

1. Introduction
Genetic epilepsy with febrile seizures plus (GEFS+) is a complex autosomal dominant and sporadic epilepsy syndrome characterized by the presence of a variety of clinical seizure types including childhood febrile seizures and later development of afebrile seizures. GEFS+ is usually diagnosed with a heterogeneous spectrum of seizure types in several members from different generations, and the GEFS+ seizure types vary among affected family members (Scheffer and Berkovic, 1997). Febrile seizures associated with a high fever begin in infancy and usually stop by age 6. GEFS+ seizures usually remit by the time the child reaches the age of 10 or 12 but can persist beyond this age, even into adulthood. Patients with GEFS+ may also develop other seizure types, such as absence, myoclonic, atonic (drop attacks), focal (partial), and generalized tonic-clonic seizures (GTCS) (Singh et al., 1999). Ictal electroencephalogram (EEG) patterns are dictated by seizure types, whereas interictal EEGs usually show normal background with generalized spike-wave discharges. Most individuals with GEFS+ have normal cognition and few behavioral comorbidities.
The etiopathology of GEFS+ is complex with strong evidence for a genetic predisposition. Mutations in several genes have been identified to be associated with GEFS+ (Baulac et al., 2001; Parihar and Ganesh, 2013; Macdonald et al., 2010; Carvill et al., 2014; Le et al., 2017). The most commonly associated genes are the sodium channel subunit genes (SCN1A, SCN1B, SCN2A), GABAA receptor subunit genes (GABRA1, GABRG2, GABRB3, GABRD) and several nonchannel genes (PCDH19, STXBP1). To better understand the molecular basis of GEFS+ in humans, we developed a mouse model harboring a human mutation (c.983A > T, p. Lys328Met (K328M)) in the GABAA receptor γ2 subunit gene (GABRG2). The K328M mutation was one of the first mutations identified in a GABR gene (GABRG2) in a large French family in which multiple individuals displayed GEFS+ seizures (Baulac et al., 2001).
GABAA receptors are the primary mediators of fast inhibitory synaptic neurotransmission in the central nervous system. They are pentameric, ligand-gated chloride channels that are assembled from a panel of 19 subunits (α1-α6, β1-β3, γ1-γ3, δ, ε, π, θ, and ρ1-ρ3) and are most commonly composed of 2α, 2β, and 1γ subunits in the mammalian brain (Barnard et al., 1998). Expression of γ2 subunits is widespread and abundant in the embryonic and neonatal brain, especially in the cerebral cortex, hippocampus, and other regions involved in generating seizures (Laurie et al., 1992; Hortnagl et al., 2013). The γ2 subunit plays an important role during brain development and has a critical role in GABAA receptor trafficking and clustering at synapses. Dysfunction of GABAA receptors can lead to a variety of seizure types (Macdonald et al., 2010). GABRG2 mutations have been associated with febrile seizures as well as a spectrum of epilepsy syndromes including childhood absence epilepsy (CAE), GEFS+ , Dravet syndrome, and myoclonic astatic epilepsy (MAE) (Kang and Macdonald, 2016).
The amino acid residue K328 is in the highly conserved extracellular region between membrane-spanning domains M2 and M3 (the M2/M3 loop) of the GABAA receptor γ2 subunit. The effects of the mutant γ2 (K328M) subunit on GABAA receptor functions were examined in in vitro studies using brief rapid application of GABA to lifted transfected HEK293T cells. Mutant γ2(K328M) subunit-containing α1β2γ2 receptors were shown to have accelerated deactivation and unchanged amplitude (Bianchi et al., 2002; Hales et al., 2006). These results were consistent with reports from cultured hippocampal neurons, that the presence of the mutant γ2(K289M, corresponding to the K328M in the immature peptide) subunit accelerated deactivation of synaptic currents without affecting peak currents, surface expression, or localization of GABAA receptor γ2 subunits at synaptic or extrasynaptic sites in transfected hippocampal neurons, or the amplitude of either synaptic or tonic GABAA receptor currents (Eugene et al., 2007).
It has been shown that membrane expression of γ2(K328M) subunits may rapidly decrease on exposure to elevated temperature (Kang et al., 2006), which is consistent with another study demonstrating that both the number of GABAA receptor clusters and the frequency of miniature inhibitory postsynaptic currents (mIPSCs) were decreased with elevation of temperature in hippocampal neurons expressing γ2(K328M) subunits (Bouthour et al., 2012). These observations were not replicated in another study, however, which reported that the K328M mutation did not affect trafficking or aggregation of GABAA receptor γ2(K328M) subunits at synaptic or extrasynaptic sites in transfected hippocampal neurons (Eugene et al., 2007). Although these in vitro studies provided very valuable information for uncovering the underlying mechanisms for the mutant γ2(K328M) subunit causing febrile seizures, both transfected heterologous HEK293T cells and cultured neurons have limitations. HEK cells lack synaptic specializations making them less relevant for study of synaptic GABA signaling, whereas transfected cultured neurons suffer from nonbiological stoichiometric ratios of receptor subunits that may not reflect the true nature of heterozygosity in febrile seizure patients. Thus, the causal role of γ2(K328M) can only be established directly by determining its effect on neurons in brain.
To understand better how the GABRG2 K328M mutation causes epilepsy in patients, we generated and characterized a knock-in (KI) mouse model, the heterozygous Gabrg2+/K328M mouse, corresponding to the heterozygous GABRG2 983A > T mutation in humans with GEFS+. We showed that Gabrg2+/K328M mice recapitulated most features of GEFS+ patients, including febrile seizures and afebrile seizures with multiple seizure semiologies, a distinctive generalized slow spike-and-wave EEG discharge pattern, and associated mild behavioral comorbidities.
2. Results
2.1. Generation of Gabrg2+/K328MKI mice
To study the effects of the human GEFS+ GABRG2 983A > T mutation, on GABAA receptors in vivo, we generated mice with a targeted 983A > T mutation in GABRG2 at the endogenous Gabrg2 locus using conventional homologous recombination in embryonic stem (ES) cells derived from the C57BL/6 J mouse (Fig. 1A). The correctly targeted ES cell clones were identified by long-range PCR with one of the primers (A or D) located outside of the homologous arms to exclude clones with random vector insertion (Fig. 1A). Chimeric mice were bred with Flpe transgenic mice to screen for germline transmission and to excise the PGK-Neo selection cassette. Upon Cre-mediated recombination, exon 8 was removed and replaced by exon 8* with the 983A > T mutation, resulting in generation of the K328M KI mouse, Gabrg2+/K328M (for details and rationale, see methods). The leftover FRT and loxP sites allowed for convenient genotyping (Fig. 1B). Presence of the 983A > T mutation was confirmed by PCR/sequencing of the targeted allele.
Fig. 1. Generation of the Gabrg2+/K328M KI mice.
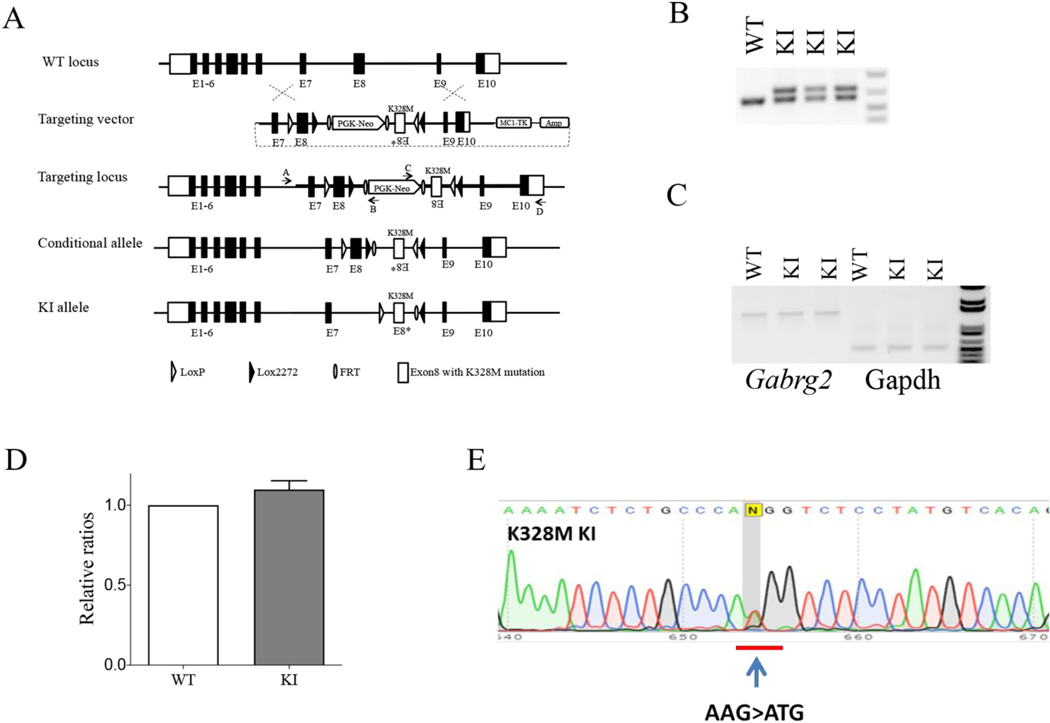
A. Scheme of the targeting strategy for generating K328M KI mice. With the intention to study its developmental impact on GEFS+, a KI vector was constructed to generate a conditional KI mouse. Homologous recombination of the targeting vector at the Gabrg2 gene locus resulted in a “conditional KI allele” in which the Gabrg2 exon 8 genomic DNA was replaced by the exon 8, neomycin-resistance gene cassette (PGK-Neo) and an inverted exon 8* containing 983A > T mutation. The PGK-Neo was removed by breeding with Flpe mice to generate mice with “conditional allele”, then were bred with Cre mice to generate mice with KI allele. B. Genotyping of KI mice. PCR amplified a 605 bp fragment from the WT Gabrg2 allele and a 689 bp fragment from the mutant Gabrg2 KI allele due to the insertion of FRT, loxP sequences and other modifications. C. Semi-quantitative RT-PCR showed that the KI allele did not affect Gabrg2 gene expression, using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control. D. Band intensities of the RT-PCR products were first normalized to GAPDH for quantification, and then to WT levels (P > 0.05, n-3 with bars referring to mean ± SEM). E. Sequencing chromatogram of RT-PCR derived from total brain RNA showed the presence of the 983A > T mutation and corresponding equal level of expression.
An optimal KI mouse model must satisfy the key requirement that the KI allele does not alter its expression. To determine if the 983A > T mutation and other modifications, such as insertion of FRT and loxP sites in the Gabrg2 locus, affected transcription of Gabrg2, semi-quantitative RT-PCR was carried out with total RNAs isolated from whole brain. The results revealed that expression levels of Gabrg2 mRNA from whole brain were at the same levels in KI mice and wild-type (WT) littermates (Fig. 1C, D). The sequencing chromatogram of the RT-PCR products of total RNA isolated from the KI mice showed the presence of the 983A > T mutation and a similar intensity of peak A (WT mRNA) and peak T (mutant mRNA) nucleotides (Fig. 1E), which was consistent with the chromatogram pattern derived from template with equal amounts of PCR products harboring A and T at position 983 (data not shown). These results indicated that expression levels of Gabrg2 mRNA from whole brain were the same in KI and WT littermates. Additionally, sequence analysis using RT-PCR included the entire coding region of the Gabrg2 transcript from the Gabrg2+/K328M mouse brain confirmed there were no other unintended DNA sequence changes.
2.2. Gabrg2+/K328M KI mice had multiple seizure semiologies consistent with GEFS+
Having generated Gabrg2+/K328M KI mice, we determined whether they showed a similar GEFS+ syndrome phenotype by using systematic home cage observation and synchronized video-EEG analyses. The clinical phenotype of patients with GEFS+ is diverse and ranges from mild, familial febrile seizures to afebrile GTCS as well as other seizure types, including absence, myoclonic, and atonic seizures. The GEFS+ syndrome has also been associated with Dravet syndrome and Doose syndrome (Scheffer and Berkovic, 1997; Singh et al., 1999). We observed multiple types of spontaneous seizures with variable severity in the KI mice (see supplementary video), but not in their WT littermates (except for infrequent, brief atypical absence-like seizures, as is expected in C57BL6 mice (Arain et al., 2012) (Reid et al., 2011) (Letts et al., 2014).
In adult KI mice, multiple seizure types were observed and the most common ones include atypical absence, typical absence, and myoclonic seizures (Fig. 2A). Atypical absence seizures were associated with behavioral arrest that usually lasted from a few seconds to several minutes. They had irregular ictal high-amplitude SWDs (3–5 Hz) that were not always synchronized with the onset of behavioral arrest (Fig. 2B, Video), as is consistent with atypical absence seizures (Cortez et al., 2001). Typical absence seizures were also seen, generally lasting for a few seconds, and were associated with symmetrical, high-amplitude spike-wave discharges (SWDs) with rhythmic 6–8 Hz on ictal EEG. The onset was accompanied by time-locked EEG activity and sudden behavioral arrest (Fig. 2C, Video), as is consistent with typical absence seizures (Letts et al., 2014; Jarre et al., 2017). On average, K328M mice had 56 atypical and 12 typical absence seizures per day, indicating a 5-fold higher occurrence of atypical absence seizures. Myoclonic seizures were also observed, but they were very subtle and infrequent in adults. They entailed a sudden, brief muscle contraction accompanied by a single, high-amplitude sharp wave on EEG (Fig. 2D, Video). GTCS were not observed in the adult KI mice but were observed in pre-weaned KI pups (Video). Without Video-EEG, however, we can’t exclude the possibility that younger mice may have other seizure types. In general, afebrile absence, atypical absence, and myoclonic seizures were observed in adult Gabrg2+/K328M KI mice, and GTCS progressively disappeared in the adulthood. Taken together, these seizure semiologies were consistent with those in humans with GEFS+.
Fig. 2. Gabrg2+/K328M KI mice had spontaneous seizures.
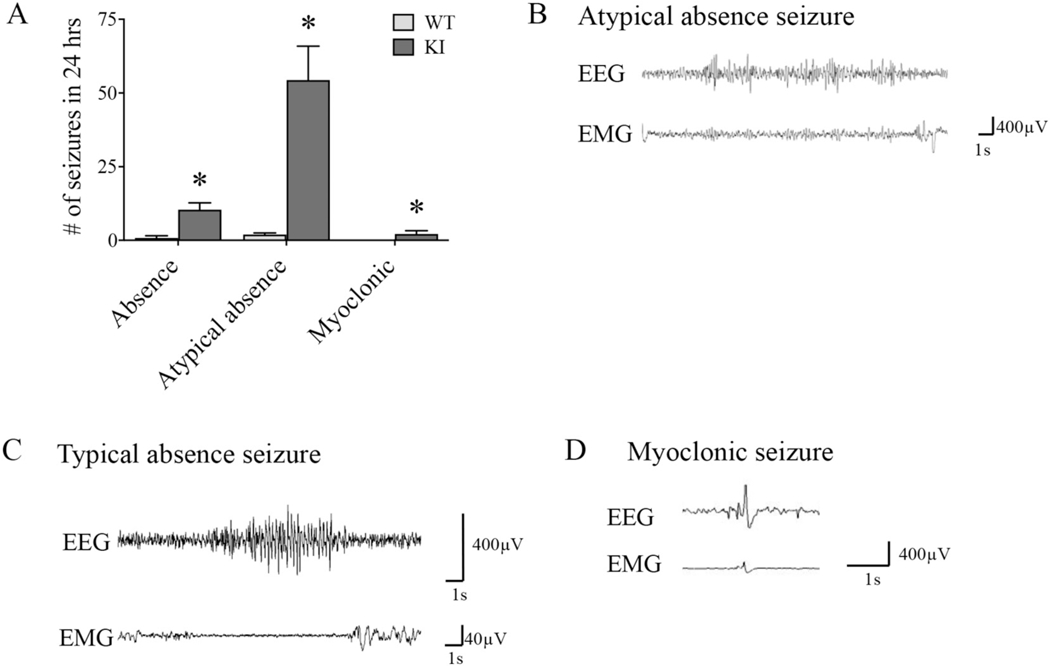
A. Average number of seizures in Gabrg2+/K328M KI (n = 11) and WT (n = 4) mice in a 24-h period. Representative EEG traces for B. atypical absence, C. typical absence, D. myoclonic seizures seen in KI mice. Typical absence seizures were time-locked with behavioral arrest, while prolonged atypical absence seizures were not always time-locked with the behavioral onset and termination. Myoclonic seizures were very brief and had spikes in both EEG and electromyogram (EMG) channels. Wildtype mice exhibits a low baseline incidence of spontaneous SWDs as previously reported. See supplementary video for examples of KI video-EEG recordings for each seizure type.
2.3. Gabrg2+/K328M KI mice displayed a reduced the threshold for pentylenetetrazol (PTZ)-induced seizures
To examine whether mutant γ2(K328M) subunits affect seizure susceptibility, we compared PTZ-induced seizure threshold for Gabrg2+/K328M KI mice to their WT littermates. PTZ is a non-competitive GABAA receptor antagonist that has been used as a central nervous system stimulant. One-month-old mice were injected intraperitoneally with varying doses of PTZ, and the latencies to myoclonic seizures and GTCS were recorded. We found that at a dose of 30 mg/kg, PTZ induced myoclonic and GTCS seizures in Gabrg2+/K328M mice compared to absence-like seizures in most of WT mice, whereas a dose of 60 to 80 mg/kg PTZ was required to induce GTCS or seizure-related death in WT mice. Survival curves for PTZ-induced myoclonic seizures and GTCS were plotted (Fig. 3A, B) and showed that Gabrg2+/K328M mice had a lower seizure threshold than WT littermates. These results indicated that mutant γ2(K328M) subunits affected brain network excitability and significantly lowered the PTZ-induced seizure threshold.
Fig. 3.
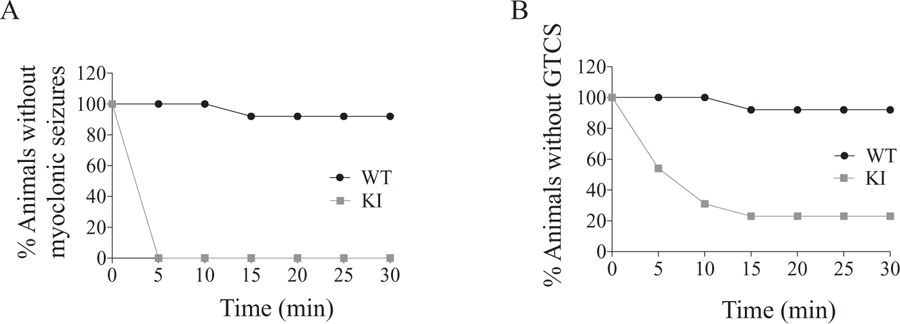
Gabrg2+/K328M mice had lowered threshold to pentylenetetrazol-induced seizures. The KI mice and their WT littermates were injected intraperitoneally with pentylenetetrazol (PTZ) at a dose of 30 mg/kg. The susceptibility to PTZ-induced myoclonic seizures and GTCS were assessed by survival curves. The Mantel-Cox method was used to analyze the difference between WT and KI mice. A. Myoclonic seizures were observed in 13 of 13 KI mice and 1 of 13 WT mice within 30 min of drug administration (Mantel-Cox p < 0.0001). B. GTCS were observed in 10 of 13 KI mice and 1 of 13 WT mice had GTCS within 30 min of drug administration. (Mantel-Cox p = 0.0002).
2.4. Gabrg2+/K328M KI pre-weaned mice had premature sudden death
Sudden Unexplained Death in EPilepsy (SUDEP) is commonly observed in patients with epilepsy. We first observed that many pups died in their home cage prior to weaning. In the first two-week postnatal period, sporadic deaths for WT and KI pups occurred at similar rates (mostly observed on the first day after birth); however, in the third post-natal week (especially from P15-P17) KI mice died at a much higher rate (14%, 35 died vs 210 survived) than WT mice (0.4%, 1 died vs 243 survived), consistent with SUDEP in humans (Supplemental Fig. 1). Systematic home cage video monitoring showed that the dead mice had frequent GTCS before their death. The GTCS and sudden death resolved mostly at ages older than P17.
2.5. Gabrg2+/K328M KI mice had age-dependent hyperthermic seizures
The GABRG2 K328M mutation was originally identified in a large French family composed of 17 affected members, of which 13 had histories of febrile seizures. We thus examined whether elevated temperature caused hyperthermic seizures in Gabrg2+/K328M KI mice. To evaluate hyperthermic seizures, we put KI mice and WT littermates in an incubator at 42 °C for 30 min. The presence of and latencies to myoclonic seizures and GTCS were determined. Since EEG was not used to detect hyperthermia-induced seizures, some seizures without convulsive movements would have been missed.
Almost none of the WT mice (n = 1/113, 0.9%) had hyperthermic myoclonic seizures or GTCS at any age tested, whereas Gabrg2+/K328M KI mice had hyperthermic myoclonic seizures and GTCS in an age-dependent fashion (Fig. 4, video).
Fig. 4.
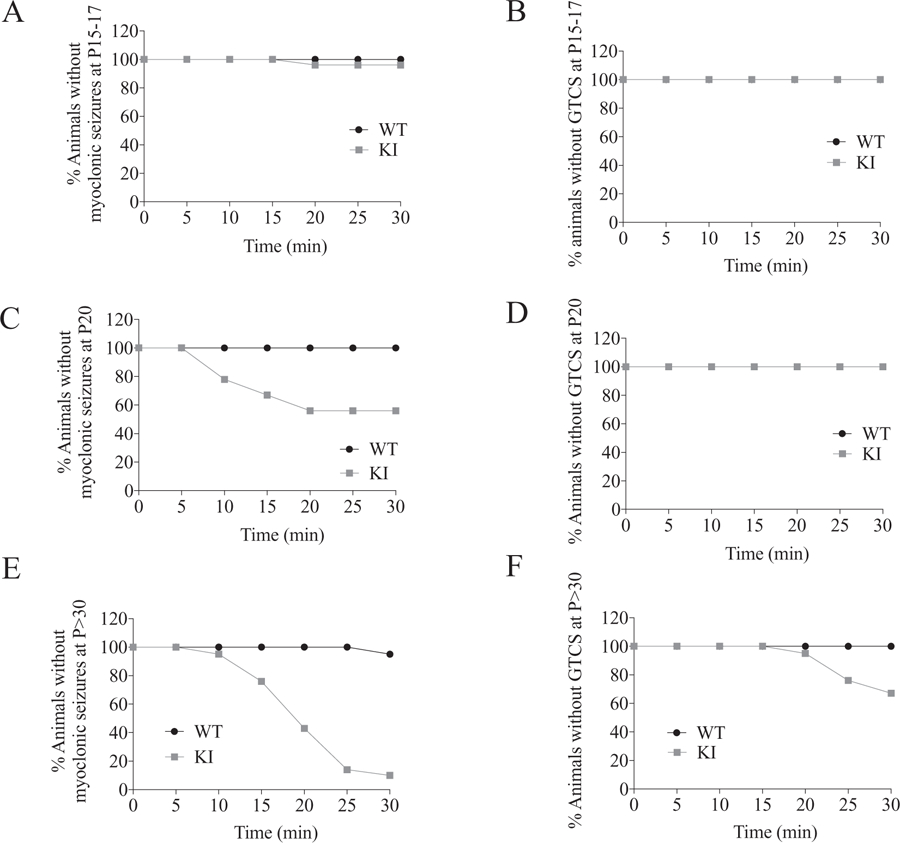
Gabrg2+/K328M KI mice had age-dependent hyperthermic seizures. Mice were placed in a 42 °C incubator for 30 min. The percentage of mice remaining free of myoclonic seizures (A, C, E) or GTCS (B, D, F) during hyperthermia to 42 °C was plotted against duration of hyperthermia. The Mantel-Cox method was used to analyze the difference between WT and KI mice. A. For P15–17 mice, myoclonic seizures were observed in 1 of 27 KI mice and none of 24 WT mice within 30 min of hyperthermia (n = 24 WT and 27 KI, p = 0.34). B. No GTCS was observed for both KI and WT mice. C. For P20 mice, myoclonic seizures were observed in 4 of 9 KI mice and none of 13 WT mice (n = 13 WT and 9 KI, p = 0.008). D. No GTCS was observed for both KI and WT mice. E. For mice older than P30 mice, myoclonic seizures were observed in 19 of 21 KI mice and 1 of 21 WT mice (n = 21 WT and 21 KI, p < 0.0001). F. GTCS observed in 7 of 21 KI mice and none of 21 WT mice (n = 21 WT and 21 KI, p = 0.004).
Young KI mice (P15-P17) had minimal (n = 1/27, 3.7%) hyperthermic myoclonic seizures (Fig. 4A) and no (0/27, 0%) hyperthermic GTCS with 30 min of hyperthermia (Fig. 4B). P20 KI mice had frequent (n = 4/9, 44.4%) hyperthermic myoclonic seizures (Fig. 4C) and no (0/9, 0%) hyperthermic GTCS (Fig. 4D). In contrast, most P30 or older KI mice (n = 19/21, 90%) had hyperthermic myoclonic seizures (Fig. 4E), and many P30 and older KI mice (n = 7/21, 33%) had hyperthermic GTCS (Fig. 4F). These results demonstrated that the K328M mutation caused febrile seizures in an age-dependent manner and elevated temperature alone was sufficient to induce seizures in Gabrg2+/K328M KI mice at weaning ages, with a transition from afebrile to febrile seizures at ~P20.
2.6. Gabrg2+/K328M KI mice had hyperactivity and normal cognition
GEFS+ is a genetic disorder with heterogenous clinical phenotypes and varying severity from the most common and mildest simple febrile seizures to the most severe disorder, the epileptic encephalopathies Dravet syndrome and Doose syndrome. Some patients with GEFS+ have febrile seizures that persist in childhood as late as puberty, past the usual cutoff of 6 years old for simple febrile seizures. Patients with simple febrile seizures have normal cognition (with only rare long-term consequences), whereas Dravet and Doose patients usually have impaired cognition and behavioral problems including autism spectrum disorder. It has been difficult to precisely define the behavioral status of patients with GEFS+ or FS+ in humans bearing the GABRG2 K328M mutation due to the small number of patients and limited clinical data. Therefore, the mouse model of GEFS+ offers a unique opportunity to examine its behavioral comorbidities experimentally.
We carried out several behavioral studies in adult mice to examine potential effects of mutant γ2(K328M) subunits on anxiety, hyperactivity, socialization, and spatial learning/memory, and to assess whether behavioral comorbidities in Gabrg2+/K328M KI mice recapitulate those seen in GEFS+ patients. We found that Gabrg2+/K328M KI mice had a hyperactivity phenotype without other changes in cognition. In a 60-min open-field test, Gabrg2+/K328M KI mice traveled significantly farther than WT mice (Fig. 5A). Gabrg2+/K328M KI mice also had an increased number of vertical counts (or rearing) (Fig. 5B). Together, these data indicate KI mice were hyperactive, consistent with the report that attention-deficit/hyperactivity disorder (ADHD) is one of the most frequently diagnosed comorbidities of GEFS+ patients.
Fig. 5.
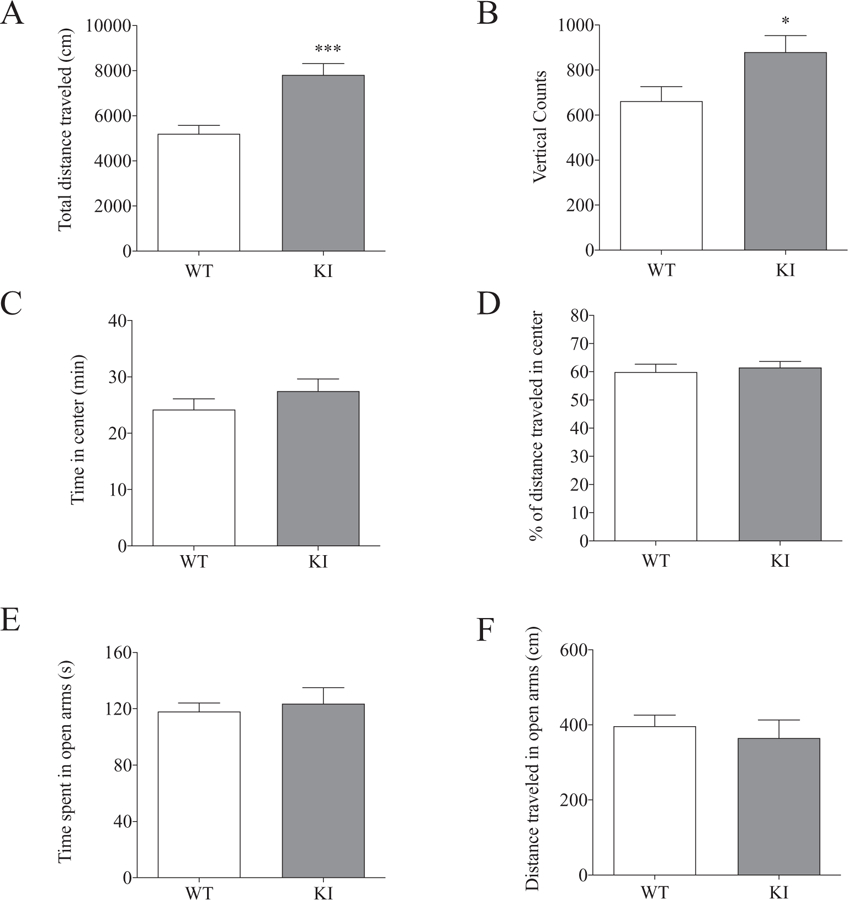
Gabrg2+/K328M KI mice exhibited hyperactivity but not anxiety. Hyperactivity was assessed using the open field test and KI mice had a hyperactive phenotype. Data was collected from 60 min in the locomotor activity chambers. A. KI mice traveled significantly greater distances (WT 5195 ± 376.5 cm, n = 10; KI 7807 ± 505.7 cm, n = 11; Student’s t-test, p = 0.001). B. KI mice had more vertical jumps than WT mice (WT 662 ± 65, n = 10; KI 879 ± 74, n = 11; p = 0.04). KI mice displayed no anxiety-like behaviors in both the open field test and the elevated zero maze. C, D. KI mice spent equal time in the center of the open field chamber (WT 24.2 ± 6.1 min, n = 10; KI 27.5 ± 7.2 min, n = 11, p = 0.27) and traveled the same distance in the center, as a percentage of total distance traveled, compared to WT littermates (WT 59.71 ± 2.71, n = 10; KI 62.31 ± 2.16, n = 11, p = 0.64). E, F. In the elevated zero maize, KI mice spent similar time in the open arms (WT 118.07 ± 6.07 s, n = 11; KI 123.64 ± 11.37 s, n = 11, p = 0.67) and traveled similar distance (WT 396.78 ± 29.51 cm, n = 11; KI 365.17 ± 48.06 cm, n = 11, p = 0.58) compared to WT littermates.
Gabrg2+/K328M KI mice spent an equal amount of time and traveled the same distance in the center (or in the perimeter) of the chamber (open-field test) compared to WT mice, suggesting no anxiety phenotype (Fig. 5C, D). We further confirmed this with the elevated zero maze test, which is widely used to assess anxiety-like behaviors in rodents. Gabrg2+/K328M KI and WT mice spent similar times and traveled similar distances in the open arms of the maze (Fig. 5E, F).
To evaluate whether Gabrg2+/K328M KI mice had cognitive deficits, their spatial learning and memory were examined with the Barnes maze. Both Gabrg2+/K328M KI mice and WT mice showed similar learning curves, represented as time to find the escape hole in the learning trials (Fig. 6A). These results demonstrated KI mice had normal spatial learning. After completing five days of learning trials, probe trials were performed to test how well the mice remembered the location of the escape hole. As expected from their learning curves, both Gabrg2+/K328M KI mice and WT mice spent similar times (escape latency) to find the target hole, which indicated that KI mice had no spatial memory deficit (Fig. 6B). In addition, Gabrg2+/K328M KI mice committed a similar numbers of errors by entering non-target hole zones compared to WT mice (Fig. 6C). These results clearly suggested that the Gabrg2+/K328M mice had normal spatial cognition.
Fig. 6.
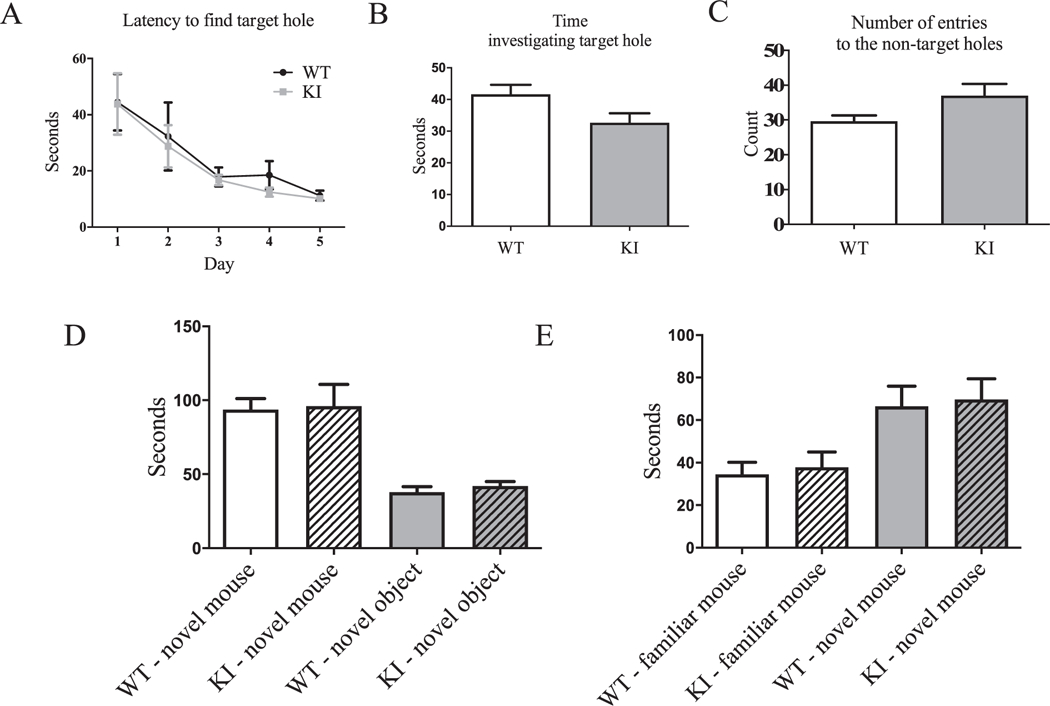
Gabrg2+/K328M KI mice had no spatial learning or memory deficits and had normal social interaction. The Barnes maze test showed normal spatial learning and memory in the KI mice compared to WT littermates. Five days of learning trials demonstrated KI mice have no spatial learning deficit. A. The time (latency) it takes each animal to find the target hole for each day was plotted. Two-way ANOVA for repeated measures showed no genotype effect (WT n = 11, KI n = 11). Five min probe trial for spatial memory performed on day 5 after the learning trials showed normal spatial memory in KI mice. B. Time spent in the vicinity of the covered target hole (WT 44.34 ± 3.24 s, n = 11; KI 34.47 ± 3.47 s, n = 11, Student’s t-test, p = 0.38). C. Number of non-target hole zones entered (errors) were plotted (WT 29.64 ± 1.69, n = 11; KI 37.00 ± 3.36, n = 11, p = 0.064). Values were expressed as mean ± SEM. D. The three-chamber socialization test showed normal sociability. Sociability: Like WT mice, KI mice also spent significantly more time investigating the novel mouse than novel object (Student’s t-test, p = 0.0018; novel mouse: WT 93.9 ± 7.31 s, KI 96.1 ± 14.74 s; novel object: WT 37.9 ± 3.65 s, KI 42.1 ± 2.94 s). Two-way ANOVA showed a no significant genotype effect (p = 0.91). E. Social novelty: As WT mice, KI mice preferred to investigate the novel mouse than the familiar mouse (Student’s t-test, p = 0.016; familiar mouse: WT 34.5 ± 5.70 s, KI 37.9 ± 7.2 s; novel mouse: WT 66.4 ± 9.51 s, KI 69.7 ± 9.72 s). Two-way ANOVA showed a no significant genotype effect (p = 0.69). N = 11 for both WT and KI mice.
We also studied whether Gabrg2+/K328M KI mice had deficits in social behavior (Moretti et al., 2005). We used the three-chamber socialization test to examine social interaction ability of Gabrg2+/K328M KI mice. Mice were tested for their preference between exploring a novel object or a novel mouse and a familiar mouse or a novel mouse. Both WT and Gabrg2+/K328M KI mice showed a clear preference for novel mouse over exploration of a novel object (Fig. 6D) and interacting with the novel mouse over the familiar mouse (Fig. 6E). Taken together, these results indicated that Gabrg2+/K328M KI mice had no abnormalities in socialization. In summary, Gabrg2+/K328M KI mice are hyperactive with normal socialization and cognition.
2.7. Expression and trafficking of GABAA receptor α1, β3, and γ2 subunits were unchanged in brains of Gabrg2+/K328M KI mice
GABRG2 is abundantly expressed in the mammalian brain, and γ2 subunits play a critical role in GABAA receptor trafficking and clustering at synapses. Impaired biogenesis and trafficking resulting in an alteration of cell surface expression was one of the mechanisms for genetic epilepsies associated with GABAA receptor mutations (Macdonald et al., 2010).
Having confirmed that KI mice had GEFS+ seizure semiologies and altered the PTZ-induced seizure threshold, we next examined the underlying mechanisms for the seizures. To determine if the effects of the mutant γ2(K328M) subunits on GABAA receptor-mediated currents were related to its expression and surface trafficking in vivo under normal or elevated temperature conditions (42 °C), total and surface proteins from adult mice were blotted for α1, β3, and γ2 subunits and quantified. The expression of GABAA receptor subunits in cerebellum, cortex, hippocampus, and thalamus of KI mice was similar to that of WT mice (Supplemental Fig. 2A, B). The surface expression of GABAA receptor subunits was evaluated by using the membrane fraction of whole brain tissue at room temperature or elevated (42 °C) temperature. The results showed no significant difference between KI and WT mice at room temperature and elevated temperature also did not alter their expression level significantly (Supplemental Fig. 2C, D). We further examined whether mutant γ2(K328M) subunits affected the synaptosomal distribution of GABAA receptor subunits by subcellular fractionation at room temperature or at elevated (42 °C) temperatures. The results showed that in synaptosomes, the amounts of α1, β3, and γ2 GABAA receptor subunits and gephyrin, a central GABAergic synapse organizer, were not significantly altered in KI mice compared to WT littermates at room temperature or at elevated (42 °C) temperatures (Supplemental Fig. 2E, F). These results demonstrated that mutant γ2(K328M) subunits did not affect GABAA receptor biogenesis, trafficking, or synaptic distribution, suggesting that the mutant γ2 subunits can effectively compete with WT subunits to form structurally intact, but functionally impaired, GABAA receptors in brain, ultimately leading to decreased GABAergic inhibition and seizures.
2.8. Gabrg2+/K328M KI mice had reduced mIPSC amplitudes recorded from somatosensory (SS) cortex layer V/VI pyramidal neurons
The γ2 subunit is an important constituent of GABAA receptors and is widely expressed in the brain, including in neural circuits involved in seizure generation such as the cortex, hippocampus, and thalamus (Laurie et al., 1992; Hortnagl et al., 2013). Previous in vitro results showed that the γ2(K328M) subunit altered macroscopic kinetic properties of GABAA receptor currents with an accelerated current deactivation in transfected HEK293T cells (Bianchi et al., 2002; Hales et al., 2006; Eugene et al., 2007). We investigated whether the γ2(K328M) subunit affected the functional properties of GABAergic synapses on cortical layer V/VI pyramidal neurons, which involve in the corticothalamic feedback loop (Ledergerber and Larkum, 2010). Using whole-cell patch-clamp recordings from acute coronal slices, we observed decreased GABAA receptor-mediated mIPSC amplitude, unchanged mIPSC frequency (WT 4.12 ± 1.11/s; KI 3.81 ± 0.51/s; p = 0.816) and decay time (WT 58.01 ± 18.06 msec; KI 57.56 ± 10.97 msec; p = 0.984) in the Gabrg2+/K328M KI mice (Fig. 7A-C). The reduced current amplitudes of GABAA receptors containing mutant γ2(K328M) subunits in layer V/VI SS cortical neurons would be predicted to decrease interneuron GABAergic inhibitory output and increase the excitability of downstream neuronal networks, resulting in epilepsy.
Fig. 7.
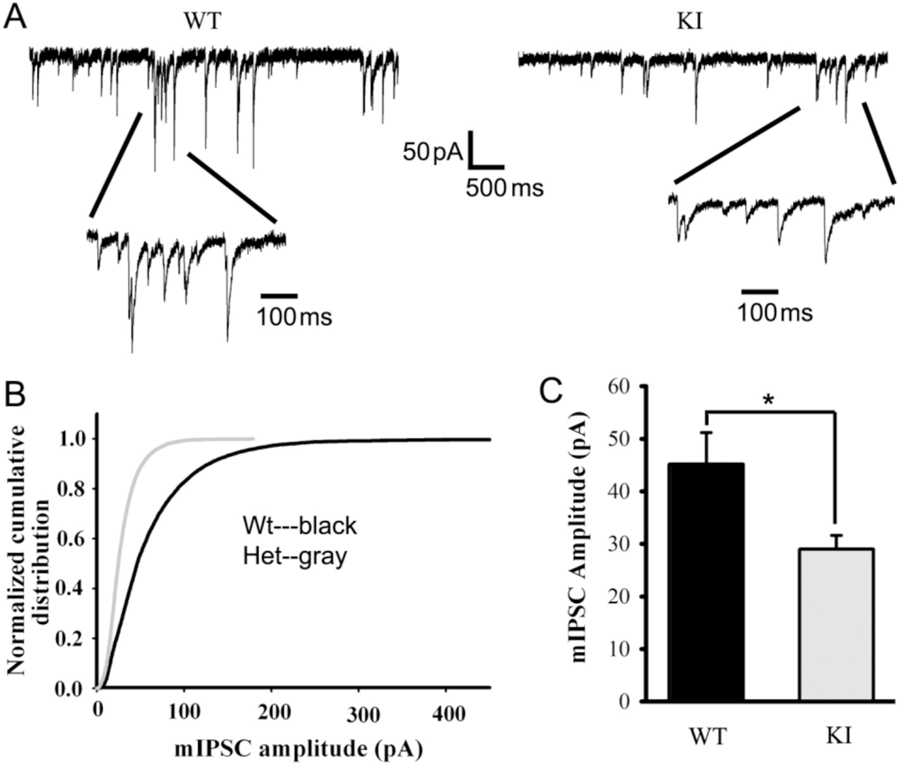
mIPSCs recorded from Gabrg2+/K328M KI mouse SS cortex layer V/VI neuron were altered. A. mIPSCs were recorded from SS cortex layer V/VI neurons (voltage-clamped at −60 mV with equal chloride concentration inside and outside cells) in thalamocortical slices from KI mice and WT littermates. The ACSF perfusion solution contained 20 μM NBQX and 1 μM TTX. B. Normalized cumulative distribution was plotted for WT and KI mouse mIPSCs. C. mIPSCs recorded from SS cortex layer V/VI neurons of WT mice were compared to those in KI mice, showing significantly reduced amplitudes (WT 45.11 ± 6.07 pA, n = 4 mice; KI 28.98 ± 2.62 pA, n = 5 mice. Data were presented as mean ± SEM. Student’s t-test p = 0.036).
The experimental results from in vitro and ex vivo experiments were quite different. The basis for the difference in effect of the mutant γ2 (K328M) subunits on GABAA receptor currents in vitro and in vivo is unclear. In the HEK cell experiments, GABAA receptor currents were recorded from receptors assembled from only α1, β3, and γ2 subunits on cells with no synapses while the currents in the brain slice experiments were recorded from receptors assembled from whichever subunits were expressed in the postsynaptic membranes of layer V/VI cortical pyramidal cells. The kinetic properties of GABAA receptor channels are determined by the subunits expressed in the receptors, and thus, the differences in current kinetic properties may be due to differences in subunit composition of the receptors from in vitro and in vivo preparations.
2.9. Gabrg2+/K328M KI mice exhibited prolonged thalamocortical oscillations recorded from VBn neurons
The excitatory and inhibitory synaptic balance critically regulates neural network activity and brain function. Disruption of this balance has been strongly associated with seizures and other neuropsychiatric disorders (Magloczky and Freund, 2005; Marin, 2012; McCormick and Contreras, 2001; Penzes et al., 2013; Staley, 2015). After showing that mutant γ2(K328M) subunits affected the functional properties of GABAergic synapses by reducing GABAA receptor-mediated mIPSC amplitude, we wanted to know whether the mutant subunits affected neural network activity. Spontaneous thalamocortical network oscillatory activity was measured in the thalamic ventrobasal nucleus (VBn) in horizontal slices. Multiunit recordings from VBn in slices showed that KI mice exhibited prolonged and high amplitude, spontaneous network oscillations (long period rhythmic neuronal firing activity with different action potential amplitudes) compared to the very short spontaneous network bursts [a very short (<100 ms) synchronous neuronal firing with similar action potential amplitudes] from WT mice (Fig. 8A and B). These results were consistent with the notion that excessive large-scale hypersynchronous neuronal activity occurs in the cortex of the brain during epileptic seizures (Fisher et al., 2005). However, these results do not exclude possible roles for other brain regions from participating in the spontaneous oscillations.
Fig. 8. Gabrg2+/K328M KI mice had longer spontaneous thalamocortical network oscillations than those from WT littermates.
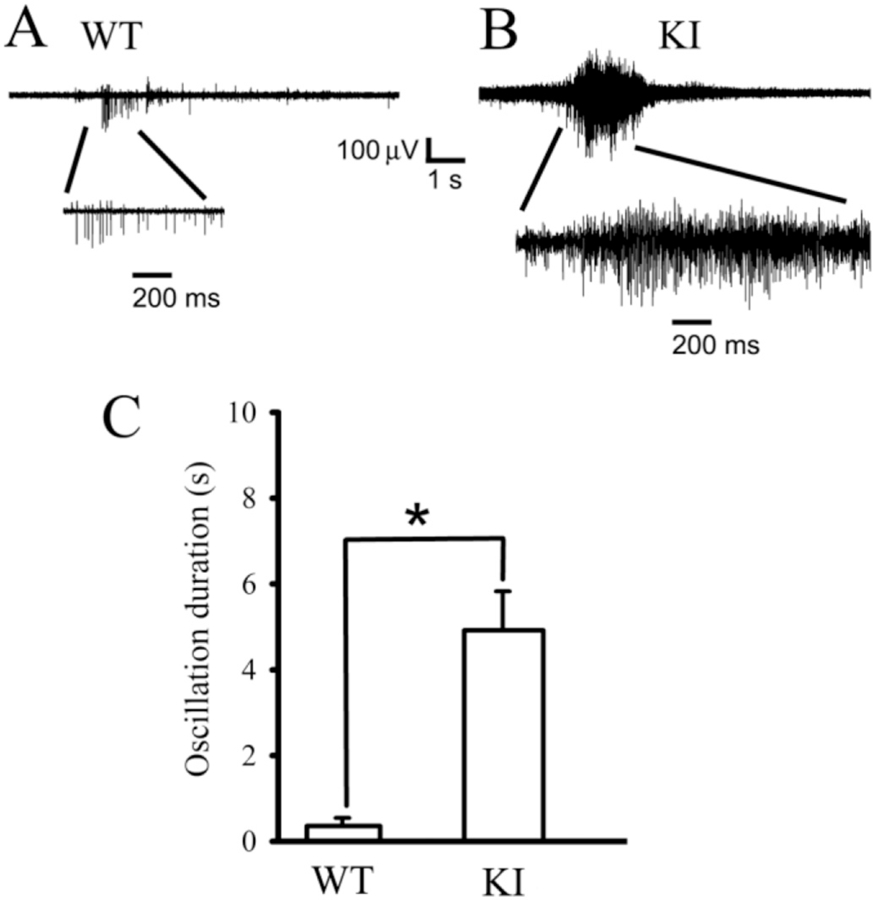
A. Extracellular multiple unit recordings showing spontaneous brief thalamocortical bursts in WT littermates. B. Spontaneous prolonged thalamocortical oscillations in KI mice. One short burst from a WT mouse and one oscillation from a KI mouse were expanded in each panel to show the multiple spikes in the burst or oscillation. Scale bars were labeled as indicated. C. Averaged oscillatory duration from both spontaneous bursts for P42 WT (0.356 ± 0.18 s, n = 5 mice) or oscillations from P42 KI (4.91 ± 0.91 s, n = 8 mice, t-test p = 0.002) mice were plotted (p = 0.002, Student’s t-test). Data were presented as mean ± SEM.
3. Discussion
3.1. The human GABRG2 K328M mutation caused GEFS+ in the Gabrg2+/K328M KI mouse
The human GABRG2 K328M mutation was identified in a large French-Canadian family with a GEFS+ pedigree (Baulac et al., 2001). Most patients with this mutation had a history of febrile seizures and many developed GTCS. In this study, we engineered a KI mouse model with the human GABRG2(K328M) mutation. We demonstrated that mutant γ2(K328M) subunits caused a GEFS+ epilepsy phenotype in Gabrg2+/K328M KI mice. Consistent with GEFS+ in humans, KI mice had both hyperthermic seizures and afebrile seizures including spontaneous GTCS, absence seizures and myoclonic seizures. This GEFS+ mouse model provided us with an opportunity to examine the mechanisms underlying GEFS+ associated with the K328M mutation and may facilitate the evaluation of new drugs and treatments.
3.2. Decreasing GABAergic inhibition contributed to GEFS+ in the Gabrg2+/K328M KI mouse
GEFS+ is a complex autosomal dominant familial and sporadic disorder with a heterogeneous genetic predisposition. Environmental factors such as fever or viral infection also play an important role in this disorder. Mutations in several genes have been found to contribute to the GEFS+ syndrome with diverse mechanisms. The most commonly associated mutations are in voltage-gated sodium channel genes, and most of these mutations are in SCN1A with autosomal dominant inheritance or with sporadic de novo mutation. The biophysical effects of the SCN1A GEFS+ mutations have been examined using in vitro heterologous expression systems, and it has been shown that these mutations altered sodium currents in either a gain-of-function or a loss-of-function manner (Escayg and Goldin, 2010). Most of the loss-of-function mutations were associated with a severe form of GEFS+, Dravet syndrome (Marini et al., 2011). However, results from in vivo mouse models suggest that dysfunction of SCN1A impairs interneuronal GABAergic inhibitory synaptic function (Ogiwara et al., 2007; Martin et al., 2010; Hawkins et al., 2011). The loss-of-function mutations in SCN1A resulted in reduced interneuron firing, which in turn led to decreased GABA release and impaired GABAergic inhibition. It is likely that impaired GABAergic signaling via a pre-synaptic mechanism is a major factor contributing to seizure generation in this mouse model and in patients with SCN1A GEFS+. It is not known if impairment of interneuronal inhibition is a general effect of SCN1A mutations causing epilepsy.
Mutations in genes encoding GABAA receptor subunits also cause genetic epilepsies (Macdonald et al., 2010). These genes include GABRA1, GABRA5, GABRA6, GABRB1, GABRB2, GABRB3, GABRG2, and GABRD. Among these GABRs, mutations in GABRG2 are most frequent and well characterized. Several missense mutations in GABRG2 were identified from patients associated with a spectrum of epilepsy syndromes including febrile seizures, CAE, GEFS+, and epileptic encephalopathies (Kang and Macdonald, 2016). The results from in vitro studies suggested an underlying common molecular and functional basis, reduction of GABAergic synaptic inhibition. All mutations, to different extents, reduced GABAA receptor channel function by diverse mechanisms including impaired expression, assembly, trafficking, GABA binding, and channel gating (Macdonald et al., 2010). The basis for the severity of epilepsy phenotypes with GABR mutations is likely related to the extent of reduction of receptor function.
While in vitro functional study combined with the genetic information suggests that these mutations may be major contributors to the epilepsy phenotypes, a causal role of these mutations can only be established directly from developing a mouse model harboring these mutations. Insights into the functional defects produced by GABR mutations are from studies of gene knock-out (KO) or KI mouse models. The Gabrg2−/− KO mouse has been generated and characterized extensively (Gunther et al., 1995). Most mice with complete loss of Gabrg2 (Gabrg2−/−) died within a few days of birth. Rare survivors can live up to P18 and exhibit sensorimotor deficits with excessive hyperactivity, impaired grasping, impaired righting reflexes, and abnormal gait (Gunther et al., 1995), whereas heterozygous Gabrg2 (Gabrg2+/−) KO mice largely have normal life span, social behavior, and motor coordination (Crestani et al., 1999). However, haploinsufficiency in Gabrg2+/− mice does lead to decreased synaptic clustering of GABAA receptors, enhanced anxiety (Crestani et al., 1999), and absence seizures in a seizure-prone genetic background (Reid et al., 2013).
A mouse harboring the GABRG2 g.G245A, p.R82Q (corresponding mutation R43Q in the mature peptide) mutation associated with febrile seizures and absence seizures was the first and only GABRG2 missense mutation KI mouse model (Tan et al., 2007). This mouse is a model of CAE and febrile seizures and recapitulates the human phenotype, including febrile seizures, early onset absence seizures, and SWDs on EEG associated with behavioral arrest (Tan et al., 2007; Petrou and Reid, 2012). In transfected HEK cells, the R82Q mutation reduced GABA-activated current due to ER retention and impaired trafficking of mutant receptors to the cell surface (Bianchi et al., 2002; Hales et al., 2006; Eugene et al., 2007). Analysis of synaptic inhibition in the Gabrg2+/R82Q KI mouse showed a small reduction in cortical, but not thalamic, GABAA receptor-mediated synaptic transmission as a possible underlying mechanism for CAE. The reduction in mutant γ2(R82Q) subunit surface expression likely contributes to reduction of mIPSCs and to the observed seizure phenotype (Tan et al., 2007).
In contrast, the mutant γ2(K328M) subunit did not alter total or cell surface expression of γ2 or its synaptosomal distribution. Electrophysiological analysis of layer V/VI somatosensory cortex pyramidal neurons in the Gabrg2+/K328M KI mouse revealed decreased amplitude of mIPSCs and prolonged spontaneous thalamocortical oscillations in the VBn of the thalamus. Like in Gabrg2+/R82Q KI mice, impaired GABAA receptor-mediated inhibitory transmission was the underlying mechanism for the observed epilepsy phenotypes in Gabrg2+/K328M KI mice.
3.3. Premature sudden death is associated with GTCS in Gabrg2+/K328M KI mice
SUDEP is the most common cause of death in many epilepsy populations. Clinical and mouse model studies suggest multiple pathophysiologic contributory factors for SUDEP including seizure-induced cardiac arrhythmia, pulmonary and respiratory dysfunction, cerebral depression, and autonomic dysfunction, but the underlying mechanisms are largely unknown (Devinsky et al., 2016). Gabrg2+/K328M KI mice displayed a spectrum of epilepsy phenotypes that changed with development from febrile seizures and afebrile seizures including the early onset GTCS to absence and myoclonic seizures in adulthood. We found that an unexpected property in Gabrg2+/K328M KI mice was seizure-induced premature sudden death, which occurred mostly from P15 to P17 compared to other genetic mouse models with sporadic death (Yu et al., 2006; Kang et al., 2015; Smart et al., 1998). Video monitoring revealed that sudden death occurred after GTCS, which was consistent with human studies that demonstrated that GTCS are the greatest risk factors for SUDEP (Devinsky et al., 2016). It was unclear why the sudden death mostly occurred in KI mice from P15 to P17. It could be related to the developmental change in neuronal chloride reversal potential which occurs around this age (Ben-Ari et al., 2011). It was not clear whether individuals from the large French-Canadian family in which the GEFS+GABRG2(K328M) mutation was identified had early onset GTCS and SUDEP. The early onset GTCS and sudden death in the Gabrg2+/K328M KI mouse could also be influenced by genetic background. The Gabrg2+/K328M KI mouse has a pure C57BL/6 J genetic background, whereas humans have diverse genetic backgrounds. It has been shown that genetic background not only plays critical roles to define epileptic phenotypes (Reid et al., 2013; Tan et al., 2007; Yu et al., 2006; Miller et al., 2014; Mistry et al., 2014), but also affects GABR gene expression (Hawkins and Kearney, 2016; Mulligan et al., 2019). Nonetheless, the Gabrg2+/K328M KI mouse model may offer a new opportunity to study the underlying mechanisms of SUDEP induced by severe seizures. Our findings could also serve as a model for seizure-management practices and prevention of SUDEP in patients harboring mutations that produce GTCS.
3.4. The short, severe epilepsy phenotype in early life did not result in long-lasting behavioral/cognitive dysfunction in Gabrg2+/K328M KI mice
Clinical phenotypes of GEFS+ patients vary from the most common and mild simple febrile seizures with normal intelligence and learning abilities to the most severe disorder, the epileptic encephalopathy Dravet syndrome, which is usually associated with severe learning and behavioral impairment. A critical issue is whether the epilepsy affects the development of cognitive and behavioral function in the Gabrg2+/K328M KI mice. We performed a suite of behavioral assessments of adult Gabrg2+/K328M KI mice and revealed mostly normal behavioral phenotypes (normal intelligence, socialization, and learning ability) and only hyperactivity. We were surprised by these results given the fact that preweaning age KI mice had severe GTCS and about 14% of them had premature sudden death. It was reported that short, repeated seizures in early-life were associated with an increase in seizure susceptibility and spontaneous seizure severity, hyperactivity, and impairment in social behaviors and recognition in adulthood (Dutton et al., 2017; Salgueiro-Pereira et al., 2019). The neural mechanisms underlying the behavioral comorbidities in epilepsy are unclear, but it has been increasingly recognized the importance of the interplay between underlying genetic mutation, epilepsy and its comorbidities. Epileptiform activity itself can affect brain development through multiple mechanisms such as alteration of neurotransmitter systems and neuronal properties (Jarero-Basulto et al., 2018). The observation that individuals with epileptic encephalopathy who are successfully treated with surgery can show improvement in cognitive function demonstrates that seizures may play an important role in the neuropsychiatric deficits (Lee et al., 2014). It is not clear why mutant γ2(K328M) subunits can cause a severe early-life epilepsy in Gabrg2+/K328M KI mice without long-lasting behavioral/cognitive effects. It is possible that these survivors might not have had severe GTCS to affect the behavioral comorbidities, even 14% of Gabrg2+/K328M KI mice had premature sudden death, likely due to severe GTCS. We speculate that either seizure timing and/or seizure duration were not severe enough to modify brain structure and function, either transiently or permanently, to have long-lasting effects in Gabrg2+/K328M KI mice. It is also conceivable that neurobehavior impairment and seizures are the result of two separate end-results of the underlying mutation in the Gabrg2+/K328M KI mouse model. Studies of Scn1a knockout mice and Arx−/YEmx1Cre mice have demonstrated that on-going seizures were unrelated to cognitive impairments (Bender et al., 2013; Simonet et al., 2015).
3.5. Febrile seizures in Gabrg2+/K328M KI mice
Most patients with the K328M mutation had histories of febrile seizures (Baulac et al., 2001). We confirmed that hyperthermia can induce seizures in Gabrg2+/K328M KI mice, and that the severity of hyperthermia-induced seizures was age-dependent after postnatal day 20. We also found that the spontaneous afebrile GTCS precede susceptibility to febrile seizures (at 42 °C) in Gabrg2+/K328M KI mice. Interestingly, the heterozygous loss-of-function Scn1a+/− mice became susceptible to temperature-induced seizures before the occurrence of spontaneous seizures. As with Gabrg2+/K328M KI mice, Scn1a+/− KI mice showed no temperature-induced seizures on P17–18, and had temperature-induced myoclonic and generalized seizures in nearly all P20–22 and P30–46 mice, whereas spontaneous seizures were observed only in mice older than P32 (Oakley et al., 2009). Spontaneous afebrile GTCS preceding febrile seizures may suggest that hyperthermia alone is not enough to induce seizures before P18 and that there is a critical age-dependent transition or additional factors that are required for febrile seizure susceptibility during Gabrg2+/K328M KI mouse development. It could also mean that younger animals may have a higher temperature threshold for febrile seizures. Given the similarity of development of spontaneous seizures and febrile seizures, it will be of interest in future studies to examine whether Scn1a and Gabrg2 interact to regulate afebrile seizure or febrile seizure genesis.
4. Conclusions
In conclusion, we showed that Gabrg2+/K328M KI mice recapitulated most features of GEFS+ patients, including febrile seizures and afebrile seizures with multiple seizure semiologies. The electrophysiological results suggest that the mutant γ2(K328M) subunit causes GEFS+ by reducing GABAergic inhibitory synaptic transmission. The Gabrg2+/K328M mouse model should be useful for further investigation of the mechanisms underlying development of GEFS+ resulting from GABAA receptor dysfunction and for testing new treatments for GEFS+.
5. Methods
All animal care and experiments were performed in accordance with the policies of the Vanderbilt University Medical Center IACUC and to the National Institutes of Health Guide for Care and Use of Laboratory. Mice were maintained in cages of up to five adults. Mice after EEG headmount surgery were single-housed. Mice were housed in either the VU Level 5 (not specific-pathogen-free) breeding facility or the Level 7 Neurobehavioral Core, both of which are maintained by the Department of Animal Care. Facilities use a 12-h light/dark cycle, corn-cob bedding, and ad libitum food and water.
5.1. Generation of the Gabrg2+/K328M KI mouse
The murine Gabrg2 983A > T mutation is in exon 8 and changes an AAG codon to ATG, thus converting a lysine to methionine at residual 328 (K328M) in the γ2 subunit. We collaborated with the Applied StemCell, Inc. (Milpitas, CA) to generate the KI mouse by using conventional homologous recombination method. With the intention to study its developmental impact on GEFS+, a KI vector was constructed to generate a conditional KI mouse based on the Cre-dependent genetic switch (FLEx switch) strategy (Schnutgen et al., 2003). Briefly, the replacement targeting vector was constructed by cloning 7.2 kb 5′ homology arm, which contains loxP flanked exon 8 (loxP-exon8-lox2272) and a 5 kb 3′ arm, which contains loxP flanked inverted exon 8 with 983A > T mutation (exon8-loxP-lox2272), into the upstream and downstream of FRT flanked PGK-Neo (the phosphoglycerate kinase I promoter driving the neomycin phosphotransferase gene) cassette, respectively, in a vector with the negative selection marker MC1-thymidine kinase cassette (Fig. 1). The loxP and lox2272 sites were positioned about 600 bp and 400 bp away, respectively, from intronexon boundary to avoid interfere with spliceosome function. The homology arm fragments were retrieved from a Gabrg2-containing BAC clone (RP23–233 N1) by a recombineering approach (Liu et al., 2003). The 983A > T point mutation in exon 8 was generated by site-directed mutagenesis (QuikChange II Site-Directed Mutagenesis Kit, Agilent Technologies). The linearized targeting vector was electroporated into C57BL/6 J mouse ES cells. The ES cells with a correctly targeted locus, identified first by long range PCR with one of primers located outside of homology arms (A or D) and followed by PCR/sequencing, were micro-injected into the C57BL/6 J albino blastocysts to produce chimeric mice. Chimeric mice were bred with Actin-Flpe C57BL/6 J transgenic mice (Rodriguez et al., 2000) to screen for germline transmission and to excise the PGK-Neo selection cassette to generate a mouse with conditional allele. If the presence of the inverted exon 8 and other modifications did not interfere with normal RNA splicing, the resultant mice harboring the conditional allele would produce normal Gabrg2 transcripts and serve as a conditional KI model. However, we found that these genetic modifications disrupted normal RNA splicing and resulted in a truncated transcript without exon 8–9 (data not shown). Therefore, we converted the “conditional allele” to a KI allele by breeding the “conditional KI mice” with CMV-Cre mice. Upon Cre-mediated recombination, exon 8 was removed and replaced by exon 8* with the 983A > T mutation, resulting in generation of the K328M KI mouse, Gabrg2+/K328M. The Flpe and Cre transgenes were subsequently bred out by back crossing with C57BL/6 J mice. The KI mice were genotyped by using a pair of primers (Forward 5’-GTGCACATACATGCACACATGCATTC-3′ and backward 5’-GAGTATGGCACCCTGCATTATTTTGTCAG-3′). The PCR fragment for the WT allele was 605 bp and the fragment for the KI mutant allele was 689 bp due to the presence of leftover FRT, loxP and other modifications.
5.2. Video-EEG recordings and analysis
Synchronized video-EEGs were recorded from 2.5 to 6.5-month-old WT and KI mice after electrode implantation (Pinnacle Technology, #8201) with a video-EEG monitoring system from Pinnacle Technology as described in (Arain et al., 2012). Briefly, mice were anesthetized with 1–3% isoflurane (vol/vol), the front edge of the head mount was placed 3.0–3.5 mm anterior of bregma and secured by four stainless steel screws and dental cement. Two EMG leads were inserted into the trapezius muscles. Mice recovered from implantation surgery for at least 1 week before EEG recording.
Video-EEG monitoring lasted for 24 h with freely moving mice in the chamber. The pre-amplifier filtered (1 Hz high pass) and amplified the signal (gain 100×), and additional filtering occurred at the data acquisition/conditioning system. The acquisition rate for EEG-EMG channels was 400 Hz and for the infrared camera was 15 frames/s. Video-EEG-EMG data were analyzed offline with Sirenia® Seizure software. The reviewer was blind to the genotypes.
5.3. Febrile seizure test
Febrile seizure tests were performed using an incubator with several modifications: (1) two thick pre-warmed Plexiglas plates was put interior to the front door and back wall to minimize external cool air from creating low temperature regions in the chamber; (2) a mesh metal shelf plate was placed to prevent mice having direct contact with the warmer incubator bottom; and (3) the front door was sealed to prevent air leaks. With the modifications, we created a temperature-controlled environment with limited airflow except for a small hole to provide air exchange in the back wall. Mouse body temperature was predetermined with a rectal probe (Physitemp Instruments Inc.) to confirm a core body temperature of 42 °C. It took about 7 min for P16, 10 min for P42 and 20 min for P84 mice to achieve a core temperature of 42 °C. During hyperthermic seizure experiments (no rectal probe), mouse tails were labeled with nontoxic dye for identification purposes. The experimental procedure was simultaneously monitored by the investigator and video recorded. The presence of and latencies to myoclonic seizures and GTCS were determined and recorded during 30 min of hyperthermia. The experimenter was blind to the genotypes.
5.4. Pentylenetetrazol (PTZ) induced seizures
Mice between 1 and 2 months old were injected intraperitoneally with PTZ at a dose of 30 mg/kg and were observed for 30 min. Latencies to myoclonic and GTCS were recorded. The susceptibility to PTZ-induced seizures was assessed by survival cure. Mantel-Cox method was used for statistics. The experimenter was blind to the genotypes.
5.5. Mouse behavioral testing
The mouse behavior experiments were performed in the VUMC Murine Neurobehavioral Core Laboratory. Three to 4-month-old Gabrg2+/K328M mice and WT littermate mice were housed in a 12-h light/dark cycle with standard rodent food and water ad libitum. Anxiety tests were performed first, and the more complex, potentially anxiety-causing tests were performed subsequently in the order described below. The same cohort was used for all tests. The experimenter was blind to the genotypes.
Open field tests were performed as previously described (Kang et al., 2015; Fukada et al., 2012; McLaughlin et al., 2012) using the standard protocol in the VUMC MNBCL. Each individual mouse was placed for 60 min in an open field activity chamber (Med Associates, 27 × 27 × 20.3 cm) with light- and air-controlled environment. Location and movement were detected by interruption of infrared beams by the mouse (16 photocells in each horizontal direction and 16 photocells elevated by 4 cm to measure rearing) and were measured by the Med Associates Activity Monitoring program. The chamber was cleaned with Vimoba (Chlorine Dioxide, Quip Labs) and wiped with paper towels between each trial. The total distance traveled and time resting in either the center (50% of area) or peripheral zone were analyzed as measurement of locomotion and anxiety. Vertical counts were analyzed as a measurement of hyperactivity.
The elevated zero maze test was used for assessing anxiety-related behaviors (Kang et al., 2015; Fukada et al., 2012; McLaughlin et al., 2012). The elevated circular platform (40 cm off the ground, 50 cm in diameter) is divided into equal fourths, with two enclosed arenas opposite each other (with 15 cm high walls) and two open arenas between the enclosed arenas. Lumens in each of the four arenas are recorded and maintained constant across all animals. Briefly, each mouse was lowered by its tail into the open arena of the maze and allowed to explore the maze for 5 min. The whole circular platform was cleaned with Vimoba and wiped with paper towels between each animal. Mouse activity was monitored via an overhead camera connected to a computer in a separate room using video acquisition and ANY-maze analysis software (Stoelting, Wood Dale, IL). The time spent and distance traveled in open versus closed arms, and number of entries into open arms were analyzed.
The three-chamber social interaction protocol was based on a previous study (Carter et al., 2011). Briefly, the apparatus is divided into 3 equal size chambers separated by high walls with a closable door. Subject mouse was moved to the middle chamber between each stage but was not removed from the apparatus until the entire protocol completed. Additional mice and objects were to be placed in the two side chambers. Habituation: Subject mouse freely explores the entire empty apparatus for 10 min. Sociability: Subject mouse freely explores a stranger mouse and empty pencil cup for 10 min. Social Novelty: Subject mouse freely explores a novel mouse and familiar mouse (used in sociability test) for 10 min. The time subject mouse spent actively investigating each stimulus was recorded by hand using ANY-maze.
The Barnes maze test was based on the protocol described in the previous studies (Harrison et al., 2006; Paylor et al., 2001). The procedure includes pretraining, training, and a memory probe. Pretraining: the mouse is placed in a black start box for 30 s and is then guided to the target hole where the mouse enters the escape box. After 30 s, the mouse is removed from the escape box and placed back in the start box for an additional three trials. The pretraining session only occurred on the first day of Barnes maze testing. The maze is not cleaned between pretraining trials. Training: similar to pretraining, the mouse is placed in the start box for 30 s. After 30 s in the start box, the mouse is allowed to freely explore the maze in search of the target hole using visual cues for a maximum of 300 s. On each training trial, 11 of the 12 holes are blocked. Mice are exposed to 4 training trials per day for 5 consecutive days, or until an asymptote is reached in a plot of latency to entry to the target hole. The maze is cleaned with Vimoba between each mouse and each trial, and the maze is rotated 90° between each trial to prevent the animals from utilizing intra-maze cues when locating the target hole. Probe: A 300 s probe test is conducted 1 h after the final training trial on the fifth day using the same parameters described during the training session, except that all 12 holes are now blocked. The time spent investigating the target hole (the hole which was previously uncovered), and number of and time spent investigating non-target holes were recorded. Data are collected using ANY-maze.
Behavioral data were analyzed with GraphPad Prism software. Independent-sample Student’s t-tests were used in the analyses of locomotor activity, elevated zero maze, and the probe trials of the Barnes maze. A two-way ANOVA was used for the three-chamber test of sociability and social novelty. A repeated-measures ANOVA was used to analyze the training trials of the Barnes maze. Post hoc and a priori Bonferroni comparisons were conducted to evaluate individual mean comparisons where appropriate. All analyses used an alpha level of 0.05 to determine statistical significance. Data were presented as mean ± SEM.
5.6. RT-PCR
Total RNAs were extracted from brain tissues of P10-P12 mice by using Direct-zol™ RNA MiniPrep Plus kit from Zymo Research. RT-PCRs were carried out by using QIAGEN OneStep RT-PCR kit as suggested by the manufacturer. The forward primer 5′- GTA GAG TGG AGC AGT TCC CAC TCA G – 3′ was located upstream of the ATG start codon, and backward primer 5′- GAC AGG CAG GGT AAT ATT TCA CTC AGT G – 3′ was located downstream of the TGA stop codon. The GAPDH primers were 5′- GTG AAG GTC GGT GTG AAC GGA TTT G – 3′ and 5′- GAT GGC ATG GAC TGT GGT CAT GAG – 3′.
5.7. Western blots
Cerebellum, cortex, hippocampus and thalamus were dissected from 1 to 3 months old mouse brains and homogenized with RIPA buffer (50 mM Tris-Cl, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate and 0.1% SDS) to obtain lysates. Proteins were fractionated by SDS/PAGE and immunoblotted with following antibodies: ATPase (1:2000, a6F, DSHB), GABAA receptor α1 subunit (1:500, 75–136, NeuroMab), GABAA receptor β3 subunit (1:500, B300–199, Novus Biological), GABAA receptor γ2 subunit (1:500, AB 5559, Millipore), gephyrin (1:1000, 3B11, Cedarlane), and GAPDH (1:6000, MAB374, Millipore) antibodies.
Surface expression of GABAA receptor subunits was evaluated by extraction of plasma membrane proteins from brain. Plasma membrane proteins were isolated by using 101Bio kit (101bio.com) with minor modification. Proteins were fractionated by SDS/PAGE and subjected to immunoblot analyses. ATPase was used as membrane protein loading control, whereas GAPDH was cytoplasmic proteins contamination control.
Synaptosomes were isolated by using the protocol in the previous study (Kang et al., 2015). Briefly, brains were dissected and homogenized in pre-chilled homogenization HEPES buffered sucrose buffer (HBS containing 0.32 M sucrose, 10 mM HEPES, pH 7.4, 2 mM EDTA) with a ratio of 10 ml buffer: 1 g of brain tissue with 10–15 strokes. The cell debris and nuclei were removed from the homogenates by spinning down at 1000 g for 10 min. The supernatant was centrifuged at 13,800 g for 20 min. The pellet (crude synaptosomal fraction) was resuspended with HBS buffer at 1:10 ratio and centrifuged at 13,800 g for 15 min. The washed pellet was then mixed with 9× vol ice cold H2O-protease inhibitors. After rapidly adjusting to 4 mM HEPES by using 1 M HEPES (pH 7.4), the suspension was then rotated for 30 min at 4 °C, followed by spinning at 25,000 g for 20 min. The pellet was taken and resuspended with 1.5 ml HBS buffer followed by discontinuous gradient sucrose ultracentrifuge. An equal volume (2.2 ml) of sucrose with different concentrations (1.4 M, 1.2 M, 1.0 M to 0.8 M) was added from bottom to top in the tube. The sucrose tube containing the 1.5 ml sample on the top layer was centrifuged at 150,000 g for 1 h. Each layer of interest was carefully collected and stored at −80 °C. The synaptosome layer (spm) was at 1.0/1.2 M sucrose interface.
5.8. Whole cell slice recording
Brain slices were prepared according to previously published methods (Zhou et al., 2011; Huang et al., 2017). Young adult mice (~1-month-old, either gender) were deeply anesthetized with isoflurane and transcardially perfused with ice-cooled dissection solution (4 °C) (mM: 2.5 KCl, 0.5 CaCl2, 10 MgSO4, 1.25 NaH2PO4, 24 NaHCO3, 11 Glucose, 214 Sucrose). Mouse brains were dissected out after decapitation and coronal brain slices (300 μm) were cut using a LEICA VT-1200S vibratome (Leica Inc) with oxygenated (bubbling with 95% O2/5% CO2) dissection solution. The brain slices were transferred to an incubation chamber with oxygenated ACSF (mM: 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, 10 Glucose, 310 mOsm, pH 7.4) for 40 min at 35–36 °C. The slices were allowed then to recover at room temperature for at least 1 h before experiments.
With an upright Eclipse FN-1IR-DIC microscope (Nikon) and a MultiClamp 700B amplifier and Digidata 1440A (Molecular devices Inc.), whole-cell recordings of layer V/VI pyramidal neurons (SS cortex) were obtained. The neurons were selected based on their apical dendrites and location above the white matter, and mIPSCs were isolated pharmacologically by including 10–20 μM NBQX and 1 μM tetrodotoxin in the ACSF (flow rate: 1–1.5 ml per min). The internal pipette solution for recordings contained (mM): 135 CsCl, 10 HEPES, 10 EGTA, 5 QX-314, 5 ATP-Mg (290–295 mOsm, pH = 7.3), and filled glass electrodes had 3–5 MΩ resistances (Zhou et al., 2011; Huang et al., 2017). Serial resistances during recording were continuously monitored, confirmed to be less than 20–25 MΩ and compensated by 70%; cell capacitances were also compensated. Recordings with access resistance variations >20% or magnitudes larger than 25 MΩ were discarded. Junction potentials were compensated when electrodes were in ACSF. The chloride ion reversal potential was maintained close to 0 mV, and neurons were clamped at −60 mV. Data were collected using the Clampex program 10.2 (Molecular devices Inc.), and synaptic currents were filtered at 2 kHz and digitized at 10 KHz. All recordings were performed at room temperature (24 °C) continuously for 20–30 min after membrane rupture, and mIPSCs were analyzed with Clampfit (Molecular Devices Inc.) using threshold detection (at least 2.5× baseline RMS with no clear synaptic events) (Zhou et al., 2011; Huang et al., 2017). Histogram and cumulative graphs were constructed with the Clampfit program (Molecular devices Inc.).
5.9. Thalamocortical oscillations
Horizontal thalamocortical brain slices (350–400 μm) were prepared using the method of Huguenard and Price (Huguenard and Prince, 1994) and were put on top of a self-made nylon mesh interface (only one side of the slice contacted the ACSF solution). One FHC glass-coated tungsten electrode (www.fh-co.com). was placed in the VBn of the thalamus to record spontaneous multi-Unit activity (MultiClamp 700B, current-clamp mode), which was band-filtered between 100 Hz and 3 kHz as reported by Huguenard and Prince (Huguenard and Prince, 1994). The experiments were performed at 31–32 °C. Data were analyzed using both Clampfit (for spike histogram and autocorrelation function) and Matlab to obtain autocorrelograms. Oscillation indices were calculated to compare different groups (Sohal et al., 2003; Sugihara et al., 1995).
Supplementary Material
Acknowledgements
The authors would like to thank Laurel G. Jackson for editing supplemental video. We would like to thank John Allison for his technical assistance in the Vanderbilt MNL. This research was supported by NIHR01NS51590 and NS107424.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2021.105296.
Declaration of Competing Interest
None.
References
- Arain FM, Boyd KL, Gallagher MJ, 2012. Decreased viability and absence-like epilepsy in mice lacking or deficient in the GABAA receptor alpha1 subunit. Epilepsia 53 (8), e161–e165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard EA, et al. , 1998. International Union of Pharmacology. XV. Subtypes of gamma-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacol. Rev 50 (2), 291–313. [PubMed] [Google Scholar]
- Baulac S, et al. , 2001. First genetic evidence of GABA(a) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat. Genet 28 (1), 46–48. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Tyzio R, Nehlig A, 2011. Excitatory action of GABA on immature neurons is not due to absence of ketone bodies metabolites or other energy substrates. Epilepsia 52 (9), 1544–1558. [DOI] [PubMed] [Google Scholar]
- Bender AC, et al. , 2013. Focal Scn1a knockdown induces cognitive impairment without seizures. Neurobiol. Dis 54, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, et al. , 2002. Two different mechanisms of disinhibition produced by GABAA receptor mutations linked to epilepsy in humans. J. Neurosci 22 (13), 5321–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouthour W, et al. , 2012. A human mutation in Gabrg2 associated with generalized epilepsy alters the membrane dynamics of GABAA receptors. Cereb. Cortex 22 (7), 1542–1553. [DOI] [PubMed] [Google Scholar]
- Carter MD, et al. , 2011. Absence of preference for social novelty and increased grooming in integrin beta3 knockout mice: initial studies and future directions. Autism Res 4 (1), 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill GL, et al. , 2014. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology 82 (14), 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, McKerlie C, Snead OC 3rd, 2001. A model of atypical absence seizures: EEG, pharmacology, and developmental characterization. Neurology 56 (3), 341–349. [DOI] [PubMed] [Google Scholar]
- Crestani F, et al. , 1999. Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat. Neurosci 2 (9), 833–839. [DOI] [PubMed] [Google Scholar]
- Devinsky O, et al. , 2016. Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol 15 (10), 1075–1088. [DOI] [PubMed] [Google Scholar]
- Dutton SBB, et al. , 2017. Early-life febrile seizures worsen adult phenotypes in Scn1a mutants. Exp. Neurol 293, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, Goldin AL, 2010. Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51 (9), 1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugene E, et al. , 2007. GABA(A) receptor gamma 2 subunit mutations linked to human epileptic syndromes differentially affect phasic and tonic inhibition. J. Neurosci 27 (51), 14108–14116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RS, et al. , 2005. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 46 (4), 470–472. [DOI] [PubMed] [Google Scholar]
- Fukada M, et al. , 2012. Loss of deacetylation activity of Hdac6 affects emotional behavior in mice. PLoS One 7 (2), e30924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther U, et al. , 1995. Benzodiazepine-insensitive mice generated by targeted disruption of the gamma 2 subunit gene of gamma-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. U. S. A 92 (17), 7749–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales TG, et al. , 2006. An asymmetric contribution to gamma-aminobutyric type a receptor function of a conserved lysine within TM2–3 of alpha1, beta2, and gamma2 subunits. J. Biol. Chem 281 (25), 17034–17043. [DOI] [PubMed] [Google Scholar]
- Harrison FE, et al. , 2006. Spatial and nonspatial escape strategies in the Barnes maze. Learn. Mem 13 (6), 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NA, Kearney JA, 2016. Hlf is a genetic modifier of epilepsy caused by voltage-gated sodium channel mutations. Epilepsy Res 119, 20–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NA, et al. , 2011. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol. Dis 41 (3), 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortnagl H, et al. , 2013. Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience 236, 345–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., et al., 2017. Overexpressing wild-type gamma2 subunits rescued the seizure phenotype in Gabrg2(+/Q390X) Dravet syndrome mice. Epilepsia 58 (8), 1451–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA, 1994. Intrathalamic rhythmicity studied in vitro: nominal T-current modulation causes robust antioscillatory effects. J. Neurosci 14 (9), 5485–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarero-Basulto JJ, et al. , 2018. Interactions between epilepsy and plasticity. Pharmaceuticals (Basel) 11 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarre G, et al. , 2017. Genetic Models of Absence Epilepsy in Rats and Mice, 2nd edition. Models of Seizures and Epilepsy, pp. 455–471. [Google Scholar]
- Kang JQ, Macdonald RL, 2016. Molecular pathogenic basis for GABRG2 mutations associated with a spectrum of epilepsy syndromes, from generalized absence epilepsy to dravet syndrome. JAMA Neurol 73 (8), 1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL, 2006. Why does fever trigger febrile seizures? GABAA receptor gamma2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J. Neurosci 26 (9), 2590–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, et al. , 2015. The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration. Nat. Neurosci 18 (7), 988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie DJ, Wisden W, Seeburg PH, 1992. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J. Neurosci 12 (11), 4151–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SV, et al. , 2017. A mutation in GABRB3 associated with Dravet syndrome. Am. J. Med. Genet. A 173 (8), 2126–2131. [DOI] [PubMed] [Google Scholar]
- Ledergerber D, Larkum ME, 2010. Properties of layer 6 pyramidal neuron apical dendrites. J. Neurosci 30 (39), 13031–13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, et al. , 2014. Outcomes of epilepsy surgery in childhood-onset epileptic encephalopathy. Brain and Development 36 (6), 496–504. [DOI] [PubMed] [Google Scholar]
- Letts VA, Beyer BJ, Frankel WN, 2014. Hidden in plain sight: spike-wave discharges in mouse inbred strains. Genes Brain Behav 13 (6), 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG, 2003. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13 (3), 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ, Gallagher MJ, 2010. Mutations in GABAA receptor subunits associated with genetic epilepsies. J. Physiol 588 (Pt 11), 1861–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magloczky Z, Freund TF, 2005. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci 28 (6), 334–340. [DOI] [PubMed] [Google Scholar]
- Marin O, 2012. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci 13 (2), 107–120. [DOI] [PubMed] [Google Scholar]
- Marini C, et al. , 2011. The genetics of Dravet syndrome. Epilepsia 52 (Suppl. 2), 24–29. [DOI] [PubMed] [Google Scholar]
- Martin MS, et al. , 2010. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J. Biol. Chem 285 (13), 9823–9834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Contreras D, 2001. On the cellular and network bases of epileptic seizures. Annu. Rev. Physiol 63, 815–846. [DOI] [PubMed] [Google Scholar]
- McLaughlin B, et al. , 2012. Haploinsufficiency of the E3 ubiquitin ligase C-terminus of heat shock cognate 70 interacting protein (CHIP) produces specific behavioral impairments. PLoS One 7 (5), e36340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AR, et al. , 2014. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav 13 (2), 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry AM, et al. , 2014. Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol. Dis 65, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, et al. , 2005. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum. Mol. Genet 14 (2), 205–220. [DOI] [PubMed] [Google Scholar]
- Mulligan MK, et al. , 2019. Identification of a functional non-coding variant in the GABA A receptor alpha2 subunit of the C57BL/6J mouse reference genome: major implications for neuroscience research. Front. Genet 10, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley JC, et al. , 2009. Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc. Natl. Acad. Sci. U. S. A 106 (10), 3994–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, et al. , 2007. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci 27 (22), 5903–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar R, Ganesh S, 2013. The SCN1A gene variants and epileptic encephalopathies. J. Hum. Genet 58 (9), 573–580. [DOI] [PubMed] [Google Scholar]
- Paylor R, et al. , 2001. Learning impairments and motor dysfunctions in adult Lhx5-deficient mice displaying hippocampal disorganization. Physiol. Behav 73 (5), 781–792. [DOI] [PubMed] [Google Scholar]
- Penzes P, et al. , 2013. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J. Neurochem 126 (2), 165–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrou S, Reid CA, 2012. The GABAAgamma2(R43Q) mouse model of human genetic epilepsy. In: Jasper’s Basic Mechanisms of the Epilepsies th, et al. , Editors. Bethesda; (MD: ). [Google Scholar]
- Reid CA, et al. , 2011. Low blood glucose precipitates spike-and-wave activity in genetically predisposed animals. Epilepsia 52 (1), 115–120. [DOI] [PubMed] [Google Scholar]
- Reid CA, et al. , 2013. Multiple molecular mechanisms for a single GABAA mutation in epilepsy. Neurology 80 (11), 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez CI, et al. , 2000. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet 25 (2), 139–140. [DOI] [PubMed] [Google Scholar]
- Salgueiro-Pereira AR, et al. , 2019. A two-hit story: seizures and genetic mutation interaction sets phenotype severity in SCN1A epilepsies. Neurobiol. Dis 125, 31–44. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic SF, 1997. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 120 (Pt 3), 479–490. [DOI] [PubMed] [Google Scholar]
- Schnutgen F, et al. , 2003. A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat. Biotechnol 21 (5), 562–565. [DOI] [PubMed] [Google Scholar]
- Simonet JC, et al. , 2015. Conditional loss of arx from the developing dorsal telencephalon results in behavioral phenotypes resembling mild human ARX mutations. Cereb. Cortex 25 (9), 2939–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, et al. , 1999. Generalized epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Ann. Neurol 45 (1), 75–81. [DOI] [PubMed] [Google Scholar]
- Smart SL, et al. , 1998. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron 20 (4), 809–819. [DOI] [PubMed] [Google Scholar]
- Sohal VS, et al. , 2003. Dynamic GABA(a) receptor subtype-specific modulation of the synchrony and duration of thalamic oscillations. J. Neurosci 23 (9), 3649–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley K, 2015. Molecular mechanisms of epilepsy. Nat. Neurosci 18 (3), 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara I, Lang EJ, Llinas R, 1995. Serotonin modulation of inferior olivary oscillations and synchronicity: a multiple-electrode study in the rat cerebellum. Eur. J. Neurosci 7 (4), 521–534. [DOI] [PubMed] [Google Scholar]
- Tan HO, et al. , 2007. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc. Natl. Acad. Sci. U. S. A 104 (44), 17536–17541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, et al. , 2006. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci 9 (9), 1142–1149. [DOI] [PubMed] [Google Scholar]
- Zhou C, et al. , 2011. Hypoxia-induced neonatal seizures diminish silent synapses and long-term potentiation in hippocampal CA1 neurons. J. Neurosci 31 (50), 18211–18222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.