(py)]Cl (4 mg, 0.01 mmol). The tube was sealed with a rubber septum, evacuated and backfilled with argon three times before alkane 2 (0.6 mL for liquids. For solid alkanes, 2.0 mmol of alkane was added to the vial before backfilled with argon) was injected into the vial. The mixture was then sequentially added Bu4NCl (11.1 mg, 0.04 mmol), CHCl3 (1.5 mL), and TFA (46 μL, 0.6 mmol) in the glove box and then sealed with an aluminium cap with a septum. The reaction vial was taken out from the glove box and stirred at 20–25 °C or 55–60 °C under light irradiation of a 300 W Xe lamp with a 280 nm filter for 20–36 h, as the time indicated. After the reaction was completed, the reaction was basified with sat NaHCO3 (aq), extracted with EtOAc, and filtered through a short pad of MgSO4. The volatiles were removed under reduced pressure to obtain the crude product. The isolated product was obtained by preparative thin-layer chromatography.
Supplementary information
Acknowledgements
We are grateful to the Canada Research Chair Foundation (to C.-J.L.), the Canada Foundation for Innovation, the FQRNT Center in Green Chemistry and Catalysis, the Natural Sciences and Engineering Research Council of Canada, and McGill University for supporting our research. We would like to acknowledge the McGill Chemistry Facility for their contribution to this work, specifically, Robin Stein on the NMR characterisation and Alexander Wahba on HRMS characterisation, and Weihua Wang for Xe lamp maintenance. We would also like to thank our group members and colleagues for their generous help in polishing the manuscript.
Author contributions
C.-J.L. and J.L. designed the project. C.-Y.H. performed the experiments. J.L. checked and repeated the protocols. C.J.L. supervised the project. C.Y.H., J.L., and C.J.L. wrote the manuscript.
Data availability
Most data generated or analysed during this study are included in this published article and its Supplementary Information. All data are available from the author(s) upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Chia-Yu Huang, Jianbin Li.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-021-24280-9.
References
- 1.Buzzetti L, Prieto A, Roy SR, Melchiorre P. Radical-based C–C bond-forming processes enabled by the photoexcitation of 4-alkyl-1,4-dihydropyridines. Angew. Chem. Int. Ed. 2017;56:15039–15043. doi: 10.1002/anie.201709571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garza-Sanchez RA, Tlahuext-Aca A, Tavakoli G, Glorius F. Visible light-mediated direct decarboxylative C–H functionalization of heteroarenes. ACS Catal. 2017;7:4057–4061. doi: 10.1021/acscatal.7b01133. [DOI] [Google Scholar]
- 3.Liu P, Liu W, Li C-J. Catalyst-free and redox-neutral innate trifluoromethylation and alkylation of aromatics enabled by light. J. Am. Chem. Soc. 2017;139:14315–14321. doi: 10.1021/jacs.7b08685. [DOI] [PubMed] [Google Scholar]
- 4.Liu W, Liu P, Lv L, Li C-J. Metal-free and redox-neutral conversion of organotrifluoroborates into radicals enabled by visible light. Angew. Chem. Int. Ed. 2018;57:13499–13503. doi: 10.1002/anie.201807181. [DOI] [PubMed] [Google Scholar]
- 5.Proctor RSJ, Davis HJ, Phipps RJ. Catalytic enantioselective Minisci-type addition to heteroarenes. Science. 2018;360:419–422. doi: 10.1126/science.aar6376. [DOI] [PubMed] [Google Scholar]
- 6.Silvi M, et al. Visible-light excitation of iminium ions enables the enantioselective catalytic beta-alkylation of enals. Nat. Commun. 2017;9:868–873. doi: 10.1038/nchem.2748. [DOI] [PubMed] [Google Scholar]
- 7.Spinnato D, et al. A photochemical organocatalytic strategy for the alpha-alkylation of ketones by using radicals. Angew. Chem. Int. Ed. 2020;59:9485–9490. doi: 10.1002/anie.201915814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, et al. Bromide-promoted visible-light-induced reductive Minisci reaction with aldehydes. ACS Catal. 2020;10:154–159. doi: 10.1021/acscatal.9b04411. [DOI] [Google Scholar]
- 9.Yu X-Y, et al. A visible-light-driven iminyl radical-mediated C–C single bond cleavage/radical addition cascade of oxime esters. Angew. Chem. Int. Ed. 2018;57:738–743. doi: 10.1002/anie.201710618. [DOI] [PubMed] [Google Scholar]
- 10.Zheng D, Studer A. Asymmetric synthesis of heterocyclic γ‐amino‐acid and diamine derivatives by three‐component radical cascade reactions. Angew. Chem. Int. Ed. 2019;131:15950–15954. doi: 10.1002/ange.201908987. [DOI] [PubMed] [Google Scholar]
- 11.Zuo Z, et al. Merging photoredox with nickel catalysis: coupling of alpha-carboxyl sp3-carbons with aryl halides. Science. 2014;345:437–440. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuo Z, MacMillan DW. Decarboxylative arylation of alpha-amino acids via photoredox catalysis: a one-step conversion of biomass to drug pharmacophore. J. Am. Chem. Soc. 2014;136:5257–5260. doi: 10.1021/ja501621q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi H, et al. Recent advances in radical C–H activation/radical cross-coupling. Chem. Rev. 2017;117:9016–9085. doi: 10.1021/acs.chemrev.6b00620. [DOI] [PubMed] [Google Scholar]
- 14.Blanksby SJ, Ellison GB. Bond dissociation energies of organic molecules. Acc. Chem. Res. 2003;36:255–263. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- 15.Jeffrey JL, Terrett JA, MacMillan DW. O–H hydrogen bonding promotes H-atom transfer from alpha C–H bonds for C-alkylation of alcohols. Science. 2015;349:1532–1536. doi: 10.1126/science.aac8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishida N, et al. Carboxylation of benzylic and aliphatic C–H bonds with CO2 induced by light/ketone/nickel. J. Am. Chem. Soc. 2019;141:19611–19615. doi: 10.1021/jacs.9b12529. [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki T, Ishida N, Murakami M. Dehydrogenative coupling of benzylic and aldehydic C–H bonds. J. Am. Chem. Soc. 2020;142:3366–3370. doi: 10.1021/jacs.9b13920. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, et al. LiBr-promoted photoredox Minisci-type alkylations of quinolines with ethers. Adv. Synth. Catal. 2019;361:5643–5647. doi: 10.1002/adsc.201901168. [DOI] [Google Scholar]
- 19.Shaw MH, et al. Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science. 2016;352:1304–1308. doi: 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu A, Guo J-J, Pan H, Zuo Z. Selective functionalization of methane, ethane, and higher alkanes by cerium photocatalysis. Science. 2018;361:668–672. doi: 10.1126/science.aat9750. [DOI] [PubMed] [Google Scholar]
- 21.Sarver PJ, et al. The merger of decatungstate and copper catalysis to enable aliphatic C(sp3)–H trifluoromethylation. Nat. Commun. 2020;12:459–467. doi: 10.1038/s41557-020-0436-1. [DOI] [PubMed] [Google Scholar]
- 22.Shen Y, Gu Y, Martin R. sp3 C–H arylation and alkylation enabled by the synergy of triplet excited ketones and nickel catalysts. J. Am. Chem. Soc. 2018;140:12200–12209. doi: 10.1021/jacs.8b07405. [DOI] [PubMed] [Google Scholar]
- 23.Du J, Chen Z, Chen C, Meyer TJ. A half-reaction alternative to water oxidation: chloride oxidation to chlorine catalyzed by silver ion. J. Am. Chem. Soc. 2015;137:3193–3196. doi: 10.1021/jacs.5b00037. [DOI] [PubMed] [Google Scholar]
- 24.Heitz DR, Tellis JC, Molander GA. Photochemical nickel-catalyzed C–H arylation: synthetic scope and mechanistic investigations. J. Am. Chem. Soc. 2016;138:12715–12718. doi: 10.1021/jacs.6b04789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawasaki T, Ishida N, Murakami M. Photoinduced specific acylation of phenolic hydroxy groups with aldehydes. Angew. Chem. Int. Ed. 2020;59:18267–18271. doi: 10.1002/anie.202008897. [DOI] [PubMed] [Google Scholar]
- 26.Niu L, et al. Visible light-induced direct alpha C–H functionalization of alcohols. Nat. Commun. 2019;10:467. doi: 10.1038/s41467-019-08413-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng H-P, Zhou Q, Wu J. Microtubing-reactor-assisted aliphatic C−H functionalization with HCl as a hydrogen-atom-transfer catalyst precursor in conjunction with an organic photoredox catalyst. Angew. Chem. Int. Ed. 2018;57:12661–12665. doi: 10.1002/anie.201804844. [DOI] [PubMed] [Google Scholar]
- 28.Ohkubo K, Fujimoto A, Fukuzumi S. Metal-free oxygenation of cyclohexane with oxygen catalyzed by 9-mesityl-10-methylacridinium and hydrogen chloride under visible light irradiation. Chem. Commun. 2011;47:8515–8517. doi: 10.1039/c1cc12534f. [DOI] [PubMed] [Google Scholar]
- 29.Ohkubo K, Mizushima K, Fukuzumi S. Oxygenation and chlorination of aromatic hydrocarbons with hydrochloric acid photosensitized by 9-mesityl-10-methylacridinium under visible light irradiation. Res. Chem. Intermed. 2013;39:205–220. doi: 10.1007/s11164-012-0643-5. [DOI] [Google Scholar]
- 30.Rohe S, Morris AO, McCallum T, Barriault L. Hydrogen atom transfer reactions via photoredox catalyzed chlorine atom generation. Angew. Chem. Int. Ed. 2018;57:15664–15669. doi: 10.1002/anie.201810187. [DOI] [PubMed] [Google Scholar]
- 31.Ackerman LKG, Martinez Alvarado JI, Doyle AG. Direct C–C bond formation from alkanes using Ni-photoredox catalysis. J. Am. Chem. Soc. 2018;140:14059–14063. doi: 10.1021/jacs.8b09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng H-P, et al. Photoinduced nickel-catalyzed chemo- and regioselective hydroalkylation of internal alkynes with ether and amide α-hetero C(sp3)–H bonds. J. Am. Chem. Soc. 2017;139:13579–13584. doi: 10.1021/jacs.7b08158. [DOI] [PubMed] [Google Scholar]
- 33.Lian P, et al. Visible-light-induced vicinal dichlorination of alkenes through LMCT excitation of CuCl2. Angew. Chem. Int. Ed. 2020;59:23603–23608. doi: 10.1002/anie.202010801. [DOI] [PubMed] [Google Scholar]
- 34.Nielsen MK, et al. Mild, redox-neutral formylation of aryl chlorides through the photocatalytic generation of chlorine radicals. Angew. Chem. Int. Ed. 2017;56:7191–7194. doi: 10.1002/anie.201702079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shields BJ, Doyle AG. Direct C(sp3)–H cross coupling enabled by catalytic generation of chlorine radicals. J. Am. Chem. Soc. 2016;138:12719–12722. doi: 10.1021/jacs.6b08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Treacy SM, Rovis T. Copper catalyzed C(sp3)–H bond alkylation via photoinduced ligand-to-metal charge transfer. J. Am. Chem. Soc. 2021;143:2729–2735. doi: 10.1021/jacs.1c00687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu P, Chen P-Y, Xu H-C. Scalable photoelectrochemical dehydrogenative cross-coupling of heteroarenes with aliphatic C–H bonds. Angew. Chem. Int. Ed. 2020;59:14275–14280. doi: 10.1002/anie.202005724. [DOI] [PubMed] [Google Scholar]
- 38.Shu C, Noble A, Aggarwal VK. Metal-free photoinduced C(sp3)-H borylation of alkanes. Nature. 2020;586:714–719. doi: 10.1038/s41586-020-2831-6. [DOI] [PubMed] [Google Scholar]
- 39.Proctor RSJ, Phipps RJ. Recent advances in Minisci-type reactions. Angew. Chem. Int. Ed. 2019;58:13666–13699. doi: 10.1002/anie.201900977. [DOI] [PubMed] [Google Scholar]
- 40.Girard SA, Knauber T, Li C-J. The cross-dehydrogenative coupling of C(sp3)-H bonds: a versatile strategy for C–C bond formations. Angew. Chem. Int. Ed. 2014;53:74–100. doi: 10.1002/anie.201304268. [DOI] [PubMed] [Google Scholar]
- 41.Li C-J. Cross-dehydrogenative coupling (CDC): exploring C–C bond formations beyond functional group transformations. Acc. Chem. Res. 2009;42:335–344. doi: 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]
- 42.Li L, et al. Photo-induced iodination of aryl halides under very mild conditions. Nat. Protoc. 2016;11:1948–1954. doi: 10.1038/nprot.2016.125. [DOI] [PubMed] [Google Scholar]
- 43.Li L, et al. Photo-induced metal-catalyst-free aromatic Finkelstein reaction. J. Am. Chem. Soc. 2015;137:8328–8231. doi: 10.1021/jacs.5b03220. [DOI] [PubMed] [Google Scholar]
- 44.Li L, et al. Simple and clean photoinduced aromatic trifluoromethylation reaction. J. Am. Chem. Soc. 2016;138:5809–5812. doi: 10.1021/jacs.6b02782. [DOI] [PubMed] [Google Scholar]
- 45.Liu W, Li J, Huang C-Y, Li C-J. Aromatic chemistry in the excited state: facilitating metal-free substitutions and cross-couplings. Angew. Chem. Int. Ed. 2020;59:1786–1796. doi: 10.1002/anie.201909138. [DOI] [PubMed] [Google Scholar]
- 46.Liu W, Li J, Querard P, Li C-J. Transition-metal-free C–C, C–O, and C–N cross-couplings enabled by light. J. Am. Chem. Soc. 2019;141:6755–6764. doi: 10.1021/jacs.9b02684. [DOI] [PubMed] [Google Scholar]
- 47.Liu W, Yang X, Gao Y, Li C-J. Simple and efficient generation of aryl radicals from aryl triflates: synthesis of aryl boronates and aryl iodides at room temperature. J. Am. Chem. Soc. 2017;139:8621–8627. doi: 10.1021/jacs.7b03538. [DOI] [PubMed] [Google Scholar]
- 48.Liu W, Yang X, Zhou Z-Z, Li C-J. Simple and clean photo-induced methylation of heteroarenes with MeOH. Chem. 2017;2:688–702. doi: 10.1016/j.chempr.2017.03.009. [DOI] [Google Scholar]
- 49.Mariano PS. Electron-transfer mechanisms in photochemical transformations of iminium salts. Acc. Chem. Res. 1983;16:130–137. doi: 10.1021/ar00088a003. [DOI] [Google Scholar]
- 50.McCallum T, et al. The photochemical alkylation and reduction of heteroarenes. Chem. Sci. 2017;8:7412–7418. doi: 10.1039/C7SC03768F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao H, et al. Photoinduced site-selective alkenylation of alkanes and aldehydes with aryl alkenes. Nat. Commun. 2020;11:1956. doi: 10.1038/s41467-020-15878-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McManus JB, Griffin JD, White AR, Nicewicz DA. Homobenzylic oxygenation enabled by dual organic photoredox and cobalt catalysis. J. Am. Chem. Soc. 2020;142:10325–10330. doi: 10.1021/jacs.0c04422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.U. Dighe S, et al. A photochemical dehydrogenative strategy for aniline synthesis. Nature. 2020;584:75–81. doi: 10.1038/s41586-020-2539-7. [DOI] [PubMed] [Google Scholar]
- 54.Yi H, et al. Photocatalytic dehydrogenative cross-coupling of alkenes with alcohols or azoles without external oxidant. Angew. Chem. Int. Ed. 2017;56:1120–1124. doi: 10.1002/anie.201609274. [DOI] [PubMed] [Google Scholar]
- 55.Zhao H, Leonori D. Minimization of back-electron transfer enables the elusive sp3 C–H functionalization of secondary anilines. Angew. Chem. Int. Ed. 2021;60:7669–7674. doi: 10.1002/anie.202100051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kariofillis SK, Doyle AG. Synthetic and mechanistic implications of chlorine photoelimination in nickel/photoredox C(sp3)–H cross-coupling. Acc. Chem. Res. 2021;54:988–1000. doi: 10.1021/acs.accounts.0c00694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fu N, Sauer GS, Lin S. Electrocatalytic radical dichlorination of alkenes with nucleophilic chlorine sources. J. Am. Chem. Soc. 2017;139:15548–15553. doi: 10.1021/jacs.7b09388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Most data generated or analysed during this study are included in this published article and its Supplementary Information. All data are available from the author(s) upon reasonable request.