

Abstract
Leading by cytotoxicity against HepG2 cells, bioactivity-guided fractionation of the EtOAc fraction from Artemisia atrovirens led to the isolation of 18 new guaianolide dimers, artematrolides A–R and lavandiolides A, B, C, H, and J. Eight compounds (1, 4, 10, 12, 13, and 19–21) were unambiguously confirmed by the single-crystal X-ray diffraction analyses, and the others were elucidated based on IR, UV, HRESIMS, 1D and 2D NMR experiments, and comparison of the experimental and calculated ECD data. Structurally, all of them were [4 + 2] Diels–Alder adducts of two monomeric guaianolides. The isolates were evaluated for their cytotoxicity against three human hepatoma cell lines, and 19 compounds demonstrated cytotoxicity against HepG2, SMMC-7721, and Huh7 cell lines. Especially, compounds 1, 12, 14, and 15 exhibited cytotoxicity with IC50 values of 4.4, 3.8, 7.6, and 6.7 μmol/L (HepG2), 9.6, 4.6, 6.6, and 6.0 μmol/L (SMMC-7721), and 7.6, 4.5, 6.9, and 5.6 μmol/L (Huh7), respectively. Notably, compound 12 showed the most promising activity against three human hepatoma cell lines and dose-dependently inhibited cell migration and invasion, induced G2/M cell cycle arrest and cell apoptosis in HepG2 cells, down-regulated the expression of BCL-2 and PARP-1, and activated PARP-1 to up-regulate the expression of cleaved-PARP-1.
KEY WORDS: Guaianolide dimers, Artematrolides A–R, Artemisia atrovirens, Cytotoxicity, Cell cycle, Apoptosis
Graphical abstract
Twenty-three guaianolide dimers were isolated from Artemisia atrovirens. Nineteen compounds demonstrated cytotoxicity against HepG2, SMMC-7721, and Huh7 cell lines. The mechanism of the most active compound 12 was investigated.

1. Introduction
Hepatocellular carcinoma (HCC) is the major type of primary liver cancer and the third leading cause of cancer-related deaths worldwide1,2. Accumulating evidence suggests that HCC threatens people's heath more and more seriously, and the primary liver cancer incidence is still on the rise at the global level3, 4, 5. Although seven antihepatoma drugs including sorafenib, regorafenib, lenvatinib, cabozantinib, nivolumab, pembrolizumab, and ramucirumab are effectively therapeutic agents clinically, there are still disadvantages involving the low response rate, serious side effects, and drug resistance. Therefore, it is desirable to search the new and effective drugs against human hepatoma. Sesquiterpenoids and their dimers are reported to exhibit diverse activities6, 7, 8, 9, such as antitumor, antiinflammation, antimalaria, neurotrophic, etc., which are a group of effective and low-toxicity natural small molecules10, 11, 12, 13. Guaianolide dimers are a class of intriguing sesquiterpenoid dimers predominantly in Asteraceae and Chloranthaceae families, especially in the Artemisia species14. The genus Artemisia (Asteraceae) includes about 380 species distributed all over the world, and 186 species in China15. Some species from the genus Artemisia, such as A. annua, A. argyi, A. capillaris, A. scoparia, and A. anomala, are used as the famous traditional Chinese medicinal herbs to treat a variety of diseases including malaria, hepatitis, cancer, eczema, diarrhea, bruise, and rheumatic disease and so on16,17. Up to now, a lot of Artemisia species had been phytochemically investigated to conclude that plants of the Artemisia genus were rich in sesquiterpenoids, especially guaianolides and their dimers18, 19, 20, 21, 22, 23, 24, and 77 dimeric guaianolides had been reported from the Artemisia plants in last 40 years, including 19 ones from A. argyi17,18,25, 26, 27, 11 ones from A. absinthium23,28, 29, 30, 31, 32, 11 ones from A. anomala20,33, 34, 35, six ones from A. rupestris36, five ones from A. sieversiana21,37, five ones from A. caruifolia38, artemyriantholides A–D from A. myriantha19, arteminolide and 8-acetylarteminolide from A. sylvatica24,39, artselenoide from A. selengensis40, artelein from A. leucodes41, and lavandiolides A‒L from A. lavandulifolia42. Based on the connecting model of the two monomeric sesquiterpenoid units, these dimeric guaianolides are classified as Diels–Alder, [2 + 2] cycloaddition, and ester linkage adducts (Supporting Information Table S1). Biogenetically, 71 dimers are derived from [4 + 2] cycloaddition of two guaiane moieties, while artelein and artesin A are formed via tandem [2 + 2]/[2 + 2] cycloadditions, and artemisianes A–D are condensed through esterification of two monomeric units. Interestingly, guaianolide-type dimers from the genus Artemisia species exhibited antitumor, antiinflammation, antivirus activities and so on. Artanomadimers A and F manifested cytotoxicity against the BGC-823 tumor cell line with IC50 values of 2.7 and 6.3 μmol/L35. Arteminolides A–D and 8-acetylarteminolide inhibited the farnesyl protein transferase (FPTase) with IC50 values of 0.7–1.025 and 1.8 μmol/L39. Artemisian B exhibited antiproliferative activity via apoptosis induction and G2/M arrest in MDA-MB-468 cells with an IC50 value of 3.2 μmol/L18. Artemisianin A displayed cytotoxicity against HT-29 cells with an IC50 value of 7.2 μmol/L and mediated cell apoptosis27. Absinthin C and isoanabsinthin showed inhibitory effects on LPS-induced NO production in BV-2 cells with IC50 values of 1.5 and 2.0 μmol/L32. Artemisianes A–D and lavandiolides A, C, G, and I demonstrated inhibitory activity on LPS-induced NO production in RAW 264.7 cells with IC50 values ranging from 0.6 to 32.1 μmol/L12,42. Caruifolin B showed anti-HIV activity by inhibiting the HIV-1-induced cytopathic effect in MT cells at 500 μg/mL38. Prompted by the diverse activities and intricate structures of guaianolide dimers in Artemisia species, phytochemical and biological investigation on this genus is an attractive topic.
To investigate structurally novel and bioactive dimeric sesquiterpenoids from natural plants, our assay suggested that the EtOH extract of A. atrovirens exhibited cytotoxicity against HepG2 cells with the inhibitory ratio of 98.9% at the concentration of 100.0 μg/mL. Previous report on essential oils from A. atrovirens by GC–MS analysis revealed its main constituents as 1,3-cyclopentadiene,5-(1,1-dimethylethyl), azulen-2-ol,1,4-dimethyl-7-(1-methylethyl), and eucalyptol, but no active compounds from A. atrovirens have been reported so far43. In our endeavor to search for novel and antihepatoma compounds from this species, 18 new guaianolide dimers and five known compounds lavandiolides A (3), B (4), C (23), H (12), and J (9), were isolated and identified with a spiro-system composed of two monomeric sesquiterpene lactone units. These five known compounds were just reported by Ye et al.42 from A. lavandulifolia during the revision of this manuscript. Nineteen compounds showed cytotoxicity, and notably four compounds (1, 12, 14, and 15) demonstrated significant cytotoxicity against three human hepatoma cell lines (HepG2, SMMC-7721, and Huh7). Herein, we described their isolation, structural elucidation, cytotoxicity, and the preliminary mechanism of the most active lavandiolide H (12).
2. Results and discussion
The EtOH extracts of the leaves of A. atrovirens were partitioned between EtOAc and H2O. The active EtOAc fraction was subjected to silica gel, MCI gel CHP 20P, Sephadex LH-20, preparative HPLC, and semi-preparative HPLC to afford 23 sesquiterpenoid dimers (Fig. 1). The structures of artematrolides A–R (1, 2, 5–8, 10, 11, 13–22) including their absolute configurations, were elucidated based on analyses of HRESIMS, 1D/2D NMR, ECD spectra, and single-crystal X-ray diffraction techniques.
Figure 1.

The structures of compounds 1–23.
Artematrolide A (1) was obtained as colorless orthorhombic crystals with a molecular formula of C30H36O6 as inferred from its (+)-HRESIMS ion at m/z 515.2411 [M+Na]+ (Calcd. for 515.2404), suggesting 13 degrees of unsaturation. Its IR spectrum displayed characteristic absorption bands assignable to hydroxy (3476 cm−1), ester carbonyl (1767 and 1744 cm−1), and olefinic (1629 cm−1) functional groups. The 1H NMR spectrum (Table 1) of compound 1 exhibited four singlet methyls at δH 1.26 (3H, s), 1.34 (3H, s), 1.50 (3H, s), and 1.92 (3H, s), two oxygenated methines at δH 5.24 (1H, d, J=9.6 Hz) and 4.38 (1H, dd, J=10.2, 10.2 Hz), and three olefinic protons at δH 6.18 (1H, d, J=3.3 Hz), 5.46 (1H, d, J=3.3 Hz), and 5.56 (1H, m). Its 13C NMR spectrum (Table 4) revealed the presence of 30 carbons classified as four methyls, eight methylenes, seven methines, and 11 nonprotonated carbons. Among these carbons, two ester carbonyls at δC 183.6 and 170.2, and six olefinic carbons at δC 148.6, 144.9, 140.9, 139.9, 125.2, and 119.4 were easily recognized in the deshielded region. The abovementioned NMR and MS features suggested a dimeric sesquiterpenoid for compound 1.
Table 1.
1H NMR spectroscopic data for compounds 1, 2, and 5–8 (δ in ppm, J in Hz).
| No. position | 1a,c | 2a,c | 5b,d | 6b,d | 7a,c | 8b,d |
|---|---|---|---|---|---|---|
| 1 | 2.34 br s | 2.33 br s | ||||
| 2 | 3.24 br s | 3.20 br s | 3.25 m | 3.23 m | 3.06 m | 3.04 m |
| 3 | 1.46 dd (9.0, 1.8) | 1.44 m | 5.44 br s | 5.42 br s | 2.53 dd (9.6, 1.5) | 1.58 m |
| 1.35 m | 1.33 m | 1.14 m | 1.39 m | |||
| 6 | 5.24 d (9.6) | 5.28 d (9.6) | 4.56 d (10.0) | 4.76 d (11.2) | 5.75 d (8.4) | 5.56 (overlapped) |
| 7 | 2.85 m | 1.90 m | 2.94 m | 2.51 m | 3.54 m | 2.96 m |
| 8 | 2.11 m | 1.94 (overlapped) | 2.23 m | 1.86 (overlapped) | 2.58 m | 2.25 m |
| 1.88 m | 1.87 m | 1.44 m | 1.43 m | 1.94 m | 1.73 m | |
| 9 | 2.00 (overlapped) | 1.94 (overlapped) | 1.81 m | 1.75 m | 4.94 dd (5.4, 1.8) | 2.00 m |
| 1.70 m | 1.61 m | 1.64 m | 1.54 m | 1.83 m | ||
| 11 | 2.24 m | 2.71 m | ||||
| 13 | 6.18 d (3.3) | 1.22 (overlapped) | 6.22 d (3.2) | 1.23 d (8.0) | 6.38 br s | 6.11 d (3.6) |
| 5.46 d (3.3) | 5.68 d (3.2) | 5.54 br s | 5.57 (overlapped) | |||
| 14 | 1.26 s | 1.22 (overlapped) | 1.06 s | 1.04 s | 2.03 s | 1.42 s |
| 15 | 1.50 s | 1.47 s | 1.84 s | 1.84 s | 1.14 s | 1.50 s |
| 2′ | 2.75 m | 2.75 br s | 2.82 m | 2.82 m | 2.74 m | |
| 2.18 (overlapped) | 2.16 (overlapped) | 2.14 m | 2.14 m | 2.11 m | ||
| 3′ | 5.56 m | 5.57 br s | 5.60 m | 5.60 m | 5.56 m | 6.16 s |
| 5′ | 2.79 m | 2.79 d (10.2) | 2.88 m | 2.88 m | 2.81 d (10.5) | 3.70 d (10.0) |
| 6′ | 4.38 dd (10.2, 10.2) | 4.37 dd (10.2, 10.2) | 4.30 dd (10.4, 10.0) | 4.29 dd (10.4, 10.0) | 3.85 dd (10.5, 10.2) | 3.65 dd (10.0, 9.6) |
| 7′ | 1.78 m | 1.77 m | 1.94 (overlapped) | 1.93 m | 1.34 m | 2.70 m |
| 8′ | 1.61 m | 1.61 m | 1.87 m | 1.87 (overlapped) | 1.27 m | 1.98 m |
| 1.57 m | 1.57 m | 1.67 m | 1.68 m | 1.20 m | 1.58 m | |
| 9′ | 2.18 (overlapped) | 2.17 (overlapped) | 2.22 m | 2.22 m | 2.00 m | 2.65 m |
| 1.91 m | 1.89 m | 2.03 m | 2.04 m | 1.79 m | 2.35 m | |
| 13′ | 1.99 (overlapped) | 1.97 m | 1.76 d (12.4) | 1.72 m | 1.92 m | 2.21 m |
| 1.39 d (12.0) | 1.36 m | 1.70 m | 1.65 m | 1.65 m | 1.61 m | |
| 14′ | 1.34 s | 1.34 s | 1.34 s | 1.34 s | 1.31 s | 2.42 s |
| 15′ | 1.92 s | 1.93 s | 1.94 (overlapped) | 1.94 s | 1.93 s | 2.33 s |
Recorded in CDCl3.
Recorded in CD3OD.
Recorded at 600 MHz.
Recorded at 400 MHz.
Table 4.
13C NMR spectroscopic data for compounds 1, 2, 5–8, 10, 11, and 13 (δ in ppm).
| No. | 1a,c | 2a,c | 5b,d | 6b,d | 7a,c | 8b,d | 10a,d | 11a,c | 13b,d |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 148.6 | 147.8 | 69.6 | 69.4 | 142.7 | 153.1 | 158.4 | 157.9 | 154.9 |
| 2 | 48.7 | 48.6 | 46.0 | 46.0 | 41.2 | 42.5 | 41.4 | 42.9 | 42.0 |
| 3 | 55.1 | 55.0 | 125.1 | 124.8 | 42.3 | 53.1 | 56.8 | 52.4 | 55.8 |
| 4 | 54.6 | 54.4 | 144.5 | 144.7 | 57.2 | 59.5 | 61.2 | 60.4 | 61.0 |
| 5 | 144.9 | 145.3 | 57.3 | 57.5 | 143.7 | 143.0 | 139.1 | 132.9 | 140.4 |
| 6 | 80.2 | 80.3 | 83.6 | 83.0 | 125.6 | 83.7 | 78.3 | 77.0 | 82.5 |
| 7 | 46.6 | 50.2 | 42.0 | 40.5 | 35.4 | 46.3 | 45.4 | 45.0 | 46.9 |
| 8 | 23.7 | 25.3 | 25.5 | 23.2 | 27.2 | 22.8 | 21.7 | 28.4 | 23.8 |
| 9 | 37.9 | 38.4 | 45.4 | 45.6 | 81.0 | 37.4 | 39.8 | 39.7 | 37.7 |
| 10 | 71.4 | 70.8 | 72.5 | 72.4 | 129.2 | 71.9 | 73.0 | 73.7 | 71.4 |
| 11 | 139.9 | 42.2 | 139.8 | 39.6 | 136.1 | 140.6 | 139.2 | 141.0 | 140.0 |
| 12 | 170.2 | 178.7 | 170.9 | 181.1 | 165.6 | 170.7 | 169.4 | 169.7 | 170.4 |
| 13 | 119.4 | 12.8 | 119.4 | 9.3 | 129.4 | 118.3 | 119.9 | 122.7 | 118.7 |
| 14 | 28.6 | 28.8 | 25.8 | 25.8 | 21.1 | 25.5 | 25.2 | 29.0 | 26.5 |
| 15 | 20.4 | 20.3 | 11.5 | 11.5 | 13.6 | 16.1 | 13.5 | 12.7 | 17.4 |
| 1′ | 72.1 | 72.0 | 72.2 | 72.1 | 73.0 | 131.8 | 72.8 | 72.8 | 72.4 |
| 2′ | 39.3 | 39.1 | 38.5 | 38.5 | 39.8 | 196.8 | 39.4 | 39.7 | 38.8 |
| 3′ | 125.2 | 125.1 | 124.9 | 124.8 | 124.8 | 134.6 | 124.6 | 125.0 | 124.6 |
| 4′ | 140.9 | 140.8 | 140.6 | 140.6 | 141.0 | 172.9 | 140.9 | 141.1 | 140.6 |
| 5′ | 52.0 | 51.9 | 51.6 | 51.6 | 54.1 | 52.0 | 53.2 | 53.8 | 53.3 |
| 6′ | 81.4 | 81.3 | 81.2 | 81.2 | 81.3 | 81.7 | 81.0 | 81.1 | 81.7 |
| 7′ | 55.7 | 55.7 | 54.6 | 54.6 | 50.2 | 53.8 | 49.4 | 50.3 | 56.4 |
| 8′ | 21.1 | 21.0 | 20.8 | 20.8 | 22.7 | 23.9 | 22.7 | 23.0 | 19.5 |
| 9′ | 33.3 | 33.2 | 32.6 | 32.6 | 34.5 | 36.1 | 33.0 | 34.3 | 33.7 |
| 10′ | 62.1 | 61.9 | 62.2 | 62.2 | 63.0 | 153.4 | 62.6 | 62.6 | 62.6 |
| 11′ | 55.9 | 55.5 | 53.8 | 53.6 | 52.8 | 56.4 | 54.4 | 54.7 | 56.8 |
| 12′ | 183.6 | 183.6 | 179.8 | 179.8 | 181.0 | 179.6 | 180.8 | 180.9 | 182.5 |
| 13′ | 42.8 | 42.6 | 34.5 | 35.0 | 34.7 | 34.3 | 35.1 | 34.6 | 41.0 |
| 14′ | 22.7 | 22.6 | 21.4 | 21.3 | 22.8 | 19.8 | 22.5 | 22.8 | 21.3 |
| 15′ | 18.8 | 18.6 | 17.5 | 17.5 | 18.6 | 18.9 | 18.6 | 18.7 | 17.4 |
Recorded in CDCl3.
Recorded in CD3OD.
Recorded at 150 MHz.
Recorded at 100 MHz.
The planar structure of 1 involving units A and B was mainly accomplished by analyzing the 2D NMR data (Fig. 2). The 1H–1H COSY spectrum revealed four isolated spin-coupling systems of H-6/H-7/H2-8/H2-9, H-2/H2-3, H-6′/H-7′/H2-8′/H2-9′, and H2-2′/H-3′. The HMBC spectrum showed correlations from H2-13 to C-7, C-11, and C-12, from H3-14 to C-1, C-9, and C-10, from H3-15 to C-3, C-4, and C-5, from H-8 to C-6, C-10, and C-11, and from H-2 to C-1, C-3, C-4, and C-5 in unit A; and correlations from H3-14′ to C-1′, C-9′, and C-10′, from H3-15′ to C-3′, C-4′, and C-5′, from H-7′ to C-5′, C-6′, C-8′, C-9′, C-12′, and C-13′, and from H2-13′ to C-7′, C-11′, and C-12′ in unit B. From the above analyses, both units A and B were deduced as guaianolide-like moieties similar to arglabin19. The linkage of units A and B through two C–C single bonds of C-2−C-11′ and C-4−C-13′ was established by the key HMBC correlations of H2-13′ with C-3, C-4, C-5, and C-15, of H-2 with C-7′, C-11′, C-12′, and C-13′, of H-3 with C-11′ and C-13′, and of H3-15 with C-13′.
Figure 2.

Selected 2D NMR correlations of compounds 1, 2, 5–8, 10, 11, and 13–22.
The relative configuration of 1 was determined by interpretation of ROESY spectrum (Fig. 3) and coupling constants. With the fact that H-7 of guaiane-type sesquiterpenoids always maintained the α-orientation37,44,45, the large coupling constant between H-6 and H-7 (J = 9.6 Hz) inferred that they were in the anti-axial configuration, i.e., H-6 was assigned to be β-orientated46. In the ROESY spectrum, the cross peaks of H-7/H-3/H3-14 indicated that CH2-3 and CH3-14 were α-orientated. Similarly, the ROESY correlation of H-7′ with H-5′ and the large coupling constant (JH-6′/H-7′ = 10.2 Hz) assigned the α-orientation of H-5′ and H-7′ and β-orientation of H-6′. To our delight, suitable crystals of compound 1 were obtained from an optimized solvent system (MeOH–CH2Cl2, 10:90, v/v), which facilitated the single-crystal X-ray diffraction experiment with Cu Kα radiation (Fig. 4). Consequently, the absolute configuration of compound 1 was unambiguously determined as 2R,4R,6S,7S,10S,1′R,5′R,6′S,7′S,10′S,11′R.
Figure 3.

Key ROESY correlations of compounds 1, 2, 5–8, 10, 11, and 13–22.
Figure 4.

ORTEP drawings of compounds 1, 4, 10, 12, 13, and 19–21.
Artematrolide B (2) was isolated as white powders with a molecular formula of C30H38O6 from the (+)-HRESIMS ion at m/z 517.2561 [M+Na]+ (Calcd. for 517.2561), indicating 12 degrees of unsaturation. The 1H and 13C NMR data of 2 (Table 1, Table 4) highly resembled those of 1, and the main difference was that the terminal olefinic carbon (δH 6.18 and 5.46, δC 139.9 and 119.4) in 1 was replaced with an ethylidene (δH 2.24 and 1.22, δC 42.2 and 12.8) in 2. Taking its molecular weight (two Da higher than 1) into consideration, compound 2 was reasonably deduced as the 11,13-dihydro derivative of 1. The above deduction was confirmed by the 1H–1H COSY correlations of H3-13/H-11/H-7, and HMBC correlations from H3-13 (δH 1.22, overlapped) to C-7 (δC 50.2) and C-12 (δC 178.7), and from H-11 (δH 2.24, m) to C-6 (δC 80.3) and C-8 (δC 25.3). In the ROESY spectrum (Fig. 3), the cross peaks of H-7/H-11 and H-6/H3-13 verified the β-orientation of the methyl at C-11. The absolute configuration of 2 was confirmed by means of ECD calculation. The calculated ECD spectrum matched well with the experimental one as shown in Fig. 5, confirming the absolute configuration as 2R,4R,6S,7S,10S,11R,1′R,5′R,6′S,7′S,10′S,11′R.
Figure 5.

Experimental and calculated ECD spectra of compounds 2, 5–8, 11, 14–18, and 22.
Artematrolide C (5) was assigned to a molecular formula of C30H36O6 by the (+)-HRESIMS ion at m/z 493.2577 [M+H]+ (Calcd. for 493.2585). Detailed interpretation of its 1H and 13C NMR data (Table 1, Table 4) proposed that compound 5 maintained the similar units A and B with compound 1, and the differences in their NMR data of the cyclopentene moiety (unit A) indicated a different connecting model of two parts. In the HMBC spectrum (Fig. 2), the correlations from H-2 to C-7ʹ, C-11ʹ, and C-12ʹ, and from H2-13ʹ to C-1, C-4, C-5, and C-6 supported the connections of C-2‒C-11′ and C-5‒C-13′, instead of C-2‒C-11′ and C-4‒C-13′ in compound 1. From a biosynthetic point of view, compound 5 was connected via a [4 + 2] Diels–Alder cycloaddition of arglabin as the dienophile19 and the conjugated Δ2,4(5)-cyclopentadiene unit as the diene. In the ROESY spectrum (Fig. 3), the correlations of H-7/H-1 and H-6/H3-14 manifested the α-orientation of H-1, and β-orientation of H-6 and CH3-14. Similarly, H-5′ and H-6′/H-2 were respectively assigned as α- and β-orientated by the ROESY correlations of H-5ʹ/H-7ʹ and H-6′/H-8′b/H-2. The α-orientation of CH2-13′ was defined by the ROESY correlation of H-7ʹ/H-13ʹa, which was consistent with that in compound 1. Its absolute configuration was confirmed as 1R,2S,5R,6S,7S,10R,1′R,5′R,6′S,7′S,10′S,11′R by comparing the experimental ECD spectrum with the calculated one (Fig. 5).
Artematrolide D (6) was deduced as the 11,13-dihydro derivative of compound 5 from its chemical composition with two additional hydrogens than compound 5, and their highly similar NMR data (Table 1, Table 4) except for positions at C-11 and C-13. Compared with compound 5, compound 6 showed the presence of a methine (δC 39.6) and a methyl (δC 9.3) but with the absence of a terminal olefinic group. This methyl group at C-11 was clearly assigned as β-orientated based on the 1H–1H COSY correlation of H-11/H-7, HMBC correlations (Fig. 2) from H3-13 to C-7 and C-12, and ROESY correlation of H-6/H3-13 (Fig. 3). Its absolute stereochemistry was assigned to be 1R,2S,5R,6S,7S,10R,11R,1′R,5′R,6′S,7′S,10′S,11′R by the high agreement between the experimental and calculated ECD spectra (Fig. 5).
Artematrolide E (7) had a molecular formula of C30H34O5 deduced by the (+)-HRESIMS ion at m/z 475.2473 [M+H]+ (Calcd. for 475.2479), revealing 14 hydrogen deficiency indices. The 1H NMR data of 7 (Table 1) displayed signals of four singlet methyl groups at δH 1.14 (3H, s), 1.31 (3H, s), 1.93 (3H, s), and 2.03 (3H, s), an exocyclomethylene at δH 6.38 (1H, br.s) and 5.54 (1H, br.s), two oxygenated methines at δH 4.94 (1H, dd, J=5.4, 1.8 Hz) and 3.85 (1H, dd, J=10.5, 10.2 Hz), and two olefinic protons at δH 5.75 (1H, d, J=8.4 Hz) and 5.56 (1H, m). The 13C NMR (DEPT) spectrum (Table 4) revealed 30 carbon resonances attributable to two ester carbonyls (δC 181.0, 165.6), terminal olefinic carbons (δC 136.1 and 129.4), trisubstituted olefinic carbons (δC 143.7, 125.6, 141.0, and 124.8), tetrasubstituted olefinic carbons (δC 142.7 and 129.2), four methyls, six sp3 methylenes, six sp3 methines (two oxygenated), and four sp3 quaternary carbons. With the characteristic signals of two sets of lactone groups at δC 165.6 (C-12) and 181.0 (C-12ʹ), it was suggested that compound 7 should be a dimeric sesquiterpene lactone.
By interpretation of the 2D NMR data (1H–1H COSY and HMBC, Fig. 2) of compound 7, four spin-coupling systems of H-6/H-7/H2-8/H-9 and H-2/H2-3 in unit A, and H-6′/H-7′/H2-8′/H2-9′ and H2-2′/H-3′ in unit B were readily furnished by the 1H–1H COSY correlations. The HMBC correlations from H2-13 to C-7, C-11, and C-12; from H3-14 to C-1, C-9, and C-10; from H3-15 to C-3, C-4, and C-5; from H-7 to C-5, C-9, C-12, and C-13; from H-9 to C-1, C-7, C-12, and CH3-14 in unit A; and from H3-14′ to C-1′, C-9′, and C-10′, from H3-15′ to C-3′, C-4′, and C-5′, from H-7′ to C-5′, C-6′, C-8′, and C-9′, and from H-13′ to C-12′ in unit B were observed. Therefore, unit A was deduced as a six-membered lactone guaianolide moiety, and the unit B was determined to be similar to arglabin19. Moreover, the HMBC correlations of H2-13′/C-1, C-2, and C-3, of H-3/C-11′ and C-13′, and of H3-15/C-11′ indicated that units A and B were connected via two C–C single bonds between C-2−C-13′ and C-4−C-11′. Consequently, the planar structure of compound 7 was proposed as illustrated in Fig. 1.
The relative configuration of compound 7 was partially established by analysis of the ROESY data (Fig. 3). With the fact that C-7 of guaiane-type sesquiterpenoids always maintained the S configuration37,44,45, the ROESY correlations of H-7/H-9, H-9/H3-14, H3-15/H-6, H3-15/H-7′ and H-7/H-3b indicated that H-7, H-9, CH3-15, and CH2-3 were α-orientated, and the ROESY correlations of H-7′/H-5′/H-8′a assigned H-5′, H-8′a to be in α-orientation, while the ROESY correlations of H-6′/H-8′b/H-13′ deduced H-6′ and CH2-13′ to be in β-configuration. This assignment was in accordance with the biogenetic origin of dimeric sesquiterpenes isolated from this genus. The absolute configuration of compound 7 was determined to be 2S,4S,7R,9S,1′R,5′R,6′S,7′S,10′S,11′R by comparison of its experimental ECD spectrum with the calculated one (Fig. 5).
Artematrolide F (8) was proposed to have a molecular formula of C30H34O6 according to the (+)-HRESIMS ion at m/z 491.2413 [M+H]+ (Calcd. for 491.2428). The 1H NMR spectrum (Table 1) showed the signals of four methyl groups at δH 1.42, 1.50, 2.33, and 2.42 (each 3H, s), an exocyclomethylene at δH 6.11 (1H, d, J=3.6 Hz) and 5.57 (1H, overlapped), two oxygenated methines at δH 5.56 (1H, overlapped) and 3.65 (1H, dd, J=10.0, 9.6 Hz), and an olefinic proton at δH 6.16 (1H, s). The 13C NMR spectrum (Table 4) of compound 8 exhibited 30 carbon resonances ascribe to four methyls, seven methylenes (including one exomethylene), seven methines, and twelve quaternary carbons. Further analyses of 1D and 2D NMR data gave rise to the construction of units A (identical with that in 1) and B (dehydroleucodin19). The HMBC correlations (Fig. 2) of H2-13′/C-1, C-2, and C-3; of H3-15/C-11′ revealed that units A and B were connected via two C–C single bonds between C-2 and C-13′ and between C-4 and C-11′, identical to that in 7. The ROESY correlations (Fig. 3) of H-3a with H-8′a, of H-8′a with H-7′ and the characteristic chemical shift of H-3a (δH 1.58) suggested the endo stereochemistry of compound 8 with the 2,4-linked form47. In addition, the cross peaks of H-7/H-3b, H-7/H3-14, H3-14/H-9b, H-7′/H-8′a, H-7′/H-5′, and H-7′/H3-15 in the ROESY spectrum indicated that CH2-3, CH3-14, and H-5′ were α-orientated, while the cross peaks of H-6/H-9a, H-6′/H-8′b, H-6′/H2-13′ revealed that H-6, H-6′, and CH2-13′ were β-orientated. By means of ECD calculation (Fig. 5), its absolute configuration was established as 2S,4S,6S,7S,10S,5′S,6′S,7′S,11′R.
Artematrolide G (10) was obtained as colorless monoclinic crystals with a molecular formula of C30H36O6 deduced by the (+)-HRESIMS ion at m/z 493.2538 [M+H]+ (Calcd. for 493.2544). The 1H and 13C NMR data (Table 2, Table 4) of 10 showed high similarity with those of 1 with the main difference around the cyclopentene moiety, suggesting a different connecting model between two sesquiterpenoid parts. In the HMBC spectrum (Fig. 2), the correlations of H2-13′/C-1 and C-3, of H-2/C-11′, of H-3/C-11′ and C-13′, and of H3-15/C-11′ demonstrated the linkages of C-2−C-13′ and C-4−C-11′ in 10. The coupling constant (10.4 Hz) of JH-6/H-7 indicated the trans-axial orientation of H-6 and H-7, and H-6 was deduced as β-orientated. In the ROESY spectrum (Fig. 3), the correlations of H-7 with H3-14 and H-6/H-3b verified the α-orientation of CH3-14 and β-orientation of CH2-3. Its structure was consolidated by the single-crystal X-ray diffraction analysis to be 2R,4R,6S,7S,10S,1′R,5′R,6′S,7′S,10′S,11′R (Fig. 4).
Table 2.
1H NMR spectroscopic data for compounds 10, 11, and 13–16 (δ in ppm, J in Hz).
| No. position | 10a,d | 11a,c | 13b,d | 14a,c | 15b,d | 16a,c |
|---|---|---|---|---|---|---|
| 2 | 3.19 m | 3.05 (overlapped) | 2.90 m | 5.86 d (5.4) | 6.41 d (5.6) | 5.85 d (5.7) |
| 3 | 2.75 m | 2.58 dd (9.0, 1.2) | 2.36 dd (8.3, 1.6) | 6.00 d (5.4) | 5.86 d (5.6) | 6.34 d (5.7) |
| 1.43 m | 1.35 m | 1.25 dd (8.3, 2.4) | ||||
| 5 | 2.99 d (10.2) | 2.96 d (10.0) | 2.31 d (9.9) | |||
| 6 | 4.87 d (10.4) | 4.70 d (6.6) | 5.29 d (9.6) | 4.06 dd (10.2, 9.6) | 4.17 dd (10.0, 9.6) | 4.18 dd (9.9, 9.6) |
| 7 | 2.98 m | 3.05 (overlapped) | 2.91 m | 3.43 m | 2.87 m | 3.26 m |
| 8 | 2.19 m | 1.87 m | 2.24 m | 2.27 m | 2.26 m | 2.23 m |
| 1.71 m | 1.74 m | 1.87 (overlapped) | 1.34 m | 1.60 m | 1.48 m | |
| 9 | 2.02 m | 1.92 m | 1.91 m | 1.90 m | 2.05 m | 1.88 m |
| 1.90 m | 1.74 m | 1.87 (overlapped) | 1.84 (overlapped) | 1.81 m | 1.84 m | |
| 13 | 6.22 d (3.2) | 6.29 d (1.2) | 6.14 d (3.6) | 6.08 d (3.3) | 6.05 d (3.4) | 6.10 d (3.6) |
| 5.51 d (3.2) | 5.70 d (1.2) | 5.60 d (3.6) | 5.36 d (3.3) | 5.50 d (3.4) | 5.37 d (3.6) | |
| 14 | 1.37 s | 1.43 s | 1.44 s | 1.45 s | 1.44 s | 1.30 s |
| 15 | 1.41 s | 1.34 s | 1.47 s | 1.44 s | 1.49 s | 1.35 s |
| 2′ | 2.73 m | 2.73 br d | 2.80 m | 2.72 br d | 2.80 m | 2.77 dd (17.9, 3.3) |
| 2.09 m | 2.12 m | 2.12 m | 2.12 br d | 2.13 (overlapped) | 2.15 (overlapped) | |
| 3′ | 5.55 m | 5.56 m | 5.61 m | 5.57 br s | 5.61 m | 5.55 m |
| 5′ | 2.93 d (10.2) | 2.82 d (10.2) | 2.81 m | 2.83 d (10.2) | 2.78 m | 2.82 d (10.2) |
| 6′ | 3.88 dd (10.4, 10.2) | 3.90 dd (10.2, 10.2) | 4.30 dd (10.4, 10.4) | 3.99 dd (10.2, 10.2) | 4.32 dd (10.4, 10.4) | 4.06 dd (10.2, 9.0) |
| 7′ | 1.90 m | 1.58 m | 1.83 m | 2.36 m | 1.82 m | 1.94 m |
| 8′ | 1.31 m | 1.43 m | 1.71 m | 1.63 m | 1.69 m | 1.72 m |
| 1.16 m | 1.29 m | 1.46 m | 1.33 m | 1.29 m | 1.69 m | |
| 9′ | 1.81 m | 2.10 m | 2.18 m | 2.02 m | 2.13 (overlapped) | 2.14 (overlapped) |
| 1.74 m | 1.81 m | 1.88 m | 1.83 m | 1.84 m | 2.00 m | |
| 13′ | 1.98 m | 1.98 dd (12.6, 4.2) | 2.49 dd (12.4, 4.4) | 1.84 (overlapped) | 2.51 d (12.3) | 2.40 d (11.7) |
| 1.70 m | 1.77 m | 1.45 m | 1.75 d (12.0) | 1.40 d (12.3) | 1.40 d (11.7) | |
| 14′ | 1.25 s | 1.33 s | 1.33 s | 1.29 s | 1.32 s | 1.34 s |
| 15′ | 1.96 s | 1.95 s | 1.97 s | 1.95 s | 1.97 s | 1.92 s |
Recorded in CDCl3.
Recorded in CD3OD.
Recorded at 600 MHz.
Recorded at 400 MHz.
Compound 11 had the same molecular formula and similar 1H and 13C NMR data (Table 2, Table 4) with 10, indicating the closely related structures. The same planar structure of 11 with 10 was determined by detailed interpretation of its 1H–1H COSY and HMBC experiments (Fig. 2). Compared with 10, the chemical shifts of C-3 in 11 was changed to δC 52.4 (vs. 56.8 in 10), and C-8 and C-14 were de-shielded to δC 28.4 (vs. 21.7 in 10) and 29.0 (vs. 25.2 in 10), respectively, suggesting the varied stereochemistry. The coupling constant (6.6 Hz) of JH-6/H-7 indicated the cis-axial orientation of H-6 and H-7, i.e., H-6 was deduced as α-orientated. Furthermore, the cross peaks of H-6/H3-15 and H3-15/H-7′, H-7′/H-5′ indicated that H-5′ and CH3-15 was α-configuration, while the correlations of H3-14/H-2, H3-14/H-3b, H-6′/H2-13′, H-6′/H-8′b, and H-8′b/H-13′a in the ROESY spectrum revealed that CH2-3, CH3-14, and CH2-13′ was β-orientated (Fig. 3). By comparing the experimental and calculated ECD spectrum (Fig. 5), its absolute configuration was assigned as 2R,4R,6R,7S,10R,1′R,5′R,6′S,7′S,10′S,11′R.
Compound 13 had a molecular formula of C30H36O6 from the (+)-HRESIMS ion at m/z 493.2069 [M+H]+ (Calcd. for 493.2068). The 13C NMR data of 13 were very similar to those of 12, and the main differences were observed at C-1, C-3, C-5, C-6, C-7′, C-12′, and C-13′ (Table 4 and Table S2). By detailed interpretation of its 1H–1H COSY and HMBC data (Fig. 2), the identical planar structure of 13 with 12 was constructed. In the ROESY spectrum (Fig. 3), the correlations of H-7/H-3b, H-7/H-8a, H-7′/H2-13′, H-7′/H-8′a, H-8′a/H-13′b verified the α-orientation of CH2-3 and CH2-13′, while the ROESY correlation of H-6/H-8b together with the big coupling constant (JH-6/H-7 = 9.6 Hz) assigned H-6 as β-orientated. The structure of 13 was confirmed though a single crystal X-ray diffraction experiment with Cu Kα radiation [Flack parameter of 0.03(6)], and the absolute configuration was confirmed as 2S,4S,6S,7S,10S,1′R,5′R,6′S,7′S,10′S,11′S (Fig. 4). Consequently, the structure of 13 was established and named as artematrolide I.
Artematrolide J (14) was assigned a molecular formula of C30H36O6 from the (+)-HRESIMS ion at m/z 493.2594 [M+H]+ (Calcd. for 493.2585). By detailed interpretation of the 1D and 2D NMR data (Table 2, Table 5), compound 14 was proposed as a guaianolide dimer similar with compound 13. Differently, the two units of compound 14 were deduced to be linked via C-1‒C-11′ and C-4‒C-13′ based on the HMBC correlations of H2-13′/C-3, C-4, C-5, and C-15, of H-2/C-11′, of H-5/C-11′ and C-13′, and of H3-15/C-13′ (Fig. 2). In the ROESY spectrum, cross signals detected for H-7/H-5, H-7/H-8a, H-5/H-2, H-5/H-3, H-7′/H-5′, and H-7′/H-8′a suggested the α-orientation of H-5, H-5′, and the ethenylene (C-2–C-3), the correlation of H-7′/H-13′, H-8′a/H-13′a revealed the same orientation H-7′ and CH2-13′, while the cross signals of H-6/H-8b, H-8b/H3-14, H-6/H3-14, and H-6′/H-8′b indicated the β-orientation of H-6, H-6′, and CH3-14 (Fig. 3). By virtue of ECD calculation, the absolute configuration of compound 14 was determined as 1S,4S,5S,6S,7S,10R,1′R,5′R,6′S,7′S,10′S,11′R (Fig. 5).
Table 5.
13C NMR spectroscopic data for compounds 14–22 (δ in ppm).
| No. | 14a,c | 15b,d | 16a,c | 17a,c | 18a,d | 19a,c | 20a,d | 21a,d | 22a,c |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 71.6 | 63.2 | 63.8 | 64.3 | 62.8 | 73.5 | 71.5 | 70.5 | 74.0 |
| 2 | 133.4 | 136.8 | 133.2 | 133.5 | 136.4 | 49.2 | 46.9 | 47.7 | 49.3 |
| 3 | 140.8 | 136.3 | 142.0 | 141.2 | 137.7 | 44.7 | 45.4 | 46.0 | 44.8 |
| 4 | 52.0 | 61.4 | 57.1 | 57.7 | 61.1 | 153.3 | 142.8 | 152.0 | 154.3 |
| 5 | 68.3 | 66.2 | 66.6 | 66.6 | 66.8 | 133.6 | 134.5 | 135.7 | 133.0 |
| 6 | 80.3 | 79.0 | 80.3 | 79.1 | 79.3 | 77.3 | 82.4 | 75.1 | 76.5 |
| 7 | 43.8 | 43.4 | 43.6 | 44.0 | 43.2 | 45.2 | 49.7 | 39.6 | 45.8 |
| 8 | 23.9 | 22.6 | 23.6 | 23.0 | 23.6 | 21.7 | 21.8 | 25.4 | 28.0 |
| 9 | 38.2 | 36.3 | 35.0 | 37.0 | 34.6 | 39.1 | 41.0 | 41.1 | 39.6 |
| 10 | 75.0 | 72.1 | 73.1 | 73.1 | 72.6 | 73.5 | 73.8 | 75.1 | 73.5 |
| 11 | 141.1 | 140.9 | 141.1 | 140.6 | 140.6 | 43.5 | 38.8 | 139.7 | 142.3 |
| 12 | 170.8 | 170.5 | 171.1 | 170.2 | 170.5 | 179.0 | 179.1 | 171.7 | 170.3 |
| 13 | 118.6 | 118.2 | 118.8 | 119.5 | 118.8 | 9.7 | 10.8 | 118.2 | 119.2 |
| 14 | 33.0 | 21.8 | 30.0 | 23.6 | 29.8 | 27.1 | 28.5 | 31.6 | 27.5 |
| 15 | 18.6 | 15.8 | 15.0 | 15.1 | 16.6 | 13.8 | 14.4 | 13.0 | 13.9 |
| 1′ | 72.4 | 72.3 | 72.2 | 72.3 | 131.8 | 72.9 | 72.4 | 72.8 | 72.9 |
| 2′ | 39.3 | 38.7 | 39.1 | 39.1 | 195.9 | 39.6 | 39.4 | 39.5 | 39.6 |
| 3′ | 125.5 | 124.8 | 125.2 | 125.2 | 135.8 | 125.4 | 125.3 | 124.5 | 125.4 |
| 4′ | 140.6 | 140.6 | 141.0 | 141.1 | 170.2 | 140.8 | 140.4 | 141.1 | 140.8 |
| 5′ | 53.0 | 53.1 | 52.0 | 52.1 | 53.8 | 53.8 | 54.0 | 53.5 | 53.9 |
| 6′ | 82.2 | 81.7 | 80.0 | 79.8 | 83.0 | 83.2 | 82.8 | 82.3 | 83.2 |
| 7′ | 54.0 | 56.9 | 52.5 | 52.6 | 59.2 | 52.4 | 52.8 | 49.8 | 52.5 |
| 8′ | 22.7 | 21.2 | 21.4 | 21.6 | 25.3 | 22.6 | 21.0 | 23.7 | 22.8 |
| 9′ | 33.4 | 33.6 | 32.6 | 32.7 | 38.8 | 33.4 | 34.3 | 33.6 | 33.4 |
| 10′ | 62.4 | 62.5 | 62.1 | 62.0 | 152.0 | 63.4 | 62.6 | 63.4 | 63.5 |
| 11′ | 56.8 | 56.7 | 57.4 | 57.5 | 56.6 | 55.5 | 55.7 | 54.9 | 55.6 |
| 12′ | 184.8 | 182.2 | 179.0 | 178.4 | 180.2 | 186.2 | 185.6 | 183.8 | 186.1 |
| 13′ | 43.9 | 40.7 | 35.5 | 34.8 | 41.6 | 35.5 | 35.1 | 34.7 | 35.4 |
| 14′ | 22.6 | 21.3 | 22.6 | 22.7 | 21.4 | 22.8 | 22.4 | 22.8 | 22.8 |
| 15′ | 18.8 | 17.4 | 18.8 | 18.8 | 20.2 | 18.9 | 18.5 | 18.6 | 18.9 |
Recorded in CDCl3.
Recorded in CD3OD.
Recorded at 150 MHz.
Recorded at 100 MHz.
Artematrolide K (15) was assigned the same molecular formula of C30H36O6 as that of compound 14 by the (+)-HRESIMS ion at m/z 493.2550 [M+H]+ (Calcd. for 493.2561). The 1H and 13C NMR data (Table 2, Table 5) of compound 15 was highly resembled artemyriantholide D19, indicating the closely related structures. By further interpretation of its 1H–1H COSY and HMBC spectra (Fig. 2), compound 15 was proposed the same planar structure with artemyriantholide D. The differences in their NMR data should be ascribed to the changed stereochemistry in structures. Compared with artemyriantholide D, the chemical shifts of C-1, C-9, and C-14 were shifted from δC 62.8, 34.7, and 29.8 in artemyriantholide D to 63.2, 36.3, and 21.8 in compound 15. In the ROESY spectrum (Fig. 3), the correlations of H-5 and H-7 with H3-14 suggested the α-orientation of CH3-14, and compound 15 was deduced as the 10-epimer of artemyriantholide D. The absolute configuration of compound 15 was elucidated as 1R,4R,5S,6S,7S,10S,1′R,5′R,6′S,7′S,10′S,11′S via quantum chemical calculation (Fig. 5).
Artematrolide L (16) maintaining a molecular formula of C30H36O6 was deduced from the (+)-HRESIMS ion at m/z 493.2583 [M+H]+ (Calcd. for 493.2585). Detailed analyses of its 1H and 13C NMR data (Table 2, Table 5) suggested that compound 16 was an isomer of artemyriantholide D, and the main differences were located at C-1 (δC 63.8), C-2 (δC 133.2), C-3 (δC 142.0), and C-4 (δC 57.1) in contrast to δC 62.8, 135.8, 138.3, and 61.2 in artemyriantholide D. In the ROESY spectrum, the cross peaks of H-7/H-5, H-7/H-8a, H-7′/H-5′, H-7′/H-8′a, and H-7′/H3-15 proposed the α-orientation of H-5 and H-5′, while the cross peaks of H-6/H-2, H-6/H-3, H-6/H-8b, H-6/H3-14, H-6′/H-8′b, and H-6′/H2-13′ manifested the β-orientation of H-6, H-6′, CH3-14, CH2-13′, and the ethenylene (C-2–C-3, Fig. 3). Thus, compound 16 was determined as the 11′-epimer of artemyriantholide D. The calculated ECD curve for compound 16 well matched that of the experimental one (Fig. 5), which allowed the assignment of its absolute configuration as 1R,4R,5S,6S,7S,10R,1′R,5′R,6′S,7′S,10′S,11′R.
Artematrolide M (17) had the same molecular formula of C30H36O6 with compound 16 by the (+)-HRESIMS ion at m/z 493.2551 [M+H]+ (Calcd. for 493.2561). Analyses of the 1D NMR data (Table 3, Table 5) suggested that compound 17 had the same planar structure with compound 16. By comparison with 16, the chemical shift of C-14 was significantly shielded from δC 30.0 in 16 to 23.6 in 17, suggesting the varied stereochemistry at C-14. In the ROESY spectrum (Fig. 3), the correlation of H-7/H3-14, H-7/H-5, H-7/H-8a was readily detected, supporting the α-configuration of H-7, H-5 and CH3-14, while the cross peaks of H-6/H-8b, H-6/H-3, H-6/H-2 together with the large coupling constant (JH-6/H-7 = 9.6 Hz) indicated the β-orientation of H-6 and the ethenylene (C-2–C-3), by which compound 17 was determined as the 10-epimer of compound 16. The relative configuration of unit B was confirmed by the ROESY experiment, which was the same as that of compound 16. By means of ECD calculation (Fig. 5), the absolute configuration of 17 was assigned as 1R,4R,5S,6S,7S,10S,1′R,5′R,6′S,7′S,10′S,11′R.
Table 3.
1H NMR spectroscopic data for compounds 17–22 (δ in ppm, J in Hz).
| No. | 17a,b | 18a,c | 19a,b | 20a,c | 21a,c | 22a,b |
|---|---|---|---|---|---|---|
| 2 | 6.23 d (5.4) | 5.99 d (5.6) | 2.43 dd (9.6, 1.4) | 2.23 m | 2.69 m | 2.46 m |
| 1.77 dd (9.6, 3.2) | 1.58 m | 1.40 m | 1.79 dd (9.6, 3.2) | |||
| 3 | 6.34 d (5.4) | 5.80 d (5.6) | 2.66 m | 2.54 m | 2.67 m | 2.68 m |
| 5 | 2.05 m | 3.13 d (10.0) | ||||
| 6 | 4.26 dd (10.2, 9.6) | 4.04 dd (10.0, 9.6) | 5.01 d (3.6) | 4.42 d (11.2) | 5.50 br d | 5.06 d (4.2) |
| 7 | 2.70 m | 3.35 m | 2.26 m | 2.23 m | 3.37 m | 2.85 m |
| 8 | 2.17 m | 2.29 m | 1.71 m | 1.70 m | 1.86 m | 1.86 (overlapped) |
| 1.62 m | 1.45 m | 1.33 m | 1.49 m | 1.81 m | 1.62 m | |
| 9 | 2.08 m | 1.90 m | 1.91 m | 1.82 m | 1.88 (overlapped) | 2.03 (overlapped) |
| 1.79 m | 1.82 m | 1.59 m | 1.65 m | 1.60 m | 1.55 m | |
| 11 | 2.80 m | 2.67 m | ||||
| 13 | 6.14 d (3.3) | 6.09 d (3.4) | 1.20 d (7.1) | 1.16 d (8.0) | 6.19 d (2.6) | 6.08 br s |
| 5.41 d (3.3) | 5.36 d (3.4) | 5.46 d (2.6) | 5.54 br s | |||
| 14 | 1.38 s | 1.32 s | 1.38 s | 1.39 s | 1.31 s | 1.42 s |
| 15 | 1.36 s | 1.45 s | 1.84 s | 1.88 s | 1.86 s | 1.83 s |
| 2′ | 2.77 dd (17.9, 2.2) | 2.74 dd (17.7, 3.7) | 2.69 m | 2.72 m | 2.75 m | |
| 2.16 m | 2.11 dd (17.7, 2.4) | 2.08 m | 2.10 m | 2.03 m | ||
| 3′ | 5.56 m | 6.18 s | 5.57 m | 5.54 m | 5.52 m | 5.58 m |
| 5′ | 2.82 m | 3.25 d (10.0) | 2.91 m | 2.82 d (10.4) | 2.91 d (10.4) | 2.92 d (10.2) |
| 6′ | 4.06 dd (10.2, 9.6) | 3.94 dd (10.4, 10.0) | 3.99 dd (10.2, 10.2) | 3.96 dd (10.4, 10.0) | 3.82 dd (10.4, 10.0) | 4.00 dd (10.2, 10.2) |
| 7′ | 1.93 (overlapped) | 2.34 m | 2.24 m | 2.04 m | 2.84 m | 2.25 m |
| 8′ | 1.69 m | 1.89 m | 1.16 m | 1.42 m | 1.17 m | 1.18 m |
| 1.66 m | 1.17 m | 1.10 m | 1.23 m | 1.09 m | 1.15 m | |
| 9′ | 2.14 m | 2.30 m | 2.28 m | 2.13 m | 2.46 m | 2.35 m |
| 2.02 m | 2.22 m | 1.93 m | 1.66 m | 1.99 m | 1.98 m | |
| 13′ | 2.25 d (12.1) | 2.69 d (11.8) | 1.89 m | 1.77 m | 1.88 (overlapped) | 1.91 dd (12.0, 3.8) |
| 1.58 d (12.1) | 1.31 m | 1.82 dd (12.0, 3.2) | 1.72 m | 1.59 m | 1.84 m | |
| 14′ | 1.35 s | 2.41 s | 1.30 s | 1.26 s | 1.32 s | 1.31 s |
| 15′ | 1.92 s | 2.31 s | 1.96 s | 1.91 s | 1.93 s | 1.96 s |
Recorded in CDCl3.
Recorded at 600 MHz.
Recorded at 400 MHz.
Artematrolide N (18) was obtained as a white amorphous powder with a molecular formula of C30H34O6 by the (+)-HRESIMS ion at m/z 491.2435 [M+H]+ (Calcd. for 491.2428). By comparison with compound 16, one methylene (C-2′) and two epoxidized carbons (C-1′ and C-10′) in compound 16 were replaced by a carbonyl (δC 195.9) and two non-protonated sp2 carbons (δC 131.8 and 152.0) in compound 18. Thus, a 1,4-dien-3-one partial structure was identical with that in compound 8, which was supported by the consistent correlations from H-3′ to C-1′, from H3-15′ to C-3′, from H-5′ to C-2′, C-3′, and C-10′, and from H3-14′ to C-1′ in the HMBC spectrum (Fig. 2). The relative configuration of compound 18 was determined based on the ROESY experiment, the cross peaks of H-7/H-5, H-7/H-8a, H-7′/H-5′, H-7′/H-8′a in the ROESY spectrum suggested the α-orientation of H-5 and H-5′, while the cross peaks of H-6/H-2, H-6/H-3, H-6/H-8b, H-2/H3-14, H-6′/H-8′b indicated the β-orientation of H-6, H-6′, CH3-14, and the ethenylene (C-2–C-3, Fig. 3). Moreover, the β-orientation of CH2-13′ was confirmed by the correlation between H-6′ and H2-13′ in the ROESY spectrum. By comparing the experimental and calculated ECD spectra (Fig. 5), the absolute configuration of compound 18 was determined as 1R,4R,5S,6S,7S,10R,5′S,6′S,7′S,11′R.
Artematrolide O (19) had a molecular formula of C30H38O6 from the (+)-HRESIMS ion at m/z 517.2558 [M+Na]+ (Calcd. for 517.2561), indicating 12 degrees of unsaturation. Comprehensive analyses of its 1H and 13C NMR spectroscopic data (Table 3, Table 5) indicated close resemblances to artemyriantholide C19. Its planar structure and partial relative configuration were reported, but its absolute configuration has not been determined. In the ROESY spectrum, the cross peaks of H-7/H-6, H-7/H-9a, H-6/H-9a, H-7/H-11, and H-6/H-11, together with the small coupling constant (JH-6/H-7 = 3.6 Hz) indicated that H-6 and H-11 were α-orientated, while correlation of H3-14 with H-9b assigned the β-orientation of CH3-14. Similarly, the correlation of H-7′ with H-5′ and the large coupling constant (JH-6′/H-7′ = 10.2 Hz) assigned the α-orientation of H-5′ and H-7′, and β-orientation of H-6′. Owing to the lack of ROESY correlations, the relative configuration of the additional cyclohexene moiety could not be confirmed. A single-crystal X-ray diffraction for compound 19 was performed using the Cu Ka radiation [Flack parameter of 0.06 (4)] (Fig. 4). Therefore, the structure of compound 19 was established, and its absolute configuration was assigned to be 1S,3R,6R,7S,10R,11R,1′R,5′R,6′S,7′S,10′S,11′R.
Artematrolide P (20) possessed the same molecular formula with 19 by the (+)-HRESIMS ion at m/z 517.2556 [M+Na]+ (Calcd. for 517.2561). Detailed analyses of the 1D and 2D NMR data (Table 3, Table 5) revealed that compound 20 possessed the same planar structure with compound 19. In the 13C NMR (DEPT) spectrum, the chemical shifts of C-6, C-7, and C-11 were shifted from δC 77.3, 45.2, and 43.5 in compound 19 to 82.4, 49.7, and 38.8 in compound 20, respectively, suggesting the different configuration. In the 1H NMR spectrum, H-6 was present at δH 4.42 with a large coupling constant (JH-6/H-7 = 11.2 Hz), obviously different from 19 (δH 5.01, d, J = 3.6 Hz), indicating the β-orientation of H-6 in 20. The ROESY spectrum supported this deduction by the correlations of H-7 with H-8a, H-6 with H-8b (Fig. 3). A single-crystal X-ray diffraction analysis of compound 20 by utilizing Cu Kα radiation established its absolute configuration as 1S,3R,6S,7S,10R,11R,1′R,5′R,6′S,7′S,10′S,11′R (Fig. 4).
Artematrolide Q (21) was assigned to a molecular formula of C30H36O6 by the (+)-HRESIMS ion at m/z 493.2575 [M+H]+ (Calcd. for 493.2585). By detailed inspection of its 1D and 2D NMR data (Table 3, Table 5), the same planar structure with artemyriantholide B was determined19. By comparison with artemyriantholide B, the chemical shifts of C-4, C-6, and C-7 were shifted from δC 142.6, 82.9, and 50.9 in artemyriantholide B to δC 152.0, 75.1, and 39.6 in compound 21, respectively. In the ROESY spectrum (Fig. 3), the correlation of H-6/H-7 was readily observed, indicating the α-orientation of H-6. The absolute configuration of 21 was elucidated as 1S,3R,6R,7S,10S,1′R,5′R,6′S,7′S,10′S,11′R via the single-crystal X-ray diffraction experiment (Fig. 4).
Artematrolide R (22) had a molecular formula of C30H36O6 as deduced from the (+)-HRESIMS ion at m/z 515.2405 [M+Na]+ (Calcd. for 515.2404). The 1H and 13C NMR data (Table 3, Table 5) of compound 22 were very similar to those of compound 19 with the main differences at C-8, C-11, C-12, and C-13. The terminal olefinic carbons were easily recognized in compound 22 from the characteristic protons and carbons (δH 6.08 and 5.54, δC 142.3 and 119.2), instead of a methine and a methyl in compound 19. Taking its molecular formula that was two hydrogens less than compound 19 into consideration, compound 22 was deduced as the 11,13-dehydro derivative of compound 19, which was confirmed by the correlations from H2-13 to C-7 and C-12 in the HMBC experiment (Fig. 2). The correlations of H-7/H-6, H-7/H-8a, H-7/H-9a, and H-9a/H-2b in the ROESY spectrum, and the small coupling constant (JH-6/H-7 = 4.2 Hz) indicated that H-6 and CH2-3 were α-orientated, while the cross signal of H3-14 with H-8b assigned the β-orientation of CH3-14. The correlation of H-7′ with H-5′ and the large coupling constant (JH-6′/H-7′ = 10.2 Hz) characterized the α-configuration of H-5′ and H-7′, and β-orientation of H-6′. Similarly, the correlation between H-6′ and H2-13′ manifested CH2-13′ as β-orientated. Further analysis of its ROESY spectrum (Fig. 3) suggested the same relative configuration of compound 22 with compound 19. The absolute configuration of compound 22 was determined by comparing the calculated ECD spectrum with the experimental one (Fig. 5). Hence, the structure of 22 was established and named as artematrolide R.
Compounds 3, 4, 9, 12, and 23 were identified as lavandiolides A, B, J, H, and C by comparison of their NMR data with the data in Ye's paper42, and the structures of lavandiolides B, H were confirmed by single-crystal X-ray diffraction in our report.
The EtOH extract and EtOAc fraction of A. atrovirens were tested for their inhibitory activity on HepG2 cells with inhibitory ratios of 98.9% and 95.9% at 100.0 μg/mL (Fig. 6). The EtOAc fraction was separated into three subfractions (Fr. 1−Fr. 3), of which Fr. 2 showed obvious cytotoxicity with the inhibitory ratio of 98.6%, more potent than two other subfractions [Fr. 1 (55.9%) and Fr. 3 (41.5%)] at 100.0 μg/mL.
Figure 6.
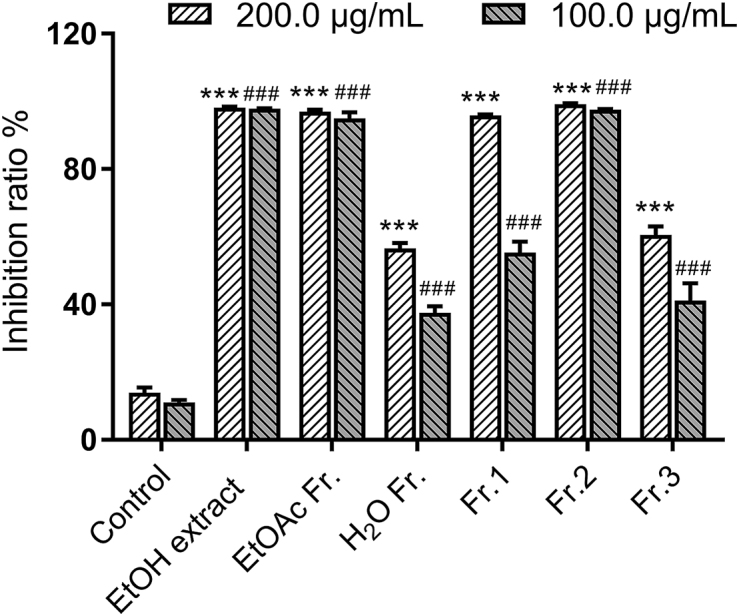
Cytotoxic activities of the EtOH extract and each fraction of A. atrovirens against HepG2 cells at 200.0 and 100.0 μg/mL. Sorafenib with an IC50 value of 6.0 μg/mL was used as the positive control. ∗∗∗P < 0.001 versus the control (200.0 μg/mL) group, ###P < 0.001 versus the control (100.0 μg/mL) group, n = 3.
The obtained compounds (1–23) from Fr. 2 were evaluated on three human hepatoma cell lines (HepG2, SMMC-7721, and Huh7), and the results were shown in Table 6. For HepG2 cells, five compounds (1, 3, 5, 12, and 13) exhibited cytotoxicity with IC50 values of 4.4, 5.5, 3.3, 3.8, and 5.3 μmol/L, which were superior to the positive control, sorafenib (IC50 7.7 μmol/L); six compounds (7, 14–16, 19, and 22) exhibited cytotoxic activity with IC50 values of 6.0, 7.6, 6.7, 8.8, 7.1, and 6.4 μmol/L, which were comparable to sorafenib.
Table 6.
Cytotoxicity of compounds 1–23 from A. atrovirens.
| No. | IC50 (μmol/L) |
||
|---|---|---|---|
| HepG2 | SMMC-7721 | Huh7 | |
| 1 | 4.4 ± 0.2 | 9.6 ± 0.3 | 7.6 ± 0.3 |
| 2 | 35.7 ± 1.0 | 86.6 ± 4.4 | 35.1 ± 0.4 |
| 3 | 5.5 ± 0.3 | 22.5 ± 2.7 | 5.4 ± 0.4 |
| 4 | 21.1 ± 2.9 | 85.4 ± 7.0 | 85.0 ± 3.9 |
| 5 | 3.3 ± 0.8 | 18.7 ± 1.9 | 5.7 ± 0.2 |
| 6 | 116.9 ± 4.3 | 8.9 ± 0.1 | 4.5 ± 0.2 |
| 7 | 6.0 ± 0.1 | 43.8 ± 1.4 | 18.6 ± 0.2 |
| 8 | 12.7 ± 1.9 | 8.9 ± 0.3 | 5.9 ± 0.3 |
| 9 | 33.4 ± 6.2 | 111.9 ± 8.8 | 70.3 ± 2.6 |
| 10 | 10.3 ± 3.1 | 24.4 ± 0.2 | 8.2 ± 0.3 |
| 11 | 58.6 ± 7.2 | 12.9 ± 0.3 | 9.1 ± 0.3 |
| 12 | 3.8 ± 0.4 | 4.6 ± 1.1 | 4.5 ± 0.1 |
| 13 | 5.3 ± 0.2 | 17.2 ± 1.3 | 4.0 ± 0.1 |
| 14 | 7.6 ± 0.2 | 6.6 ± 0.1 | 6.9 ± 0.1 |
| 15 | 6.7 ± 0.1 | 6.0 ± 0.3 | 5.6 ± 0.2 |
| 16 | 8.8 ± 0.2 | 24.4 ± 1.3 | 16.5 ± 0.5 |
| 17 | 19.5 ± 0.8 | 22.3 ± 0.1 | 8.4 ± 0.1 |
| 18 | 25.7 ± 2.2 | 11.4 ± 1.9 | 16.3 ± 0.1 |
| 19 | 7.1 ± 0.1 | 75.4 ± 2.3 | 99.3 ± 7.3 |
| 20 | 13.0 ± 0.3 | 21.6 ± 2.0 | 10.4 ± 0.2 |
| 21 | 28.8 ± 0.2 | 10.1 ± 0.5 | 16.7 ± 0.5 |
| 22 | 6.4 ± 0.1 | 47.2 ± 1.3 | 36.3 ± 0.4 |
| 23 | 33.4 ± 0.9 | 21.9 ± 0.4 | 20.6 ± 0.3 |
| Sorafenib | 7.7 ± 0.4 | 9.9 ± 0.8 | 8.3 ± 0.4 |
Data were expressed as means ± SD (n = 3).
For SMMC-7721 cells, three compounds (12, 14, and 15) indicated cytotoxicity with IC50 values of 4.6, 6.6, and 6.0 μmol/L, which were more potent than sorafenib (IC50 9.9 μmol/L); five compounds (1, 6, 8, 18, and 21) exhibited cytotoxicity with IC50 values of 9.6, 8.9, 8.9, 11.4, and 10.1 μmol/L, and were similar with sorafenib.
For Huh7 cells, seven compounds (3, 5, 6, 8, 12, 13, and 15) displayed cytotoxicity with IC50 values of 5.4, 5.7, 4.5, 5.9, 4.5, 4.0, and 5.6 μmol/L, which manifested those compounds were more potent than the positive control, sorafenib (IC50 8.3 μmol/L); six compounds (1, 10, 11, 14, 17, and 20) possessed cytotoxicity with IC50 values of 7.6, 8.2, 9.1, 6.9, 8.4, and 10.4 μmol/L, and were comparable to sorafenib. Compounds 7, 19, and 22 manifested cytotoxicity only to HepG2 cells with IC50 values of 6.0, 7.1, and 6.4 μmol/L. Compounds 6 and 8 showed inhibitory activity on both SMMC-7721 and Huh7 cells.
Interestingly, compounds 3, 5, and 13 showed inhibitory activity on both HepG2 and Huh7 cells, which were superior to sorafenib. Four compounds (1, 12, 14, and 15) exhibited inhibitory activity on the three cell lines with IC50 values of 4.4, 3.8, 7.6, and 6.7 μmol/L (HepG2), 9.6, 4.6, 6.6, and 6.0 μmol/L (SMMC-7721), and 7.6, 4.5, 6.9, and 5.6 μmol/L (Huh7), respectively. Compound 12 showed the highest cytotoxicity against three human hepatoma cell lines, which were superior to sorafenib.
To understand the effects of compound 12 on hepatoma metastasis, the potential impact of compound 12 on HCC metastasis was investigated by using cell migration and invasion assays. The results indicated that compound 12 suppressed HepG2 cell migration and invasion in a dose-dependent manner. Comparing with the control cells, the migration ratio of HepG2 cells was reduced to 84.9%, 69.2% and 37.5% at 1.0, 2.0 and 4.0 μmol/L, respectively. The invasion rate was decreased to 85.6% (1.0 μmol/L), 64.6% (2.0 μmol/L) and 43.0% (4.0 μmol/L). Consistently, these results were indicative of a potential effect of compound 12 on HCC migration (Fig. 7).
Figure 7.

Inhibitory effects of compound 12 on HepG2 cell migration and invasion. HepG2 cells were treated with different concentrations (0.0, 1.0, 2.0 and 4.0 μmol/L) of 12 for 48 h. (A) Representative photographs of Transwell assay showed migrated and invaded cells after incubation. (B) Histogram of migrated and invaded cells after incubation. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001, n = 3.
To investigate the cytotoxic mechanism of compound 12, the cell cycle progression and apoptosis effects on HepG2 cells were analyzed by flow cytometry. The composition of cells in various phases varied according to concentration when the cells were treated with different concentrations (0.0, 2.0, 4.0 and 8.0 μmol/L) of compound 12, and the cell ratio in different stages varied following the alterations of concentration. After being treated with different concentrations of compound 12, the percentage of cells in the G2/M phase increased from 13.3% to 16.6% (2.0 μmol/L), 19.3% (4.0 μmol/L) and 25.1% (8.0 μmol/L) independently by comparison to the control cells. These results demonstrated that compound 12 effectively induced a cell cycle arrest in G2/M phase (Fig. 8). Next, we investigated the expression of cell cycle regulators which control the G2/M transition. As shown in Fig. 8C, the expression of CYCLINB1 and CDC2 were suppressed with increasing concentration of compound 12 (0.0, 2.0, 4.0, and 6.0 μmol/L). Furthermore, the apoptosis effects on HepG2 cells were detected by flow cytometry. Compared to the control cells, increasing concentration of compound 12 led to enhancement in the apoptotic effect varying from 17.2% (2.0 μmol/L) to 35.9% (4.0 μmol/L), and 65.0% (8.0 μmol/L) as displayed in Fig. 9. Annexin V and propidium iodide (Annexin V/PI) double staining revealed that compound 12 induced accumulation of cells in early- (Annexin V+/PI−) and late-stage (Annexin V+/PI+) apoptosis in a dose dependent manner. Moreover, the level of apoptosis-related proteins BCL-2 and PARP-1 which are indicators of apoptosis was estimated by using western blot assay. The results indicated that compound 12 down-regulated the expression of BCL-2 and PARP-1 in a dose-dependent manner and activated PARP-1 to up-regulate the expression of cleaved-PARP-1. Thus, these results suggested that compound 12 inhibited the growth of HepG2 cells through arresting the cells cycle in G2/M phase and inducing cell apoptosis. In Ye's paper42, lavandiolide H (12) did not showed any anti-inflammatory activity. Even through lavandiolide H (12) was reported in Ye's paper, our manuscript first reported the cytotoxicity and preliminary mechanism of lavandiolide H (12).
Figure 8.

Effects of compound 12 on cell cycle arrest using HepG2 cells. HepG2 cells were treated with different concentrations (0.0, 2.0, 4.0, and 8.0 μmol/L) of 12 for 48 h. (A) and (B) Flow cytometric analysis and cell cycle quantification of HepG2 cells. (C) and (D) Western blot and statistical results of CYCLINB1 and CDC2. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001, n = 3.
Figure 9.

Apoptosis effects of HepG2 cells induced by compound 12. HepG2 cells were treated with different concentrations (0.0, 2.0, 4.0, and 8.0 μmol/L) of 12 for 48 h. (A) and (B) Flow cytometric analysis and cell apoptosis quantification of HepG2 cells. (C) and (D) Western blot and statistical results of BCL-2, PARP-1, and cleaved-PARP-1. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001, n = 3.
3. Conclusions
In summary, 18 new guaianolide dimers and five known compounds lavandiolides A (3), B (4), C (23), H (12), and J (9) with a spiro-system composed of two monomeric sesquiterpene lactone units were isolated and identified from A. atrovirens guided by cytotoxicity against HepG2 cell line. Their structures were elucidated based on extensive analyses of NMR spectroscopic data, X-ray analyses, and ECD spectra. Structurally, these compounds were involved in seven different kinds of connecting model. Five compounds showed higher activities against HepG2 cells with IC50 values superior to sorafenib, three compounds exhibited cytotoxicity against SMMC-7721 cells with IC50 values better than sorafenib, and seven compounds showed higher potential effects against Huh7 cells with IC50 values superior to sorafenib. Notably, four compounds (1, 12, 14, and 15) exhibited significant inhibitory activity on the three human hepatoma cell lines, and compound 12 showed the highest activity against three human hepatoma cell lines with IC50 values of 3.8, 4.6, and 4.5 μmol/L. The mechanism-of-action investigation revealed that compound 12 dose-dependently inhibited cell migration and invasion, induced G2/M cell cycle arrest and cell apoptosis in HepG2 cells, down-regulated the expression of BCL-2 and PARP-1, and activated PARP-1 to up-regulate the expression of cleaved-PARP-1. Our findings provide a series of new guaianolide dimers as candidate molecules against hepatoma. The synthesis, structure modification, structure–activity relationship, and in-depth mechanism of the active sesquiterpenoid dimers are ongoing in our laboratory, and will be reported in due course.
4. Experimental
4.1. General experimental procedures
4.2. Plant materials
Artemisia atrovirens Hand.-MaZZ. was collected in July 2018 from Kunming, China, and identified by Professor Dr. Ligong Lei (CAS Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences, China). A voucher specimen (No. 20180716-01) was deposited in the Laboratory of Antivirus and Natural Medicinal Chemistry, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China.
4.3. Extraction and isolation
The powdered and air-dried A. atrovirens (60 kg) was extracted twice with EtOH at room temperature (four days each time). After evaporation of the organic solvent in vacuo, the residue was suspended in H2O and extracted with EtOAc. The EtOAc-soluble fraction (3.4 kg) was subjected to a silica gel column chromatography (Si CC, 17 kg, 30 cm × 145 cm) and eluted with a gradient of acetone–petroleum ether (PE; 10:90 to 100:0, v/v) to afford three fractions [Fr. 1 (1.0 kg), Fr. 2 (450 g), and Fr. 3 (1.7 kg)]. Fr. 2 (450 g) was separated by silica gel column chromatography (Si CC, 3.6 kg, 20 cm × 45 cm) and eluted with a stepwise gradient of EtOAc–PE (10:90 to 50:50, v/v) to yield four subfractions (Fr. 2-1‒Fr. 2-4). Fr. 2-1 (91 g) was fractionated by MPLC on an MCI gel CHP 20P column (490 g, 5 cm × 50 cm) eluting with a gradient solvent of H2O–MeOH (50:50, 30:70, 10:90, and 0:100) to provide four subfractions (Fr. 2-1-1‒Fr. 2-1-4). Fr. 2-1-3 (26 g) was applied to Si CC (260 g, 6.0 cm × 25 cm) using EtOAc−PE as eluents (10:90 to 40:60) to yield five subfractions (Fr. 2-1-3-1‒Fr. 2-1-3-5). Fr. 2-1-3-4 (6.3 g) was chromatographed over Si CC (126 g, 4.5 cm × 25 cm, acetone−PE, 15:85 to 30:70) to produce three subfractions (Fr. 2-1-3-4a‒4c). Compound 1 (380 mg) was obtained from Fr. 2-1-3-4b (1.9 g) by recrystallization in MeOH–CH2Cl2 (10:90) and the residual part was further purified by preparative HPLC (H2O–CH3CN, 44:56, 10.0 mL/min) to yield compounds 2 (13 mg, tR = 21.0 min), 14 (58 mg, tR = 32.0 min), and 21 (11 mg, tR = 25.2 min). Fr. 2-1-3-5 (2.3 g) was separated into three subfractions (Fr. 2-1-3-5a‒5c) on Si CC (46 g, 2.5 cm × 25 cm) using a gradient of EtOAc−CHCl3 (2:98 to 10:90). The obtained subfraction Fr. 2-1-3-5a (340 mg) was purified by preparative HPLC (H2O–CH3CN, 44:56, 10.0 mL/min) and semi-preparative HPLC (H2O–MeOH, 20:80, 3.0 mL/min) to afford compounds 7 (14 mg, tR = 31.5 min), 19 (9 mg, tR = 23.5 min), 20 (85 mg, tR = 22.0 min), and 22 (9 mg, tR = 25.0 min). Compound 15 (45 mg, tR = 22.0 min) was obtained from Fr. 2-1-3-5b (600 mg) by preparative HPLC separation (H2O–CH3CN, 48:52, 10.0 mL/min) and semi-preparative HPLC (H2O–MeOH, 33:67, 3.0 mL/min). Fr. 2-2 (203.5 g) was separated on an MCI gel CHP 20P column with H2O–MeOH (50:50, 30:70, 10:90, and 0:100) to provide four subfractions (Fr. 2-2-1‒Fr. 2-2-4). Fr. 2-2-2 (39.5 g) was fractionated with Si CC (395 g, 6.0 cm × 40 cm) employing EtOAc−PE (20:80 to 50:50) to give four subfractions (Fr. 2-2-2-1‒Fr. 2-2-2-4). Separation of Fr. 2-2-2-2 (7.4 g) on a silica gel column (74 g, 4.0 cm × 20 cm) with acetone−PE (15:85, 20:80, 30:70) gave three fractions (Fr. 2-2-2-2a‒2c). Further purification of Fr. 2-2-2-2a by Sephadex LH-20 CC (140 g, 2.5 cm × 175 cm, MeOH–CHCl3, 50:50) followed by preparative TLC (EtOAc‒CHCl3, 50:50) yielded compound 6 (24 mg). Fr. 2-2-2-2b (2.8 g) was purified by silica gel CC (EtOAc‒CHCl3, 15:85), preparative HPLC (H2O–CH3CN, 52:48, 10.0 mL/min), and semipreparative HPLC (H2O–MeOH, 37:63, 3.0 mL/min) to give compounds 10 (8 mg, tR = 23.5 min), 11 (5 mg, tR = 22.7 min), 12 (75 mg, tR = 31.5 min), and 13 (23 mg, tR = 29.5 min). Further purification of Fr. 2-2-2-2c (1.4 g) by Sephadex LH-20 CC (120 g, 2.5 cm × 150 cm, MeOH–CHCl3, 50:50) afforded two main fractions (Fr. 2-2-2-2c-1‒Fr. 2-2-2-2c-2). Fr. 2-2-2-2c-1 was purified by semipreparative HPLC on an Eclipse XDB-C18 column (H2O–MeOH, 35:65, 3.0 mL/min) to produce 9 (16 mg, tR = 27.5 min), 16 (45 mg, tR = 26.0 min), and 17 (12 mg, tR = 23.0 min). Semipreparative HPLC purification of Fr. 2-2-2-2c-2 (106 mg) on an Eclipse XDB-C18 column (H2O–MeOH, 40:60, 3.0 mL/min) yielded 8 (40 mg, tR = 20.5 min) and 18 (10 mg, tR = 18.0 min). Fr. 2-2-2-3 (2.5 g) was purified by silica gel CC (acetone‒PE, 20:80), preparative HPLC (H2O–CH3CN, 45:55, 10.0 mL/min), and semipreparative HPLC (H2O–CH3CN, 53:47, 3.0 mL/min) to give compounds 3 (12 mg, tR = 32.0 min), 4 (74 mg, tR = 27.3 min), and 5 (75 mg, tR = 19.8 min), and the residual part was further purified by Sephadex LH-20 CC (48 g, 1.4 cm × 150 cm, MeOH–CHCl3, 50:50) and semi-preparative HPLC (H2O–CH3CN, 60:40) to yield compound 23 (13 mg, tR = 16.7 min).
4.3.1. Artematrolide A (1)
Colorless orthorhombic crystals (MeOH–CH2Cl2, 10:90); mp 239–240 °C; (c 0.028, MeOH); IR νmax 3476, 1767, 1744, 1629, 1404, 1257, 1121, 1031 cm−1; CD λmax (Δε) 203 (−0.25), 224 (+1.64), 259 (−0.17) nm; 1H and 13C NMR data (Table 1, Table 4); (+)-HRESIMS m/z 515.2411 [M+Na]+ (Calcd. for C30H36O6Na, 515.2404).
4.3.2. Artematrolide B (2)
White amorphous powder; (c 0.050, MeOH); IR νmax 3510, 3437, 1776, 1755, 1630, 1357, 1256, 1165, 1018 cm−1; CD λmax (Δε) 228 (+0.84), 203 (−0.39) nm; 1H and 13C NMR data (Table 1, Table 4); (+)-HRESIMS m/z 517.2561 [M+Na]+ (Calcd. for C30H38O6Na, 517.2561).
4.3.3. Artematrolide C (5)
White amorphous powder; (c 0.045, MeOH); IR νmax 3450, 1766, 1640, 1440, 1288, 1092 cm−1; CD λmax (Δε) 213 (+0.65), 199 (+0.29) nm; 1H and 13C NMR data (Table 1, Table 4); (+)-HRESIMS m/z 493.2577 [M+H]+ (Calcd. for C30H37O6, 493.2585).
4.3.4. Artematrolide D (6)
White amorphous powder; (c 0.058, MeOH); IR νmax 3449, 1772, 1636, 1439, 1236, 1082, 1009 cm−1; CD λmax (Δε) 213 (+1.06), 200 (+0.51) nm; 1H and 13C NMR data (Table 1, Table 4); (+)-HRESIMS m/z 495.2726 [M+H]+ (Calcd. for C30H39O6, 495.2741).
4.3.5. Artematrolide E (7)
White amorphous powder; (c 0.083, MeOH); UV (MeOH) λmax (log ε) 240 (2.65), 260 (2.59) nm; IR νmax 3446, 1759, 1637, 1384, 1226, 1184 cm−1; CD λmax (Δε) 205 (−0.19), 215 (−0.12), 233 (−0.10), 259 (−0.35) nm; 1H and 13C NMR data (Table 1, Table 4); (+)-HRESIMS m/z 475.2473 [M+H]+ (Calcd. for C30H35O5, 475.2479).
4.3.6. Artematrolide F (8)
White amorphous powder; (c 0.159, MeOH); UV (MeOH) λmax (log ε) 206 (3.30), 231 (2.90), 256 (3.14) nm; IR νmax 3444, 1766, 1687, 1640, 1618, 1321, 1150 cm−1; CD λmax (Δε) 224 (+0.02), 246 (+0.47) nm; 1H and 13C NMR data (Table 1, Table 4); (+)-HRESIMS m/z 491.2413 [M+H]+ (Calcd. for C30H35O6, 491.2428).
4.3.7. Artematrolide G (10)
Colorless monoclinic crystals (MeOH–CH2Cl2, 15:85); mp 200–201 °C; (c 0.047, MeOH); IR νmax 3453, 1760, 1653, 1633, 1258, 1055 cm−1; CD λmax (Δε) 214 (+2.18), 255 (−0.13) nm; 1H and 13C NMR data (Table 2, Table 4); (+)-HRESIMS m/z 493.2538 [M+H]+ (Calcd. for C30H37O6, 493.2544).
4.3.8. Artematrolide H (11)
White amorphous powder; (c 0.020, MeOH); IR νmax 3439, 1760, 1632, 1446, 1221, 1061 cm−1; CD λmax (Δε) 198 (−1.09), 211 (−0.22), 222 (−0.75), 265 (+0.13) nm; 1H and 13C NMR data (Table 2, Table 4); (+)-HRESIMS m/z 493.2516 [M+H]+ (Calcd. for C30H37O6, 493.2526).
4.3.9. Artematrolide I (13)
Colorless monoclinic crystals (MeOH–CH2Cl2, 10:90); mp 205–206 °C; (c 0.098, MeOH); IR νmax 3440, 1760, 1631, 1443, 1312, 1090 cm−1; CD λmax (Δε) 218 (−0.50), 241 (−0.05) nm; 1H and 13C NMR data (Table 2, Table 4); (+)-HRESIMS m/z 493.2069 [M+H]+ (Calcd. for C30H37O6, 493.2068).
4.3.10. Artematrolide J (14)
White amorphous powder; (c 0.035, MeOH); IR νmax 3446, 1760, 1739, 1631, 1400, 1272, 1133 cm−1; CD λmax (Δε) 208 (+3.75), 229 (−0.27) nm; 1H and 13C NMR data (Table 2, Table 5); (+)-HRESIMS m/z 493.2594 [M+H]+ (Calcd. for C30H37O6, 493.2585).
4.3.11. Artematrolide K (15)
White amorphous powder; (c 0.078, MeOH); IR νmax 3442, 1761, 1631, 1451, 1339, 1177, 1016 cm−1; CD λmax (Δε) 207 (−1.88), 235 (+0.07) nm; 1H and 13C NMR data (Table 2, Table 5); (+)-HRESIMS m/z 493.2550 [M+H]+ (Calcd. for C30H37O6, 493.2561).
4.3.12. Artematrolide L (16)
White amorphous powder; (c 0.063, MeOH); IR νmax 3442, 1763, 1632, 1402, 1221, 1064 cm−1; CD λmax (Δε) 196 (+1.48), 230 (−0.30) nm; 1H and 13C NMR data (Table 2, Table 5); (+)-HRESIMS m/z 493.2583 [M+H]+ (Calcd. for C30H37O6, 493. 2585).
4.3.13. Artematrolide M (17)
White amorphous powder; (c 0.115, MeOH); IR νmax 3445, 1766, 1632, 1406, 1344, 1219, 1015 cm−1; CD λmax (Δε) 196 (+1.28), 229 (−0.19) nm; 1H and 13C NMR data (Table 3, Table 5); (+)-HRESIMS m/z 493.2551 [M+H]+ (Calcd. for C30H37O6, 493.2561).
4.3.14. Artematrolide N (18)
White amorphous powder; (c 0.083, MeOH); UV (MeOH) λmax (log ε) 232 (2.93), 256 (3.12) nm; IR νmax 3441, 1762, 1686, 1638, 1618, 1219, 1146 cm−1; CD λmax (Δε) 212 (−1.62), 239 (+0.39) nm; 1H and 13C NMR data (Table 3, Table 5); (+)-HRESIMS m/z 491.2435 [M+H]+ (Calcd. for C30H35O6, 491.2428).
4.3.15. Artematrolide O (19)
Colorless orthorhombic crystals (MeOH–CH2Cl2, 10:90); mp 195–196 °C; (c 0.055, MeOH); IR νmax 3498, 1775, 1721, 1677, 1632, 1329, 1261, 1153 cm−1; CD λmax (Δε) 199 (−3.81), 222 (+1.63) nm; 1H and 13C NMR data (Table 3, Table 5); (+)-HRESIMS m/z 517.2558 [M+Na]+ (Calcd. for C30H38O6Na, 517.2561).
4.3.16. Artematrolide P (20)
Colorless orthorhombic crystals (MeOH–CHCl3, 15:85); mp 190–192 °C; (c 0.030, MeOH); IR νmax 3498, 1783, 1728, 1655, 1632, 1383, 1242, 1037 cm−1; CD λmax (Δε) 198 (−1.04), 217 (+1.30) nm; 1H and 13C NMR data (Table 3, Table 5); (+)-HRESIMS m/z 517.2556 [M+Na]+ (Calcd. for C30H38O6Na, 517.2561).
4.3.17. Artematrolide Q (21)
Colorless orthorhombic crystals (MeOH–CHCl3, 5:95); mp 190–191 °C; (c 0.063, MeOH); IR νmax 3486, 1762, 1751, 1664, 1641, 1386, 1337, 1271 cm−1; CD λmax (Δε) 201 (−3.18), 219 (−0.06), 233 (−0.54), 263 (+0.12) nm; 1H and 13C NMR data (Table 3, Table 5); (+)-HRESIMS m/z 493.2575 [M+H]+ (Calcd. for C30H37O6, 493.2585).
4.3.18. Artematrolide R (22)
White amorphous powder; (c 0.059, MeOH); IR νmax 3499, 1765, 1733, 1668, 1630, 1382, 1294, 1136 cm−1; CD λmax (Δε) 201 (−4.05), 223 (+0.44), 239 (+0.08), 261 (+0.27) nm; 1H and 13C NMR data (Table 3, Table 5); (+)-HRESIMS m/z 515.2405 [M+Na]+ (Calcd. for C30H36O6Na, 515.2404).
4.4. Cytotoxicity assays
4.5. Flow cytometry assays
HepG2 cells were seeded in 6-well plates (2 × 105 cells/well). After adherence, cells were treated with a series of concentrations of 12 (0.0, 2.0, 4.0 and 8.0 μmol/L) for 48 h. Then cells were analyzed in cell flow cytometry assay. In cell cycle assay, cells were collected and fixed in 70% EtOH at −20 °C overnight, washed with PBS, resuspended in PBS containing RNase A (200 μg/mL) for 15 min in 37 °C, and then incubated with propidium iodide (100 μg/mL) for 30 min, and analyzed by flow cytometry subsequently. In apoptosis assay, cells were harvested and suspended in binding buffer, and stained with fluorochrome Annexin V/PI for 15 min. Cell cycle and apoptosis assays were analyzed by using a BD AccuriC6 flow cytometer (BD Biosciences, San Jose, CA, USA)48.
4.6. Cell migration and invasion assays
In order to evaluate cell migration and invasion, HepG2 cells were analyzed by Transwell assay (Corning, USA). For cell migration assay, HepG2 cells (2 × 105/mL) were suspended and plated on the upper chambers with serum-free DMEM overnight and treated with compound 12 for 48 h. Then, cells in the upper chambers were wiped by cotton swabs, the migrated cells were fixed in 70% ethanol and stained with crystal violet solution (0.1%) for 30 min. The upper chambers were washed with PBS twice and dried. Then, images were taken by imaging system (Olympus IX73)49.
For cell invasion assay, matrigel was diluted to 1:50 in pre-cool DMEM medium on ice and added to the upper chamber before seeding cells. The subsequent procedures were the same as above.
4.7. Western blot
HepG2 cells were treated with compound 12 for 48 h and lysed in RIPA buffer to extract total protein, and protein concentration was quantified by BCA protein assay kit. Protein samples were separated using SDS-PAGE and transferred to PVDF membrane. The membranes were incubated with specific primary antibodies at 4 °C overnight, subsequently. The membranes were incubated with homologous secondary antibodies and detected by ECL solution (Advansta, USA) and photographed by using multispectral imaging system (UVP, USA)50.
4.8. Theoretical ECD calculation
The ECD calculations for compounds 2, 5–8, 11, 14–18, and 22 were performed with the Gaussian 09 program package. Their relative configurations of those compounds were determined based on their ROESY experiments. Their structures were pre-optimized with MM2 method, and further optimized by the DFT calculation at the b3lyp/6-31G(d,p) level in the gas phase. Frequency calculation was performed at the same level to exclude imaginary frequencies. ECD calculation was performed using the TD-DFT methodology at the b3lyp/6-31G(d,p) level in methanol. Solvent effects were taken into consideration using the SCRF method with the IEFPCM model.
4.9. X-ray crystallographic analyses
Crystals of compounds 1, 4, 10, 12, 13, 19–21 were obtained by using the solvent vapor diffusion method. The single-crystal X-ray diffraction data were recorded on a Bruker D8 QUEST instrument (Cu Kα radiation). Crystals were kept at 100.(2) K during data collection. The crystallographic data of those compounds in standard CIF format were deposited at the Cambridge Crystallographic Data Centre. The data can be accessed free of charge at http://www.ccdc.cam.ac.uk/.
Crystallographic data for 1: C30H36O6, M = 492.59, a = 8.9597(2) Å, b = 13.6560(3) Å, c = 20.5728(4) Å, α = 90°, β = 90°, γ = 90°, V = 2517.16(9) Å3, T = 100.(2) K, space group P212121, Z = 4, μ(Cu Kα) = 0.722 mm−1, 43,964 reflections measured, 4948 independent reflections (Rint = 0.0243). The final R1 values were 0.0306 [I > 2σ(I)]. The final wR(F2) values were 0.1081 [I > 2σ(I)]. The final R1 values were 0.0316 (all data). The final wR(F2) values were 0.1103 (all data). The goodness of fit on F2 was 1.111. Flack parameter = −0.009(17). CCDC 1999120.
Crystallographic data for 4: C30H38O6, M = 494.60, a = 9.7182(2) Å, b = 15.6711(3) Å, c = 16.9253(3) Å, α = 90°, β = 94.6810(10)°, γ = 90°, V = 2569.04(9) Å3, T = 100.(2) K, space group P1211, Z = 4, μ(Cu Kα) = 0.708 mm−1, 52,514 reflections measured, 10,083 independent reflections (Rint = 0.0307). The final R1 values were 0.0263 [I > 2σ(I)]. The final wR(F2) values were 0.0661 [I > 2σ(I)]. The final R1 values were 0.0268 (all data). The final wR(F2) values were 0.0666 (all data). The goodness of fit on F2 was 1.051. Flack parameter = 0.04(3). CCDC 1999121.
Crystallographic data for 10: C30H36O6, M = 492.59, a = 8.7039(2) Å, b = 16.5287(4) Å, c = 9.2353(2) Å, α = 90°, β = 97.9230(10)°, γ = 90°, V = 1315.95(5) Å3, T = 100.(2) K, space group P1211, Z = 2, μ(Cu Kα) = 0.691 mm−1, 24,243 reflections measured, 5160 independent reflections (Rint = 0.0243). The final R1 values were 0.0256 [I > 2σ(I)]. The final wR(F2) values were 0.0662 [I > 2σ(I)]. The final R1 values were 0.0257 (all data). The final wR(F2) values were 0.0662 (all data). The goodness of fit on F2 was 1.061. Flack parameter = 0.07(3). CCDC 1999123.
Crystallographic data for 12: 2(C30H36O6)·H2O, M = 1003.19, a = 9.2451(3) Å, b = 32.7201(9) Å, c = 9.2897(3) Å, α = 90°, β = 114.6360(10)°, γ = 90°, V = 2554.35(14) Å3, T = 100.(2) K, space group P1211, Z = 2, μ(Cu Kα) = 0.736 mm−1, 47963 reflections measured, 10064 independent reflections (Rint = 0.0271). The final R1 values were 0.0256 [I > 2σ(I)]. The final wR(F2) values were 0.0657 [I > 2σ(I)]. The final R1 values were 0.0257 (all data). The final wR(F2) values were 0.0657 (all data). The goodness of fit on F2 was 1.054. Flack parameter = 0.040(18). CCDC 2004025.
Crystallographic data for 13: C30H36O6, M = 492.59, a = 16.5004(4) Å, b = 8.9087(2) Å, c = 18.6173(4) Å, α = 90°, β = 108.0790(10)°, γ = 90°, V = 2601.58(10) Å3, T = 100.(2) K, space group P1211, Z = 4, μ(Cu Kα) = 0.699 mm−1, 57,080 reflections measured, 10,194 independent reflections (Rint = 0.0603). The final R1 values were 0.0400 (I > 2σ(I)). The final wR(F2) values were 0.1029 (I > 2σ(I)). The final R1 values were 0.0407 (all data). The final wR(F2) values were 0.1037 (all data). The goodness of fit on F2 was 1.035. Flack parameter = 0.03(6). CCDC 2042988.
Crystallographic data for 19: C30H38O6, M = 494.60, a = 8.8693(2) Å, b = 13.6784(3) Å, c = 21.2902(5) Å, α = 90°, β = 90°, γ = 90°, V = 2582.88(10) Å3, T = 100.(2) K, space group P212121, Z = 4, μ(Cu Kα) = 0.704 mm−1, 48,650 reflections measured, 5081 independent reflections (Rint = 0.0251). The final R1 values were 0.0265 [I > 2σ(I)]. The final wR(F2) values were 0.0688 [I > 2σ(I)]. The final R1 values were 0.0266 (all data). The final wR(F2) values were 0.0689 (all data). The goodness of fit on F2 was 1.048. Flack parameter = −0.003(17). CCDC 1999124.
Crystallographic data for 20: C30H38O6·CHCl3, M = 613.97, a = 10.4221(3) Å, b = 12.2544(3) Å, c = 23.5810(6) Å, α = 90°, β = 90°, γ = 90°, V = 3011.68(14) Å3, T = 100.(2) K, space group P212121, Z = 4, μ(Cu Kα) = 3.102 mm−1, 54935 reflections measured, 5892 independent reflections (Rint = 0.0346). The final R1 values were 0.0285 [I > 2σ(I)]. The final wR(F2) values were 0.0754 [I > 2σ(I)]. The final R1 values were 0.0286 (all data). The final wR(F2) values were 0.0755 (all data). The goodness of fit on F2 was 1.067. Flack parameter = 0.045(3). CCDC 1999125.
Crystallographic data for 21: C30H36O6, M = 492.59, a = 8.9299(2) Å, b = 13.2860(4) Å, c = 21.1616(6) Å, α = 90°, β = 90°, γ = 90°, V = 2510.67(12) Å3, T = 100.(2) K, space group P212121, Z = 4, μ(Cu Kα) = 0.724 mm−1, 24592 reflections measured, 4932 independent reflections (Rint = 0.0367). The final R1 values were 0.0361 [I > 2σ(I)]. The final wR(F2) values were 0.0943 [I > 2σ(I)]. The final R1 values were 0.0362 (all data). The final wR(F2) values were 0.0944 (all data). The goodness of fit on F2 was 1.065. Flack parameter = 0.05(3). CCDC 1999126.
Acknowledgments
This study was supported by the Yunnan Wanren Project (YNWR-KJLJ-2019-002), and the Program of Yunling Scholarship (Ji-Jun, Chen), the Reserve Talents of Young and Middle-aged Academic and Technical Leaders in Yunnan Province (Changan, Geng).
Author contributions
Ji-Jun Chen designed and guided all the experiments and revised the manuscript. Lihua Su conducted the separation, structural identification, and wrote the manuscript. Xintian Zhang performed the mechanism experiments. Yunbao Ma and Xiaoyan Huang carried out cytotoxicity assays. Changan Geng conducted the ECD calculations and revised the manuscript. Jing Hu designed the pharmacological test and analyzed the corresponding data. Tianze Li doublechecked the data and revised the manuscript. Shuang Tang, Cheng Shen and Zhen Gao participated datum analysis. Xuemei Zhang assisted the chemical experiments. All authors read and approved the final manuscript.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2020.12.006.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Yang J.D., Roberts L.R. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7:448–458. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Y.Y., Wang W., Fang B., Ma F.Y., Zheng Q., Deng P.Y. Anti-tumor effect of germacrone on human hepatoma cell lines through inducing G2/M cell cycle arrest and promoting apoptosis. Eur J Pharmacol. 2013;698:95–102. doi: 10.1016/j.ejphar.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 3.Poupon R., Fartoux L., Rosmorduc O. Therapeutic advances in hepatocellular carcinoma. Bull Acad Natl Med. 2008;192:23–31. [PubMed] [Google Scholar]
- 4.Chai F.N., Ma W.Y., Zhang J., Xu H.S., Li Y.F., Zhou Q.D. Coptisine from Rhizoma coptidis exerts an anti-cancer effect on hepatocellular carcinoma by up-regulating miR-122. Biomed Pharmacother. 2018;103:1002–1011. doi: 10.1016/j.biopha.2018.04.052. [DOI] [PubMed] [Google Scholar]
- 5.Gan L., Liu Z.J., Sun C. Obesity linking to hepatocellular carcinoma: a global view. Biochim Biophys Acta, Rev Cancer. 2018;1869:97–102. doi: 10.1016/j.bbcan.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Zhan Z.J., Ying Y.M., Ma L.F., Shan W.G. Natural disesquiterpenoids. Nat Prod Rep. 2011;28:594–629. doi: 10.1039/c0np00050g. [DOI] [PubMed] [Google Scholar]
- 7.Zhang R., Tang C.P., Liu H.C., Ren Y.M., Ke C.Q., Yao S. Tetramerized sesquiterpenoid ainsliatetramers A and B from Ainsliaea fragrans and their cytotoxic activities. Org Lett. 2019;21:8211–8214. doi: 10.1021/acs.orglett.9b02909. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y., Shen Y.H., Jin H.Z., Fu J.J., Hu X.J., Qin J.J. Ainsliatrimers A and B, the first two guaianolide trimers from Ainsliaea fulvioides. Org Lett. 2008;10:5517–5520. doi: 10.1021/ol802249z. [DOI] [PubMed] [Google Scholar]
- 9.Xie Y.G., Zhong X.L., Xiao Y.Z., Zhu S.L., Muhammad I., Yan S.K. Vieloplains A−G, seven new guaiane-type sesquiterpenoid dimers from Xylopia vielana. Bioorg Chem. 2019;88:102891. doi: 10.1016/j.bioorg.2019.03.065. [DOI] [PubMed] [Google Scholar]
- 10.Hilmi F., Gertsch J., Bremner P., Valovic S., Heinrich M., Sticher O. Cytotoxic versus anti-inflammatory effects in HeLa, Jurkat T and human peripheral blood cells caused by guaianolide-type sesquiterpene lactones. Bioorg Med Chem. 2003;11:3659–3663. doi: 10.1016/s0968-0896(03)00346-8. [DOI] [PubMed] [Google Scholar]
- 11.Rozenblat S., Grossman S., Bergman M., Gottlieb H., Cohen Y., Dovrat S. Induction of G2/M arrest and apoptosis by sesquiterpene lactones in human melanoma cell lines. Biochem Pharmacol. 2008;75:369–382. doi: 10.1016/j.bcp.2007.08.024. [DOI] [PubMed] [Google Scholar]
- 12.Xue G.M., Zhu D.R., Zhu T.Y., Wang X.B., Luo J.G., Kong L.Y. Lactone ring-opening seco-guaianolide involved heterodimers linked via an ester bond from Artemisia argyi with NO inhibitory activity. Fitoterapia. 2019;132:94–100. doi: 10.1016/j.fitote.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Lindenmeyer M.T., Hrenn A., Kern C., Castro V., Murillo R., Mueller S. Sesquiterpene lactones as inhibitors of IL-8 expression in HeLa cells. Bioorg Med Chem. 2006;14:2487–2497. doi: 10.1016/j.bmc.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 14.Ma L.F., Chen Y.L., Shan W.G., Zhan Z.J. Natural disesquiterpenoids: An update. Nat Prod Rep. 2020;37:999–1030. doi: 10.1039/c9np00062c. [DOI] [PubMed] [Google Scholar]
- 15.Ling Y.R., Humphries C.J., Gilbert M.G. Artemisia Linnaeus, Sp. P1. 2:845. 1753. In: Wu Z.Y., Peter H.R., Hong D.Y., editors. 1st ed. vols. 20–21. Science Press: Beijing and Missouri Botanical Garden Press; St. Louis: 2011. pp. 676–737. (Flora of China). [Google Scholar]
- 16.Mohamed A.E.-H.H., El-Sayed M.A., Hegazy M.E., Helaly S.E., Esmail A.M., Mohamed N.S. Chemical constituents and biological activities of Artemisia herba-alba. Rec Nat Prod. 2010;4:1–25. [Google Scholar]
- 17.Lee S.H., Lee M.Y., Kang H.M., Han D.C., Son K.H., Yang D.C. Anti-tumor activity of the farnesyl-protein transferase inhibitors arteminolides, isolated from Artemisa. Bioorg Med Chem. 2003;11:4545–4549. doi: 10.1016/j.bmc.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 18.Xue G.M., Han C., Chen C., Li L.N., Wang X.B., Yang M.H. Artemisians A−D, diseco-guaianolide involved heterodimeric [4+2] adducts from Artemisia argyi. Org Lett. 2017;19:5410–5413. doi: 10.1021/acs.orglett.7b02681. [DOI] [PubMed] [Google Scholar]
- 19.Wong H.F., Brown G.D. Dimeric guaianolides and a fulvenoguaianolide from Artemisia myriantha. J Nat Prod. 2002;65:481–486. doi: 10.1021/np0103113. [DOI] [PubMed] [Google Scholar]
- 20.Wen J., Shi H.M., Xu Z.R., Chang H.T., Jia C.Q., Zan K. Dimeric guaianolides and sesquiterpenoids from Artemisia anomala. J Nat Prod. 2010;73:67–70. doi: 10.1021/np900462u. [DOI] [PubMed] [Google Scholar]
- 21.Bohlmann F., Ang W., Trinks C., Jakupovic J., Huneck S. Dimeric guaianolides from Artemisia sieversiana. Phytochemistry. 1985;24:1009–1015. [Google Scholar]
- 22.Huang Z.S., Pei Y.H., Liu C.M., Lin S., Tang J., Huang D.S. Highly oxygenated guaianolides from Artemisia dubia. Planta Med. 2010;76:1710–1716. doi: 10.1055/s-0030-1249957. [DOI] [PubMed] [Google Scholar]
- 23.Beauhaire J., Fourrey J.L., Vuilhorgne M., Lallemand J.Y. Dimeric sesquiterpene lactones: Structure of absinthin. Tetrahedron Lett. 1980;21:3191–3194. [Google Scholar]
- 24.Lee S.H., Kim M.J., Bok S.H., Lee H., Kwon B.M., Shin J. Arteminolide, an inhibitor of farnesyl transferase from Artemisia sylvatica. J Org Chem. 1998;63:7111–7113. doi: 10.1021/jo980919p. [DOI] [PubMed] [Google Scholar]
- 25.Lee S.H., Kim H.K., Seo J.M., Kang H.M., Kim J.H., Son K.H. Arteminolides B, C, and D, new inhibitors of farnesyl protein transferase from Artemisia argyi. J Org Chem. 2002;67:7670–7675. doi: 10.1021/jo020299z. [DOI] [PubMed] [Google Scholar]
- 26.Reinhardt J.K., Klemd A.M., Danton O., De Mieri M., Smiesko M., Huber R. Sesquiterpene lactones from Artemisia argyi: absolute configuration and immunosuppressant activity. J Nat Prod. 2019;82:1424–1433. doi: 10.1021/acs.jnatprod.8b00791. [DOI] [PubMed] [Google Scholar]
- 27.Xue G.M., Zhu D.R., Han C., Wang X.B., Luo J.G., Kong L.Y., Artemisianins A.−D. New stereoisomers of seco-guaianolide involved heterodimeric [4+2] adducts from Artemisia argyi induce apoptosis via enhancement of endoplasmic reticulum stress. Bioorg Chem. 2019;84:295–301. doi: 10.1016/j.bioorg.2018.11.013. [DOI] [PubMed] [Google Scholar]
- 28.Karimov Z., Kasymov S.Z., Yagudaev M.R., Sidyakin G.P. Crystal structure of the sesquiterpene lactone absinthin. Khim Prir Soedin. 1980:729–730. [Google Scholar]
- 29.Beauhaire J., Fourrey J.L., Lellemand J.Y., Vuilhorgne M. Dimeric sesquiterpene lactone. Structure of isoabsinthin. Acid isomerization of absinthin derivatives. Tetrahedron Lett. 1981;22:2269–2272. [Google Scholar]
- 30.Beauhaire J., Fourrey J.L., Guittet E. Structure of absintholide, a new guaianolide dimer of Artemisia absinthium L. Tetrahedron Lett. 1984;25:2751–2754. [Google Scholar]
- 31.Ovezdurdyev A., Abdullaev N.D., Yusupov M.I., Kasymov S.Z. Artenolide, a new disesquiterpenoid from Artemisia absinthium. Khim Prir Soedin. 1987;1987:667–671. [Google Scholar]
- 32.Turak A., Shi S.P., Jiang Y., Tu P.F. Dimeric guaianolides from Artemisia absinthium. Phytochemistry. 2014;105:109–114. doi: 10.1016/j.phytochem.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 33.Jakupovic J., Chen Z.L., Bohlmann F. Artanomaloide, a dimeric guaianolide and phenylalanine derivatives from Artemisia anomala. Phytochemistry. 1987;26:2777–2779. [Google Scholar]
- 34.Tu P.F., Wen J., Jiang Y., Zhou S.X., inventors, Peking University, assignee . 2009. Guaianolide type sesquiterpene lactones dimers, their preparation and application. CN101585841A. [Google Scholar]
- 35.Zan K., Chai X.Y., Chen X.Q., Wu Q., Fu Q., Zhou S.X. Artanomadimers A−F: six new dimeric guaianolides from Artemisia anomala. Tetrahedron. 2012;68:5060–5065. [Google Scholar]
- 36.Zhang C., Wang S., Zeng K.W., Li J., Ferreira D., Zjawiony J.K. Nitric oxide inhibitory dimeric sesquiterpenoids from Artemisia rupestris. J Nat Prod. 2016;79:213–223. doi: 10.1021/acs.jnatprod.5b00894. [DOI] [PubMed] [Google Scholar]
- 37.Zhou X.D., Chai X.Y., Zeng K.W., Zhao M.B., Jiang Y., Tu P.F. Artesin A, a new cage-shaped dimeric guaianolide from Artemisia sieversiana. Tetrahedron Lett. 2015;56:1141–1143. [Google Scholar]
- 38.Ma C.M., Nakamura N., Hattori M., Zhu S., Komatsu K. Guaiane dimers and germacranolide from Artemisia caruifolia. J Nat Prod. 2000;63:1626–1629. doi: 10.1021/np000005+. [DOI] [PubMed] [Google Scholar]
- 39.Lee S.H., Kang H.M., Song H.C., Lee H., Lee U.C., Son K.H. Sesquiterpene lactones, inhibitors of farnesyl protein transferase, isolated from the flower of Artemisia sylvatica. Tetrahedron. 2000;56:4711–4715. [Google Scholar]
- 40.Hu J.F., Feng X.Z. Artselenoide, a new dimeric guaianolide from Artemisia selengensis. Chin Chem Lett. 1998;9:829–832. [Google Scholar]
- 41.Mallabaev A., Tashkhodzhaev B., Saitbaeva I.M., Yagudaev M.R., Sidyakin G.P. Structure of artelein, a new dimeric lactone from Artemisia leucodes. Khim Prir Soedin. 1986;1986:46–52. [Google Scholar]
- 42.Wang Q.Q., Zhang T., Ke C.Q., Tang C.P., Yao S., Lin L.G., Ye Y. Sesquiterpene lactone dimers from Artemisia lavandulifolia inhibit interleukin-1β production in macrophages through activating autophagy. Bioorg Chem. 2020;105:104451. doi: 10.1016/j.bioorg.2020.104451. [DOI] [PubMed] [Google Scholar]
- 43.Deng W.Q., Wang J., Wang X., Li Q., Wang Y. Study on chemical components of essential oils from Artemisia atrovirens. Hubei Agri Sci. 2011;50:4062–4065. [Google Scholar]
- 44.Oikawa H., Tokiwano T. Enzymatic catalysis of the Diels−Alder reaction in the biosynthesis of natural products. Nat Prod Rep. 2004;21:321–352. doi: 10.1039/b305068h. [DOI] [PubMed] [Google Scholar]
- 45.Wang S., Sun J., Zeng K.W., Chen X.G., Zhou W.Q., Zhang C. Sesquiterpenes from Artemisia argyi: Absolute configurations and biological activities. Eur J Org Chem. 2014;5:973–983. [Google Scholar]
- 46.Kawazoe K., Tsubouchi Y., Abdullah N., Takaishi Y., Shibata H., Higuti T. Sesquiterpenoids from Artemisia gilvescens and an anti-MRSA compound. J Nat Prod. 2003;66:538–539. doi: 10.1021/np020593m. [DOI] [PubMed] [Google Scholar]
- 47.Xu X.K., Ye J., Chen L.P., Zhang W.D., Yang Y.X., Li H.L. Four new isomeric sesquiterpene lactone dimers from Carpesium faberi. Tetrahedron Lett. 2015;56:6381–6384. [Google Scholar]
- 48.Li X., Kong L.M., Yang Q.H., Duan A.Z., Ju X.M., Cai B.C. Parthenolide inhibits ubiquitin-specific peptidase 7 (USP7), WNT signaling, and colorectal cancer cell growth. J Biol Chem. 2020;295:3576–3589. doi: 10.1074/jbc.RA119.011396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H.Y., Zhang C.Y., Xu L.T., Zang K., Ning Z.Y., Zhu X.Y. Bufalin suppresses hepatocellular carcinoma invasion and metastasis by targeting HIF-1α via the PI3K/AKT/mTOR pathway. Oncotarget. 2016;7:20193–20208. doi: 10.18632/oncotarget.7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu Z.Y., Wu F.L., Chen L., Xie S.Q., Li Q., Wang C.J. ETME, a novel β-elemene derivative, synergizes with arsenic trioxide in inducing apoptosis and cell cycle arrest in hepatocarcinoma cells via a p53-dependent pathway. Acta Pharm Sin B. 2014;4:424–429. doi: 10.1016/j.apsb.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.