

ABSTRACT
Parkinson's disease (PD) is a progressive neurodegenerative disease where dopaminergic neurons in the substantia nigra are lost, resulting in a decrease in striatal dopamine and, consequently, motor control. Dopaminergic degeneration is associated with the appearance of Lewy bodies, which contain membrane structures and proteins, including α‐synuclein (α‐Syn), in surviving neurons. PD displays a multifactorial pathology and develops from interactions between multiple elements, such as age, environmental conditions, and genetics. Mutations in the GBA1 gene represent one of the major genetic risk factors for PD. This gene encodes an essential lysosomal enzyme called β‐glucocerebrosidase (GCase), which is responsible for degrading the glycolipid glucocerebroside into glucose and ceramide. GCase can generate glucosylated cholesterol via transglucosylation and can also degrade the sterol glucoside. Although the molecular mechanisms that predispose an individual to neurodegeneration remain unknown, the role of cholesterol in PD pathology deserves consideration. Disturbed cellular cholesterol metabolism, as reflected by accumulation of lysosomal cholesterol in GBA1‐associated PD cellular models, could contribute to changes in lipid rafts, which are necessary for synaptic localization and vesicle cycling and modulation of synaptic integrity. α‐Syn has been implicated in the regulation of neuronal cholesterol, and cholesterol facilitates interactions between α‐Syn oligomers. In this review, we integrate the results of previous studies and describe the cholesterol landscape in cellular homeostasis and neuronal function. We discuss its implication in α‐Syn and Lewy body pathophysiological mechanisms underlying PD, focusing on the role of GCase and cholesterol. © 2020 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society
Keywords: autophagy, glycosphingolipid, lipid storage diseases, lysosomes, multilamellar bodies, neurodegeneration
Brain Cholesterol Homeostasis and Trafficking
Mammalian cells require cholesterol for membrane integrity and fluidity, and for regulation of cell membrane organization and biophysical properties. 1 , 2 Cholesterol intercalates with phospholipids in the membrane, preventing their clustering and stabilizing the membrane. 3 Cholesterol also intercalates with sphingolipids, other membrane anchor proteins and receptors, forming dynamic lipid rafts in the Golgi apparatus (GA) and plasma membrane. 4 , 5
In neurons, lipid rafts are specialized, semiordered membrane domains where vesicle trafficking and signal transduction are triggered by neurotrophic factors. 6 , 7 , 8 , 9 Neuronal lipid rafts are abundant at the synapse, where cholesterol directly interacts with neurotransmitter receptors. Cholesterol is required for the correct functioning of the synaptic transmission and synaptogenesis. Hence cholesterol homeostasis is carefully heterogeneously regulated in different brain cells. 10 , 11 , 12 , 13 In other tissues, cholesterol is synthesized within the cells and acquired from lipoproteins in the blood; however, in the brain, it must be synthesized de novo because plasma lipoproteins cannot cross the blood–brain barrier (BBB). 14 , 15 , 16
Cholesterol is primarily synthesized in astrocytes within the endoplasmic reticulum (ER) in a multistep process in which the rate‐limiting step is catalyzed by 3‐hydroxy‐3‐methyglutaryl‐coenzyme‐A reductase (HMGR). This synthesis is regulated by a transcription factor called sterol‐regulatory element‐binding protein‐2 (SREBP‐2), which enters the nucleus and binds sterol‐regulatory element‐1 (SRE‐1) in the HMGR gene, 17 inducing its expression and, consequently, cholesterol production. SREBP‐2 also induces genes of lipid/cholesterol uptake, such as LDLR. 18 This process is regulated by SREBP cleavage activating protein (SCAP), an ER membrane protein that acts as a cholesterol sensor. When cellular cholesterol levels are high, the SREBP‐2/SCAP complex is kept in the ER. However, when cholesterol levels decline, this complex is delivered to the GA, where SCAP discharges the N terminus of SREBP‐2 by proteolytic cleavage. SREBP‐2 then enters the nucleus, where it binds to SRE‐1 18 (Fig. 1). Once synthesized, cholesterol is distributed among cell membranes and organelles, and its levels vary among them; thus, intracellular cholesterol trafficking is a dynamic process, important for understanding neural function. 19 , 20
FIG. 1.
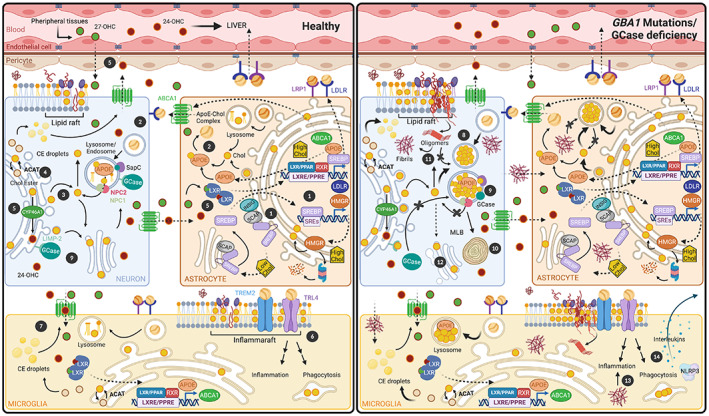
Impaired brain cholesterol trafficking induced by β‐glucocerebrosidase (GCase) deficiency. Cholesterol is primarily synthesized in astrocytes. (1) Under high cholesterol conditions, astrocytes express specific lipid transporter proteins on their membranes (ATP‐binding cassette transporter protein A1 [ABCA1]) and apolipoprotein E (ApoE) to decrease cholesterol content. In addition, sterol‐regulatory element‐binding protein (SREBP) binds to SREBP cleavage activating protein (SCAP), and both are retained in the endoplasmic reticulum (ER) bound to insulin induced gene (INSIG) to inhibit cholesterol synthesis. When cholesterol content decreases, SREBP‐SCAP is transported to the GA, where SREBP is cleaved. Then SREBP is translocated to the nucleus to activate genes required for its synthesis (3‐hydroxy‐3‐methyglutaryl‐coenzyme‐A reductase [HMGR]) and uptake (low‐density lipoprotein receptor [LDLR]) through its binding to sterol‐regulatory element‐1 (SRE‐1). HMGR can be degraded by proteasome when ER‐cholesterol is accumulated. (2) Synthesized cholesterol binds ApoE, forming an ApoE–cholesterol complex that exits the astrocyte via ABCA1, and it is internalized into the neuron by LDLR and LDLR‐related protein 1. (3) In neurons, LDLR complexes are hydrolyzed, and free cholesterol is disseminated among the plasma membrane and other organelles. (4) The excess of neuronal cholesterol is modulated by cholesterol esterification through ACAT (acetyl‐coenzyme A acetyltransferase 1) enzyme and storage in lipid droplets that are secreted through the ABCA1. (5) In the ER, the excess cholesterol is converted into 24S‐hydroxycholesterol (24‐OHC). In astrocytes, 24‐OHC and other oxysterols (such as 27‐OHC) bind LXR(Liver X Receptor) to induce the expression of APOE and ABCA1 genes. Moreover, 24‐OHC can cross the BBB. (6) ApoE–cholesterol complex from astrocyte could bind to Triggering Receptor Expressed on Myeloid Cells 2 (TREM2), Toll‐Like Receptor 4 (TLR4), and LDLR on the inflammaraft of the microglia surface to trigger inflammation and phagocytosis. (7) Oxysterols (24‐OCH and 27‐OCH) can bind LXRs to activate microglia. (8) In a pathological state of GBA1‐PD or GCase deficiency, we proposed that cholesterol is accumulating in the lysosomes independently of the cell type. (9) Cholesterol could disturb the interaction between GCase1 and its transporter (LIMP‐2) impairing GCase activity, and it also might disrupt the contact between GCase and its coactivator SapC, favoring lysosomal cholesterol buildup. (10) Cholesterol accumulation appears to lead to lysosome degeneration called multilamellar bodies (MLBs). (11) Lysosomal cholesterol accumulation could affect cholesterol pools in the rest of membrane organelles, which in turn could alter the α‐synuclein (α‐Syn) interaction with lipid rafts and contribute to α‐Syn oligomerization. Ultimately, these α‐Syn oligomers cannot be degraded by lysosomes; as a consequence, they lead to α‐Syn fibrils. (12) The α‐Syn overburden appears to affect GA tubulation and fragmentation. (13) Aberrant α‐Syn is released from neurons and transferred to microglia and astrocytes, and triggers the inflammatory response. (14) We hypothesized that GBA1 mutations increase interleukins and NLRP3 inflammasome activating the inflammatory response. Peroxisome Proliferator‐Activated Receptor (PPAR); Retinoid X Receptor (RXR); Peroxisome Proliferator Response Elements (PPRE); Liver X Receptor Response Elements (LXRE).
To deliver cholesterol to neurons, astrocytes synthesize apolipoproteins (Apos), proteins that bind lipids forming lipoproteins. 21 , 22 The most abundant Apos in the brain are ApoE, ApoJ (both bind cholesterol), and ApoA1. In response to increased cholesterol, astrocytes express ApoE and specific lipid transporters on their membranes, such as the ATP‐binding cassette transporter protein A1 (ABCA1). The ApoE–cholesterol complex exits the astrocyte via ABCA1, then is internalized into the neuron by low‐density lipoprotein (LDL) receptors (LDLR) and the LDLR‐related protein 1 (LRP1) 1 , 21 , 22 , 23 , 24 (Fig. 1). This complex is hydrolyzed within the late endosome/lysosome, resulting in free cholesterol. Subsequent trafficking into subcellular membrane compartments is facilitated by the Niemann–Pick type C1 (NPC1) and C2 (NPC2) proteins 25 (Fig. 1). Neurons use this cholesterol for building their extensive membranes of axons, dendrites, synapses, and synaptic vesicles. 1 , 2 , 21
Neurons have various mechanisms to manage excess cholesterol, including cholesterol esterification and storage in intracellular lipid droplets and secretion through the ABCA1 transporter. The main pathway to eliminate excess intracellular cholesterol is mediated by cholesterol 24‐hydroxylase (CYP46A1), which converts cholesterol to 24S‐hydroxycholesterol (24‐OHC). 26 Activated liver X receptors (LXRs) translocate 24‐OHC to the nucleus, where it inhibits cholesterol synthesis, induces the expression of ABCA1 and APOE genes, and activates cholesterol efflux to astrocytes. 27 24‐OHC and other oxysterols can cross the BBB or be delivered to the plasma via cerebrospinal fluid (Fig. 1). Oxysterol homeostasis is tightly controlled in the brain to maintain specific levels in each region. The role of these cholesterol oxidation products in neurodegeneration remains unclear; more research is needed to reveal their exact function.
Parkinson's Disease and GBA1 Mutations
Alterations in cholesterol homeostasis, biosynthesis, transport, and lipid raft organization lead to structural and functional central nervous system (CNS) neurodegenerative diseases. 9 , 28 In Parkinson's disease (PD), the relationship between abnormal cholesterol and neurodegeneration remains unclear, and present literature suggests that either an increase or decrease in brain cholesterol is associated with this disease.
PD is the most common neurodegenerative movement disorder, affecting 1.5% of people aged 65 years or older. 29 PD is characterized by the loss of dopaminergic neurons in the substantia nigra (SN), and the resulting dopamine depletion in the striatum triggers the characteristic motor symptoms. Lewy bodies (LBs), the neuropathological hallmark of PD, are composed of α‐synuclein (α‐Syn) aggregates 30 surrounded by proteins involved in ubiquitin‐proteasome degradation or in the autophagy process 31 that accumulated with aging. 32
Although the etiology of PD is unknown, many epidemiological studies indicate that the disease develops from complex interactions among age, environmental factors, and susceptibility genes that affect numerous cellular processes. 33 , 34 Evidence indicates that changes in lipid homeostasis occur in the CNS during physiological aging; this alteration is potentiated in neurodegenerative diseases, such as PD and AD. Understanding the role of cholesterol in PD will identify new targets to treat PD.
Over the past 15 years, PD research has focused on the link among various lysosomal storage diseases, α‐Syn aggregation, and dopamine loss. GBA1 encodes a lysosomal hydrolase called β‐glucocerebrosidase (GCase), which cleaves glucose moieties from the common glycosphingolipid (GSL) glucocerebroside (glucosylceramide [GlcCer]). 35 GSLs are glycolipids that consist of ceramide (Cer) and oligosaccharides. 36 GSLs and cholesterol are components of lipid rafts in membranes. 37 GBA1 mutations with prominent deficient lysosomal GCase activity cause Gaucher disease (GD), a lysosomal storage disorder. GBA1 mutations are risk factors for neuronal synucleinopathies (PD, PD dementia, or dementia with LBs). 38 These mutations are present in 7%–12% of PD cases, increasing PD risk by 20‐ to 30‐fold. 39 , 40 , 41 , 42 However, PD develops in only 10%–30% of these monoallelic or biallelic GBA1 mutant carriers by the age of 80 years. 43 , 44 , 45 Gene dose poorly correlates with PD risk; that is, there is no significantly higher incidence of PD among patients with GD and GBA1 carriers. There seems to be a link between severity of GBA1 mutations and PD phenotype and earlier disease onset. 46 , 47 Compared with patients with idiopathic PD, patients with GBA1‐associated‐PD (GBA1‐PD) show earlier disease onset and faster progression, more severe symptoms, and greater incidence of nonmotor symptoms, such as rapid eye movement, sleep disorder, hallucinations, depression and anxiety, and often, cognitive impairment and dementia. 41 , 48 , 49 , 50 A clear relationship between residual GCase activity of particular GBA1 mutations and PD risk has not been unequivocally documented. The actual lysosomal environment in aging brain might conceivably have a major impact on the catalytic capacity of a mutant enzyme.
Notably, the phenotypic disparity among patients with GD 51 or PD 52 with the same GBA1 genotype might indicate the existence of environmental or genetic modifiers. 53 , 54 These modifiers could be lysosomal genes, for example, CTSB (cathepsin B) 55 or GBA2 (discussed later). Moreover, mutations in LRRK2 (leucine‐rich repeat kinase 2), which participates in lysosomal function and inflammatory response, seem to alleviate GBA1 mutations effects in human induced pluripotent stem cell (iPSC)‐derived neurons 56 or astrocytes. 57
Currently, how GBA1 mutations increase the PD risk is unknown. GBA1 mutations decrease lysosomal GCase levels and activity because GCase is partially retained in the ER, triggering stress and leading to impaired autophagy and apoptosis. 58 , 59 , 60 GBA1 mutations cause GlcCer accumulation, which in turn causes insoluble α‐Syn oligomers to polymerize into fibrils in patient iPSC‐derived dopamine neurons. 61 , 62 This α‐Syn aggregation reduces lysosomal degradation, causing neurotoxicity. It also impairs GCase movement from the ER to the GA, decreasing its presence in lysosomes, further promoting GlcCer accumulation and producing a positive feedback loop. 62 , 63 , 64 GBA1 overexpression decreases α‐Syn aggregation in PD models, 65 indicating that increased GCase activity slows down the degenerative process. 66 , 67 In vitro models demonstrated that GCase interacts closely with the C terminus of α‐Syn. 68 Remarkably, reduced GCase activity is not limited to GBA1 carriers but is also found in the SN and putamen of idiopathic PD 69 and is found to decrease with aging even in individuals with normal GBA1. 65 Low GCase activity and high levels of the corresponding glycolipid substrates are found in postmortem brains of aged control subjects 65 , 70 , 71 and in patients with sporadic PD, 65 , 69 as well as in Gba1 +/− mice. 72
Cellular and animal PD models revealed that reducing GCase activity in early stages can potentiate preexisting α‐Syn pathology independently of brain cell type, 73 disturb physiological α‐Syn tetramers/multimers, 74 and contribute to α‐Syn spreading. 75 In turn, progressive toxic α‐Syn accumulation in lysosomes decreases GCase activity. 75
Cholesterol and α‐Syn
α‐Syn is a 140‐residue lipid‐binding protein with a typical Apo structure. 23 α‐Syn is abundantly and ubiquitously present in the brain, located predominantly in presynaptic terminals inside membranes, such as synaptic vesicles, mitochondria, and the ER. 76 , 77 The amphipathic helices of α‐Syn allow its insertion into the cell membrane, changing its curvature while maintaining its integrity. 23 , 78 Multiple steps of synaptic activity are regulated by α‐Syn, participating in synaptic vesicle cycle 79 and neurotransmitter release. 80 , 81 These α‐Syn‐mediated actions take place through regulation of the soluble NSF attachment proteins repector (SNARE) complex (proteins involved in membrane fusion) 82 and its interaction with cholesterol in the lipid rafts. 83 In dopaminergic neurons, α‐Syn is involved in regulating dopamine release through direct 84 , 85 and indirect 86 interactions with tyrosine hydroxylase, which result in reduced tyrosine hydroxylase activity and dopamine levels.
In vitro cholesterol also modulates α‐Syn expression and aggregation, enabling α‐Syn oligomers to interact with neutral charged membranes, leading to membrane disruption and cell death. 87 In intracellular membranes, α‐Syn interacts with the isooctyl chain of cholesterol through its binding domain called tilted peptide 88 (Fig. 2A). This interaction is modulated by fatty acids, GSLs, phospholipids, and gangliosides, and its alteration favors α‐Syn oligomerization inside cells or the synaptic membrane, contributing to dysfunctional neurotransmitter release. 91 In in vitro models, cholesterol‐rich regions, such as lipid rafts, can act as aggregation sites for α‐Syn, 92 and cholesterol also regulates α‐Syn binding to synaptic‐like vesicles, triggering their clustering. 93 Likewise, α‐Syn can potentially stimulate cholesterol efflux in neuronal cells, 94 creating a regulatory feedback loop between cholesterol and α‐Syn. Moreover, it is reported that extracellular α‐Syn may bind to the neuronal and glial membranes, and thus α‐Syn moves from these membranes to the inside of these cells, where in turn they can spread in a prion‐like fashion. 95
FIG. 2.
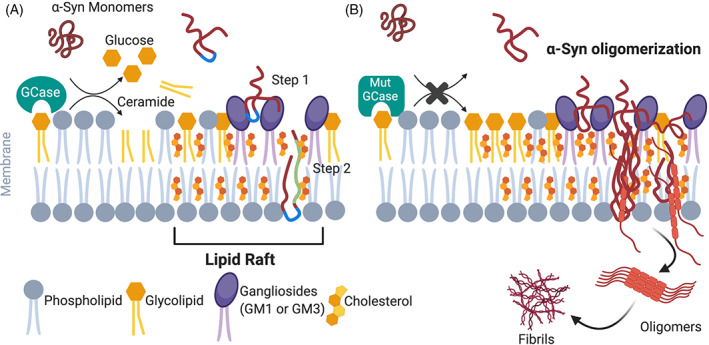
A schematic model illustrating the potential α‐synuclein (α‐Syn) interaction with lipid rafts through cholesterol in PD. Lipid rafts are enriched in cholesterol and other membrane glycolipids, such as sphingolipids. (A) Under physiological conditions, the extracellular domain (blue) of α‐Syn harbors polar amino acid residues that allow its interaction with lipid raft gangliosides, such as ganglioside 1 (GM1) expressed by neurons or GM3 expressed by astrocytes. Thus, GMs allow the attachment of α‐Syn to the surface of plasma membrane (step 2). Then α‐Syn is transferred through the membrane by forming a complex with cholesterol and its tilted peptide domain (green) (step 1). 88 , 89 (B) In the pathological state of GBA1‐PD or β‐glucocerebrosidase (GCase) deficiency, we proposed that cholesterol accumulation in the lysosomes could affect cholesterol levels in the lipid raft that in turn alter α‐Syn interactions with cholesterol. Ultimately, this alteration could favor α‐Syn oligomerization inside the cells or in the synaptic membrane, contributing to PD pathology. Noticeably, gangliosides content in lipid raft is increased in PD. 90
α‐Syn phosphorylation at serine 129 is the most significant posttranslational change related to LB pathology and is involved in α‐Syn aggregation and toxicity. 30 , 96 , 97 This phosphorylation changes the structure and its protein‐lipid binding 98 and inhibits α‐Syn–cholesterol membrane interactions, impairing its synaptic function. 99 α‐Syn also interacts with some Apos. 23 ApoE4 contributes to α‐Syn aggregation in A53T α‐Syn‐transgenic‐APOEε4 mice and accelerates cognitive impairment in patients with PD. 100 Interestingly, both APOEε4 100 and GBA1 mutations 101 are risk factors for dementia with LBs.
Cholesterol in PD
Aging adults are more susceptible to changes in lipid or cholesterol levels and to age‐related progressive diseases, including PD. Although a plethora of in vitro and in vivo studies associate cholesterol to genetically linked PD, the involvement of cholesterol remains controversial. Specifically, mutations in various PD‐related genes, such as GBA1, LRRK2, SNCA, DJ‐1, or PRKN (among others), change cholesterol levels. 58 , 64 , 102 In a PD mouse model, the cholesterol precursor lanosterol is decreased in dopaminergic neurons, 103 and cholesterol biosynthesis is reduced in PD fibroblasts, 104 suggesting possible cholesterol biosynthesis alteration in PD. In rodents, hypercholesterolemia is involved in nigral dopaminergic neurodegeneration 105 , 106 similar to other studies demonstrating that a high‐fat diet exacerbates parkinsonian pathologies. 107 , 108
Several clinical studies reported that PD is linked to hypercholesterolemia and hyperlipidemia, but these findings are controversial. 109 Various reports showed a heightened PD risk in individuals with high cholesterol, 110 , 111 whereas other studies reported a decreased PD risk. 112 , 113 , 114 , 115 , 116 Moreover, a link was reported between low cholesterol and high PD risk, 117 , 118 , 119 , 120 along with reports of a nonassociation between cholesterol levels and PD. 121 , 122 , 123 These varying data might be caused by differences in subject age and sex or other factors. Furthermore, most clinical studies determine blood cholesterol levels; however, these do not necessarily reflect cholesterol in tissues or cells (see the following section). A rigorous analysis of cholesterol impact in PD will conclusively determine which specific changes contribute to PD pathology.
Although cholesterol cannot cross the BBB, elevation of dietary cholesterol increases its metabolite, 27‐OHC, which is able to do so (Fig. 1). In human dopaminergic neuronal precursor cells, 27‐OHC is reported to regulate α‐Syn expression, 124 possibly by binding to LXR, which in turn binds the LXR response element in the α‐Syn promotor, increasing α‐Syn expression. 125 , 126 Thus, it is possible that the conflicting results of previous PD and cholesterol studies are related to oxysterols such as 27‐OHC hitherto not considered as PD risk factors. Notably, oxysterol 24‐OHC is reduced in plasma but elevated in the brains of patients with neurodegenerative diseases, including PD. 127
Cholesterol derivatives β‐sitosterol or β‐d‐glucoside can induce α‐Syn aggregation in mice 128 , 129 and in vitro reactive oxygen species (ROS) production, oxidative damage, and ultimately, neuronal death. 130 , 131 Inversely, SNCA overexpression in patient iPSC‐derived dopaminergic neurons impairs cellular cholesterol homeostasis. 132
Intracellular Cholesterol in GBA1‐PD
Because the role of intracellular cholesterol abnormalities in PD induction is tentative, it is worth considering whether the exact site of cholesterol accumulation protects cells 133 , 134 or renders them more sensitive to cell death. 135 Studies in PD mouse models indicate a dual role of intracellular cholesterol, protecting against lysosomal membrane permeabilization but also stimulating α‐Syn accumulation. 136
Research in GBA1 knockdown SH‐SY5Y cells and embryonic fibroblasts from Gba1 −/− or Gba1 +/− mice indicates increased cholesterol accumulation; this was also observed in mouse primary cortical cells treated with Conduritol B Epoxide (CBE), a GCase inhibitor. 137 In addition, fibroblasts from Gba1 −/− mice show augmented levels of cholesterol and cholesteryl esters. 137 We demonstrated in N370S‐PD fibroblasts that lysosomal cholesterol accumulation appears to lead to lysosome degeneration and increases vulnerability to cytotoxic stimuli. 58 , 59 , 138 Hence we hypothesize that higher GlcCer and cholesterol levels caused by diminished GCase activity might indirectly increase α‐Syn levels by decreasing lysosomal maturation and function. Recently, an altered lysosomal GlcCer export seems to increase risk for PD in individuals with mutations in the ATP10B gene. 139 More research is needed to elucidate whether other mechanisms exist that explain the lysosomal cholesterol accumulation in the presence of GBA1 mutations. A cholesterol binding site has been found on the GCase1‐specific transporter, lysosomal integral membrane protein‐2 (LIMP‐2) 140 , 141 (Fig. 1). This suggests that the cholesterol trafficking impairment detected in GBA1‐PD models could also interfere with the GCase transport to lysosomes. Indeed, some cholesterol accumulation occurs almost in any type of lysosomal storage disease; likely cholesterol gets stuck in dysfunctional lysosomes. Although it is known that mammalian (or mechanistic) target of rapamycin complex 1 (mTORC1) is involved in regulating cholesterol trafficking to lysosomes, 142 , 143 the exact role of mTORC1 in GCase dysfunction and autophagy is still under debate.
Lysosomal cholesterol accumulation could affect cholesterol pools in the rest of the membrane organelles; thus, it could alter how α‐Syn interacts with lipid rafts (Fig. 2B) and further downstream interactions, which are required for synaptic localization, integrity, and vesicle cycling described in mouse models. 144 The pathological accumulation of intracellular cholesterol could also affect the role of α‐Syn in modulating the SNARE complex, preventing lysosomes from fusing with autophagosomes as we previously suggested. 58 Moreover, α‐Syn overexpression appears to affect GA tubulation through Rab and SNARE proteins; this mechanism is involved in the GA fragmentation that occurs in PD and GBA1‐PD. 58 , 59 , 63 , 145 Because cholesterol could ultimately control endolysosomal membrane organization, its storage could modulate GCase activity by disrupting the contact between GCase and its coactivator SapC 146 , 147 (Fig. 1). Therefore, in GBA1‐PD models, alterations caused by cholesterol or GCase perturbation can synergize, producing a deleterious vicious cycle.
It was proposed that the accumulation of degenerating lysosomes in dopaminergic neurons, which contain α‐Syn aggregates, could form future LBs that are eventually secreted. Because of their membranous nature, these degenerating lysosomes could easily bind other neuronal membranes, thereby transmitting α‐Syn aggregates between neurons. Once inside the healthy neuron, these α‐Syn aggregates could seed the formation of new toxic aggregates in a prion‐like manner. 148 We propose that the MLBs caused by GBA1 mutations, which appear to be degenerating lysosomes full of cholesterol and other lipids, favor the aberrant interaction of cholesterol and α‐Syn. Ultimately, these MLBs could facilitate the prion‐like and transneuronal propagation of α‐Syn in GBA1‐PD (Fig. 3).
FIG. 3.
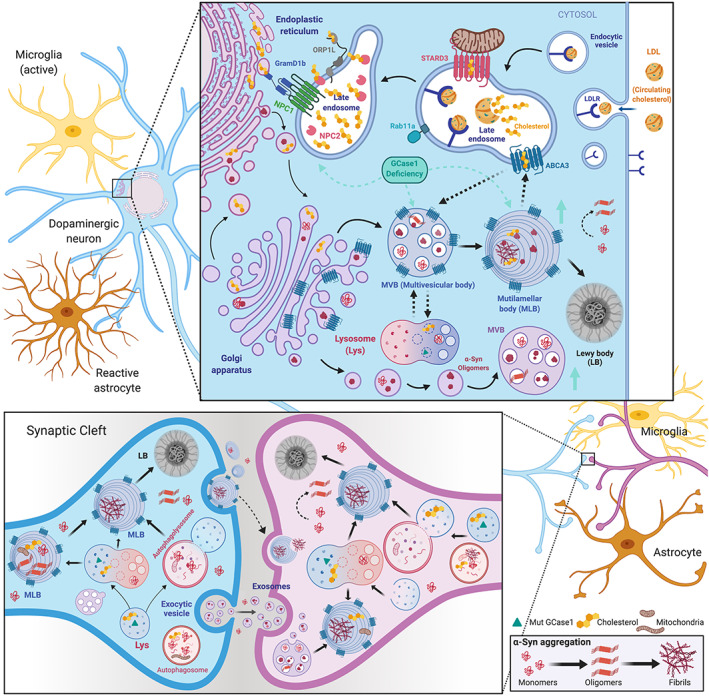
Transneuronal propagation of pathogenic α‐synuclein (α‐Syn) through multilamellar bodies (MLBs) in Parkinson's disease (PD). First, a schematic illustration representing the role of intracellular cholesterol trafficking in hypothetical MLB biogenesis is zoom visualized. The MLB biogenesis, a pathological type of lamellar body found in GBA1‐PD fibroblasts, is completely unknown. We hypothesize that Rab11a and ABCA‐like transporters could be involved. ABCA3 could ingress cholesterol or other lipids from other donor organelles into MLB for its biogenesis and, in turn, from MLBs into the endosomes. Overall, ABCA3 play a critical role in the maturation of the multivesicular bodies (MVBs) into the MLBs through fusion with the autophagolysosome by helping these vesicles become filled with lipids and cholesterol. Second, a diagram illustrates how GBA1 mutations alter the dynamics of autophagosome–lysosomes and endosomes. Accumulating cholesterol generates MVBs and MLBs containing pathogenic α‐Syn that could degenerate into Lewy bodies (LBs). Multilamellar (ML) or multivesicular (MV) organelles, as well as naked α‐Syn, could be secreted from dying neurons (blue) and, in turn, captured by healthy neurons (pink) or glial cells (yellow and orange) propagating α‐Syn pathology. Once inside the healthy neuron, α‐Syn fibrils break the ML membrane to reach the cytosol, where they seed new aggregates by recruiting monomeric α‐Syn. Notably, GCase lysosomal dysfunction increases the amount of MV endosomes that secrete α‐Syn by exosomes. Bottom right panel shows the α‐Syn aggregation process.
Mitochondrial dysfunction is also associated with GBA1 heterozygous mutations in cellular PD models through impaired mitophagy. 59 , 149 Because mitochondrial cholesterol is reported to be involved in mitophagy, 150 we speculate that cholesterol accumulation in GBA1‐PD models could cause mitochondrial dysfunction. Mitochondrial DNA disorganization is observed in NPC1 disease human fibroblasts and in control fibroblasts treated with pravastatin or U18666A, inhibitors of cholesterol synthesis or trafficking, respectively. 151 Therefore, this might also occur in GBA1‐PD models.
GBA2 and Cholesterol Metabolism
Cells contain, besides the lysosomal GCase, another beta‐glucosidase named GBA2. 152 GBA2 is located in the ER and endosomes, with its catalytic pocket facing the cytosol. 153 , 154 GlcCer is formed from Cer and UDP (Uridine DiPhosphate) ‐glucose by the enzyme GlcCer synthase in the cytosolic leaflet of the GA. Most of the generated GlcCer is transferred to the luminal membrane leaflet for further processing into complex GSLs, but some of the lipid remains at the cytosolic leaflet, where it is subject to metabolism by GBA2. 155 Interestingly, GBA2 does not only cleave GlcCer to Cer and glucose but also acts as transglucosidase, transferring the glucose from GlcCer to cholesterol. 156 Thus, GBA2 can generate from GlcCer and glucosylated cholesterol (GlcChol). 156 Importantly, GBA2 directly links in this manner GlcCer to cholesterol metabolism.
The presence of GlcChol was demonstrated in various tissues. 156 , 157 Under normal conditions, GCase degrades GlcChol in lysosomes; however, when lysosomes excessively accumulate cholesterol, as is the case in NPC disease in which cholesterol egress from lysosomes is impaired because of mutated variants of NPC1 or NPC2 protein, GCase inside lysosomes also performs transglucosylation using the abundant cholesterol as acceptor. 156 Consequently, GlcChol is markedly increased in patients with NPC and mouse models. 156 In the lipid‐laden lysosomes of NPC cells and tissues, GCase activity tends to be partly reduced, which is accompanied by increased activity of GBA2. 158 This disbalance further favors GlcChol levels. Pharmacological inhibition or genetic ablation of GBA2 in Npc −/− mice significantly increases life span and delays onset of motor impairment. 158 Based on this, excessive GBA2 activity and the related formation of toxic GlcChol might be hypothesized to contribute to neuropathology. Notably, glucosylated sitosterol, a very close analogue of GlcChol, induced PD symptoms in rodent models. 128 Recently, GBA2 was found to also form galactosylated cholesterol, a lipid that accumulates in the brain. 159
The precise role of GBA2 in neuropathology in PD‐GBA1 is far from clear. In GD, GBA2 activity is found to be increased in response to reduced GCase. 154 , 160 , 161 However, in cellular PD models 59 , 162 and SN of patients with PD 69 reduced GCase and GBA2 activities were observed. It should be kept in mind that the actual ratio of degradative lysosomal GCase and synthetic GBA2, as well as the availability of cholesterol as acceptor, will determine GlcChol levels. A careful analysis of GlcChol levels in PD‐GBA1 brain seems warranted. The various aspects of GlcChol generation have recently been reviewed. 163 , 164 , 165
Cholesterol in Neuroinflammation
Neuroinflammation and gliosis, relevant hallmarks of neurodegeneration in PD, 166 have emerged during the past few years as one of the causes of neuronal death rather than the consequence. Chronic microglial activation is detected in striatal and cortical regions of postmortem brains from patients with PD and from animal models. 167 Due to the role of microglia in tissue debris clearance, a proper lysosomal function is critical. 168 In cellular/neuronal models, PD‐GBA1 mutations impair lysosomal function promoting ROS, 59 which might lead to proinflammatory factors release from activated microglia and/or astrocytes. Ultimately, activated microglia might induce neurotoxic reactive astrocytes, promoting a faster dopaminergic neuronal death in PD. 169 In addition, aberrant α‐Syn is transferred from neurons to microglia and astrocytes, triggering a proinflammatory response. 170 , 171
α‐Syn accumulation by GCase inhibition in mice induces complement C1q 172 expression required for microglial phagocytosis. Intriguingly, genetic Gba1 ablation in dopaminergic mouse neurons activates microglia without neurodegeneration. 173 Furthermore, GlcCer accumulation in experimental and clinical GD induces complement C5a modulating inflammation. 174 Increased inflammation plasma markers are well established for GD 175 and now for patients with GBA1‐PD. 176 N370S‐GD macrophages show increased interleukins and NLRP3 (nucleotide‐binding domain, leucine‐rich repeat family pyrin domain containing 3) inflammasome hypersensitivity as a result of autophagy–lysosomal failure 177 as it occurs in MPTP (1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine)‐PD mice. 178 Notably, GBA1‐mutant mouse astrocytes exhibited dysfunction in inflammatory responses not directly associated with α‐Syn lysosomal degradation. 57
Cholesterol homeostasis and neuroinflammatory signaling are connected in neurodegeneration. 179 Indeed, Npc1 −/− mice displayed dysregulated expression of inflammatory mediators. 180 Hypercholesterolemia produces microglial activation, and high‐cholesterol diet promotes inflammatory responses. 181 Furthermore, oxysterols are involved in glial activation, 182 where LXRs can modulate cholesterol and oxysterol metabolisms through repressing neuroinflammation. 183 The inflammarafts are cholesterol‐enriched lipid domains that are thought to act as a platform mediating the cellular inflammatory response, conceivably being regulated by cholesterol and sphingolipid metabolism 184 and ApoE 185 (Fig. 1). Hence altered intracellular cholesterol related to GCase deficiency might induce a neuroinflammatory response in PD. This should be further investigated because this neuroinflammatory response may discriminate GBA1 carriers susceptible to development of clinical PD.
Overall, it is possible that a particular cholesterol or lipid profile could confer susceptibility or resistance to PD, mimicking the altered molecular mechanism of cholesterol homeostasis in patients with GBA1‐PD. Research to address this hypothesis involves determining the metabolic impairments specific to the disease and investigating interventions that can reestablish cholesterol and lipid homeostasis.
The Autophagy–Lysosomal Pathway and Cholesterol in PD
The autophagy–lysosomal pathway is in charge of bulk degradation of long‐lived or damaged proteins and organelles. An imbalance of the neuronal autophagy–lysosomal pathway function leads to excessive protein aggregation in PD. 186 GCase is important for the modulation of these pathways by maintaining effective endosomal recycling and unfolded proteins degradation.
Our studies in human GBA1‐PD‐derived fibroblasts demonstrated decreased GCase activity and changes in the subcellular localization. 58 , 59 These alterations induce ER stress and disorganization, along with GA fragmentation. Eventually, the ER stress activates the unfolded protein response that primarily leads to an increase in autophagosomes by activated Beclin1 and mTORC1 pathways. 58 , 59 , 60 However, the subsequent autophagy is ineffective due to deficient lysosomal function. This could contribute to general cellular dysfunction because it exacerbates misfolded protein aggregation along with lipid and cholesterol accumulation. 58 , 59 Future research should determine whether accumulated cholesterol and other lipids are the primary nondegraded material in GBA1‐PD or whether they represent secondary abnormalities that may trigger lipid stress and alter lipid trafficking. Moreover, this autophagic flux disruption in cellular GBA1‐PD models can hamper the degradation of worn‐out mitochondria. 149 Consequently, these mitophagy alterations lead to heightened ROS production, similar to what we found in the N370S‐PD fibroblasts, rendering affected cells more prone to cell death. 58 , 59
Oxidative stress could induce lipid peroxidation and oxidized proteins in the lysosomal membrane, reducing lysosomal efficiency to degrade cargo. As mentioned previously, 24S‐OHC is generated in the ER of neurons. Although 24S‐OHC is important for the maintenance of brain cholesterol homeostasis, it can cause neurotoxicity when it is excessively esterified by acetyl‐coenzyme A acetyltransferase 1 in the ER. 187 , 188 Therefore, further studies are necessary to determine the levels of 24‐OHC or 24‐OHC esters in GBA1‐PD models, because accumulation of the latter may disrupt the integrity of the ER membrane triggering secretion of ER luminal proteins, causing ER dysfunction and cell death. 188 Therefore, altered cholesterol homeostasis detected in N370S‐PD fibroblasts compared with control subjects 59 and Gba1‐mutant cells 137 could be a primary contributor to ER stress, dysfunction, and unfolded protein response activation instead of a direct consequence of GCase deficiency. Hence disturbance of cholesterol homeostasis may decisively impact the function and viability of neurons and initiate PD development.
Lewy Pathology
The major pathogenic mechanism of PD is presently considered to be aberrant interaction of lipids with α‐Syn, promoting its harmful aggregation. However, the relationship between this abnormal lipid–α‐Syn interaction and LB formation remains unclear. Indeed, recent studies found that the inner side of most human LBs appears to be filled with lipid‐rich structures, such as undigested membrane fragments and damaged organelles, rather than with aggregated proteins, 189 suggesting a widespread cellular dysfunction before protein accumulation. It is not currently known which precise types of α‐Syn conformation are buried in these membrane fragments and how they are involved in LB formation.
GBA1‐PD Multilamellar Bodies in LB Formation
It was shown that GBA1 carriers have more LBs that noncarriers. 190 , 191 Strikingly, altered organelles in the LBs display as packed assemblies of membranes, 189 resembling multivesicular bodies (MVBs) and multilamellar bodies (MLBs) that we observed in GBA1‐PD fibroblasts. 59 MLBs are a characteristic feature of some lysosomal storage diseases, such as NPC1 disease. Inducing an NPC1 phenotype by treating cells with the intracellular cholesterol transport inhibitor U18666A 192 , 193 or inducing lysosomal dysfunction with cationic amphiphilic drugs such as chloroquine 194 leads to the generation of numerous MLBs with concentric membrane stacks. These can represent dysfunctional lysosomes and contain undegraded phospholipids and cholesterol. It seems to be a synergistic connection between disturbances in lysosomal and mitochondrial pathways, vesicular transport, and PD pathogenesis (for a review, see Smolders and Van Broeckhoven 195 ). Many researchers have pointed out the common neurodegeneration occurrence and α‐Syn accumulation in inherited lysosomal sphingolipid disorders. Several genes implicated in inherited deficiencies in lysosomal sphingolipid metabolism have been linked to PD: GBA1 (discussed earlier), SMPD1 (sphingomyelin phosphodiesterase 1, which encodes acid sphingomyelinase that degrades sphingomyelin to phosphorylcholine and Cer), and SCARB2 (Scavenger Receptor Class B Member 2, which encodes LIMP‐2, the GCase transporter discussed previously). Less clear yet is the association of PD with mutations in the NPC1/NPC2, GLA (encoding lysosomal alpha‐galactosidase A), and GALC (encoding lysosomal galactocerebrosidase) genes. 195
Preliminary findings indicate that genes encoding other lysosomal proteins not directly involved in GSL metabolism (NAGLU, MCOLN1, ARSB, GUSB, GRN, and NEU1) have an association with PD. 195 Thus, it might be hypothesized that a lysosomal disturbance, particularly when it affects GlcCer metabolism, might increase the PD risk, either by causing secondary abnormalities in prelysosomal compartments (such as accumulation of membranous structures or impaired autophagy completion), and/or by causing formation of toxic metabolites. Regarding the latter, GBA1 deficiency is known to cause formation of glucosylsphingosine (GlcSph) via diacylation of GlcCer by the enzyme acid ceramidase. 196 GlcSph is a biologically active lipid with apparent toxic features. 165 Indeed, excessive GlcSph 197 and GlcCer 61 levels promote α‐Syn aggregation in PD models. Notably, although reduced GCase activity is found in different brain regions of patients with PD (as mentioned earlier), there is not much evidence of GCase substrate accumulation in PD brain. 74 , 198 Glycolipid buildup is more variable and lower than observed in GD. It was proposed that subtle, prolonged glycolipid anomalies generate neurodegeneration at advance ages. 147 , 199
Lamellar Bodies in Skin and Lung
Membranous structure accumulation in compartments of the endolysosomal apparatus is not unique to pathological conditions. In specialized cells of the skin and lung, similar membranous organelles called lamellar bodies are actively produced with specific physiological functions. Lamellar bodies are membrane‐enclosed compartments that contain well‐defined lamellar (membrane‐like) lipid structures. 200
In the skin, keratinocytes produce specific lamellar bodies that contain large amounts of GlcCer molecules, transported by ABCA12 (ATP‐binding cassette subfamily A, member 12), with unique Cer moieties, along with GCase. 165 The lamellar bodies are extruded to the outermost layer of skin, the stratum corneum, where GCase converts the GlcCer to Cer molecules, a modification that is essential for the generation of lipid lamellae with appropriate skin barrier features. 165 Defects in ABCA12 and GCase cause marked skin barrier abnormalities; in severe cases, these are incompatible with terrestrial life. 165 It is noteworthy that patients with PD also experience sweating and skin problems; in some cases, their skin become very dry, rough, and wrinkled. 201 , 202 The possible role of GCase in such skin manifestations warrants further research.
In the lungs, alveolar type II cells also produce specific lamellar bodies. There, the lamellar body accumulates phospholipids in multi‐bilayer membranes that form the pulmonary surfactant necessary for the pulmonary innate immune response and to reduce surface tension in the alveolar spaces. 203 The transporter ABCA3, closely related to ABCA12, 204 is localized in the lamellar body membrane 205 , 206 and is required for the incorporation of lipids in the lamellar bodies of alveolar type II cells. ABCA3 has a crucial role in trafficking phosphatidylcholine, phosphatidylglycerol, sphingomyelin, and cholesterol. ABCA3 is also expressed in the brain, specifically in oligodendrocytes, neurons, astrocytes, and microglia, but its role remains unclear. 207
Hypothesized Roles for MLBs and MVBs in the Pathogenesis of PD
We would like to put forward the possibility that the pathological MLBs observed in GBA1‐PD bear some similarity in origin to lamellar bodies. ABCA‐like transporters, commonly regulated by cholesterol levels, could play a role in MLB formation, analogous to their role in the formation of lamellar bodies in keratinocytes and alveolar type II cells. It is reported that ABCA subclass proteins might be involved as regulators of cellular lipids in neurodegeneration. 208 In addition, it is known that Rab11a is a critical protein for lamellar body biogenesis in keratinocytes. Rab11a regulates endosomal recycling of extracellular α‐Syn, 209 modulating defects in synaptic transmission caused by α‐Syn aggregation. 210 Therefore, we suggest that Rab11a participates in the biogenesis of MLBs in GBA1‐PD.
We also propose that the cholesterol needed for MLB biogenesis originates from two different routes: (1) endogenous cholesterol synthesized within the ER, transported to the cell membrane directly or through the GA; and (2) LDL‐containing cholesterol that binds to the LDLR on the cell membrane, where it is engulfed by an endocytic vesicle. These vesicles fuse with primary late endosomes, where LDL is hydrolyzed. Cholesteryl esters are then cleaved by acid lipase, and free cholesterol is released. Cholesterol is further delivered to secondary late endosomes, where NPC2 binds released cholesterol and transfers it onto NPC1, which acts as a hinge between these late endosomes and the ER, flipping the cholesterol across the membrane. 211 , 212 Finally, cholesterol is distributed via anterograde (ER to GA) and retrograde (GA to ER) trafficking throughout the cell, including all organelles and plasma membrane (Fig. 3). Alterations in these cholesterol trafficking pathways could produce cholesterol delocalization and favor the formation of MLBs. More research is needed to unwrap the participation of ABCA and Rab11a in pathological lipids, particularly those related to GBA1 mutations and PD.
It would be interesting to determine whether MLBs change in different PD stages. In the case of lysosomal storage diseases, it has been noted that after the formation of storage lysosomes, multiple abnormal vesicular structures develop during the disease course, likely because of different intracellular pathways (autophagy, endocytosis, etc.) that ultimately converge in dysfunctional lysosomes and the secondary disruption of lipid metabolism. 213 In addition, further research is needed to better understand whether MLB formation in GBA1‐PD is a cell‐autonomous process or whether they appear in specific neuronal populations that are more vulnerable. Likewise, MVBs formed by invaginations of endosomes might play a role in the process underlying the formation of the observed membranous bodies in GBA1‐PD cells. 214 Recently, these MVBs have been identified as the origin of exosomes that allow the release of intracellular cargo. As mentioned previously, it has been speculated that exosomes that contain α‐Syn aggregates may spread the disease 214 , 215 ; however, a firm association between exosomal α‐Syn and PD severity and/or progression has not yet been established.
Phospholipids appear to contribute to the spreading of α‐Syn through exosomes in neuroblastoma cells. 216 Moreover, Cers are thought to be implied in the generation of MVBs, and altered Cer levels are found in the plasma of patients with PD. 217 When GCase activity is reduced, Cer levels decrease in endosomal–lysosomal compartments, contributing to lysosomal failure to degrade α‐Syn. 218 , 219 Indeed, reduction or overexpression of GCase increases or decreases, respectively, exosomal secretion of α‐Syn in mice. 220 Exosomes may contribute to α‐Syn spreading in cellular PD models. 75 Likewise, spread of α‐Syn aggregates via extracellular vesicles is augmented in Gba1b‐mutant Drosophila. 221 Along the same line, exocytosis of the lamellated structures with α‐Syn aggregates seen in GBA1‐PD fibroblasts could propagate between neurons. 148 We therefore propose that the MLBs caused by GBA1 mutations are pathogenic and might facilitate the prion‐like and transneuronal propagation of α‐Syn in GBA1‐PD (Fig. 3). We further hypothesize that cholesterol (and other lipids) could participate in the LB formation. Special attention should be given to the potential role of cholesterol in PD pathophysiology and, in particular, its role in the formation of lamellar body‐like structures observed in GBA1‐PD cells.
Conclusion
This review describes the published evidence supporting the role of abnormal cholesterol homeostasis in PD, its link with GCase, whose gene is identified as a major risk factor for PD, and the interaction of cholesterol with α‐syn, which can cause the loss of dopaminergic neurons. We consider the role of multilamellar‐like bodies found in GBA1‐PD‐derived fibroblasts in the LB pathophysiology. There is an urgent need for more insight in the precise role of cholesterol and sphingolipid metabolism in PD pathogenesis, particularly as related to GBA1 mutations. Such knowledge might assist development of new therapeutic avenues for patients with GBA1‐PD.
Author Roles
P.G.‐S., R.M. and J.M.F.G.A., conceived and arranged this review. P.G.‐S. provided the first draft and prepared figures. R.M., P.G.‐S., and J.M.F.G.A. edited the draft and provided the final version. R.M. and J.M.F.G.A. provided financial support for the project leading to this publication. All authors approved the ultimate revised version of the manuscript and accept responsibility for their contributions.
Financial Disclosures
R.M. declares that her research lines for the previous 12 months were supported by grants from the Spanish Ministries of Innovation, Science and Universities and Health, Social Services and Equality and ISCIII, CIBERNED: PCIN2015‐098, PID2019‐111693RB‐I00, SAF2016‐78207‐R, CB06/05/0055, PI2015‐2/02, and PI2019/09‐3; Ramón Areces Foundation (172275); and European Union's Horizon 2020 research and innovation program, AND‐PD, grant agreement no. 848002. P.G.‐S. was supported by a Postdoctoral Research Contract from CIBERNED, ISCIII (CB06/05/0055). J.M.F.G.A. declares that his research lines for the previous 12 months were supported by a grant from NWO‐BBOL “Un(der)recognized glucosylceramide metabolites: their occurrence, their biological functions and their role in human pathologies” (737.016.002) and BBOL‐2007247202.
Acknowledgments
This work was supported by grants from the Spanish Ministries of Innovation, Science and Universities and Health, Social Services and Equality and ISCIII, CIBERNED: PCIN2015‐098, PID2019‐111693RB‐I00, CB06/05/0055 and PI2019/09‐3; Ramón Areces Foundation (172275); European Union's Horizon 2020 research and innovation program, AND‐PD, grant agreement no. 848002 to R.M.; and NWO (grant no. BBOL‐2007247202 to J.M.F.G.A.). Figures 1–3 were generated with BioRender.com.
Relevant conflicts of interest/financial disclosures: Nothing to report.
Funding agencies: This work was supported by grants from the Spanish Ministries of Innovation, Science and Universities and Health, Social Services and Equality and ISCIII, CIBERNED: PCIN2015‐098, PID2019‐111693RB‐I00, CB06/05/0055, and PI2019/09‐3; Ramón Areces Foundation (172275); European Union's Horizon 2020 research and innovation program, AND‐PD, grant agreement no. 848002 to R.M.; and NWO (grant no. BBOL‐2007247202 to J.M.F.G.A.).
Contributor Information
Patricia García‐Sanz, Email: pgarcia@cajal.csic.es.
Rosario Moratalla, Email: moratalla@cajal.csic.es.
References
- 1. Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol 2004;24:806–815. [DOI] [PubMed] [Google Scholar]
- 2. Dietschy JM, Turley SD. Thematic review series: brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res 2004;45:1375–1397. [DOI] [PubMed] [Google Scholar]
- 3. Lange Y, Steck TL. Cholesterol homeostasis and the escape tendency (activity) of plasma membrane cholesterol. Prog Lipid Res 2008;47:319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Friedrichson T, Kurzchalia TV. Microdomains of GPI‐anchored proteins in living cells revealed by crosslinking. Nature 1998;394:802–805. [DOI] [PubMed] [Google Scholar]
- 5. Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol 2017;18:361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fujita M, Kinoshita T. GPI‐anchor remodeling: potential functions of GPI‐anchors in intracellular trafficking and membrane dynamics. Biochim Biophys Acta ‐ Mol Cell Biol Lipids 2012;1821:1050–1058. [DOI] [PubMed] [Google Scholar]
- 7. Lingwood D, Simons K. Lipid rafts as a membrane‐organizing principle. Science 2010;327(5961):46–50. 10.1126/science.1174621 [DOI] [PubMed] [Google Scholar]
- 8. Sarkar P, Chakraborty H, Chattopadhyay A. Differential membrane dipolar orientation induced by acute and chronic cholesterol depletion. Sci Rep 2017;7(1):4484. 10.1038/s41598-017-04769-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015;6:254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Borroni MV, Vallés AS, Barrantes FJ. The lipid habitats of neurotransmitter receptors in brain. Biochim Biophys Acta ‐ Biomembr 2016;1858:2662–2670. [DOI] [PubMed] [Google Scholar]
- 11. Fester L, Zhou L, Bütow A, Huber C, et al. Cholesterol‐promoted synaptogenesis requires the conversion of cholesterol to estradiol in the hippocampus. Hippocampus 2009;19:692–705. [DOI] [PubMed] [Google Scholar]
- 12. Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol‐induced synaptogenesis in a CNS neuron. Mol Cell Neurosci 2005;29:190–201. [DOI] [PubMed] [Google Scholar]
- 13. Mauch DH, Nägier K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia‐derived cholesterol. Science 2001;294(5545):1354–1357. 10.1126/science.294.5545.1354 [DOI] [PubMed] [Google Scholar]
- 14. Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol 2001;12:105–112. [DOI] [PubMed] [Google Scholar]
- 15. Orth M, Bellosta S. Cholesterol: its regulation and role in central nervous system disorders. Cholesterol 2012;2012:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schreurs BG. The effects of cholesterol on learning and memory. Neuroscience and Biobehavioral Reviews 2010;34(8):1366–1379. 10.1016/j.neubiorev.2010.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weber LW. Maintaining cholesterol homeostasis: sterol regulatory element‐binding proteins. World J Gastroenterol 2004;10:3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shimano H, Sato R. SREBP‐regulated lipid metabolism: convergent physiology — divergent pathophysiology. Nat Rev Endocrinol 2017;13:710–730. [DOI] [PubMed] [Google Scholar]
- 19. Iaea DB, Maxfield FR. Cholesterol trafficking and distribution. Essays Biochem 2015;57:43–55. [DOI] [PubMed] [Google Scholar]
- 20. Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol 2010;11:688–699. [DOI] [PubMed] [Google Scholar]
- 21. Pfrieger FW, Ungerer N. Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res 2011;50(4):357–371. 10.1016/j.plipres.2011.06.002 [DOI] [PubMed] [Google Scholar]
- 22. Elliott DA, Weickert CS, Garner B. Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin Lipidol 2010;5:555–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Emamzadeh FN, Allsop D. α‐Synuclein interacts with lipoproteins in plasma. J Mol Neurosci 2017;63:165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arenas F, Garcia‐Ruiz C, Fernandez‐Checa JC. Intracellular cholesterol trafficking and impact in Neurodegeneration. Front Mol Neurosci 2017;10(382):eCollection. 10.3389/fnmol.2017.00382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Infante RE, Abi‐Mosleh L, Radhakrishnan A, et al. Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278‐amino acid membrane protein. J Biol Chem 2008;283:1064–1075. [DOI] [PubMed] [Google Scholar]
- 26. Russell DW, Halford RW, Ramirez DMO, Shah R, Kotti T. Cholesterol 24‐hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem 2009;78:1017–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abildayeva K, Jansen PJ, Hirsch‐Reinshagen V, et al. 24(S)‐Hydroxycholesterol participates in a liver X receptor‐controlled pathway in astrocytes that regulates apolipoprotein E‐mediated cholesterol efflux. J Biol Chem 2006;281:12799–12808. [DOI] [PubMed] [Google Scholar]
- 28. Petrov AM, Kasimov MR, Zefirov AL. Cholesterol in the pathogenesis of Alzheimer's, Parkinson's diseases and autism: link to synaptic dysfunction. Acta Naturae 2017;9:26–37. [PMC free article] [PubMed] [Google Scholar]
- 29. Blesa J, Przedborski S. Parkinson disease: animal models and dopaminergic cell vulnerability. Front Neuroanat 2014;8(155):eCollection. 10.3389/fnana.2014.00155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gómez‐Benito M, Granado N, García‐Sanz P, Michel A, Dumoulin M, Moratalla R. Modeling Parkinson's disease with the alpha‐Synuclein protein. Front Pharmacol 2020;11(356):eCollection. 10.3389/fphar.2020.00356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wakabayashi K, Tanji K, Odagiri S, Miki Y, Miki Y, Mori F, Takahashi H. The Lewy body in Parkinson's disease and related neurodegenerative disorders. Mol Neurobiol 2013;47:495–508. [DOI] [PubMed] [Google Scholar]
- 32. Michel PP, Hirsch EC, Hunot S. Understanding dopaminergic cell death pathways in Parkinson disease. Neuron 2016;90:675–691. [DOI] [PubMed] [Google Scholar]
- 33. Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 2009;18:R48–R59. [DOI] [PubMed] [Google Scholar]
- 34. Tysnes O‐B, Storstein A. Epidemiology of Parkinson's disease. J Neural Transm 2017;124:901–905. [DOI] [PubMed] [Google Scholar]
- 35. Brady RO, Kanfer JN, Bradley RM, Shapiro D. Demonstration of a deficiency of glucocerebroside‐cleaving enzyme in Gaucher's disease. J Clin Invest 1966;45:1112–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Merrill AH. Sphingolipid and Glycosphingolipid metabolic pathways in the era of Sphingolipidomics. Chem Rev 2011;111:6387–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quinn PJ. A lipid matrix model of membrane raft structure. Prog Lipid Res 2010;49:390–406. [DOI] [PubMed] [Google Scholar]
- 38. Barkhuizen M, Anderson DG, Grobler AF. Advances in GBA‐associated Parkinson's disease – pathology, presentation and therapies. Neurochem Int 2016;93:6–25. [DOI] [PubMed] [Google Scholar]
- 39. McGlinchey RP, Lee JC. Emerging insights into the mechanistic link between α‐synuclein and glucocerebrosidase in Parkinson's disease. Biochem Soc Trans 2013;41:1509–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schapira AHV. Glucocerebrosidase and Parkinson disease: recent advances. Mol Cell Neurosci 2015;66:37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sidransky E, Nalls MA, Aasly JO, Aharon‐Peretz J, et al. Multicenter analysis of Glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361:1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol 2012;11:986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anheim M, Elbaz A, Lesage S, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012;78:417–420. [DOI] [PubMed] [Google Scholar]
- 44. Mullin S, Beavan M, Bestwick J, et al. Evolution and clustering of prodromal parkinsonian features in GBA1 carriers. Mov Disord 2019;34:1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Balestrino R, Tunesi S, Tesei S, Lopiano L, Zecchinelli AL, Goldwurm S. Penetrance of Glucocerebrosidase (GBA) mutations in Parkinson's disease: a kin cohort study. Mov Disord 2020;mds.28200. 10.1002/mds.28200 [DOI] [PubMed] [Google Scholar]
- 46. Gan‐Or Z, Giladi N, Rozovski U, et al. Genotype‐phenotype correlations between gba mutations and parkinson disease risk and onset. Neurology 2008;70(24):2277–2283. 10.1212/01.wnl.0000304039.11891.29 [DOI] [PubMed] [Google Scholar]
- 47. Thaler A, Bregman N, Gurevich T, et al. Parkinson's disease phenotype is influenced by the severity of the mutations in the GBA gene. Park Relat Disord 2018;55:45–49. [DOI] [PubMed] [Google Scholar]
- 48. Neumann J, Bras J, Deas E, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain 2009;132:1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gan‐Or Z, Liong C, Alcalay RN. GBA‐associated Parkinson's disease and other Synucleinopathies. Curr Neurol Neurosci Rep 2018;18:44. [DOI] [PubMed] [Google Scholar]
- 50. Riboldi GM, Di Fonzo AB. GBA, Gaucher disease, and Parkinson's disease: from genetic to clinic to new therapeutic approaches. Cells 2019;8:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goker‐Alpan O. Divergent phenotypes in Gaucher disease implicate the role of modifiers. J Med Genet 2005;42:e37–e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Woodard CM, Campos BA, Kuo S‐H, et al. iPSC‐derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson's disease. Cell Rep 2014;9:1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schierding W, Farrow S, Fadason T, et al. Common variants Coregulate expression of GBA and modifier genes to delay Parkinson's disease onset. Mov Disord 2020;35:1346–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Do J, McKinney C, Sharma P, Sidransky E. Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener 2019;14:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Blauwendraat C, Reed X, Krohn L, et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson's disease and Lewy body dementia. Brain 2020;143:234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sanyal A, Novis HS, Gasser E, Lin S, LaVoie MJ. LRRK2 kinase inhibition rescues deficits in lysosome function due to heterozygous GBA1 expression in human iPSC‐derived neurons. Front Neurosci 2020;14(442):eCollection. 10.3389/fnins.2020.00442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sanyal A, DeAndrade MP, Novis HS, et al. Lysosome and inflammatory defects in GBA1 ‐mutant astrocytes are normalized by LRRK2 inhibition. Mov Disord 2020;35:760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. García‐Sanz P, Orgaz L, Fuentes JM, Vicario C, Moratalla R. Cholesterol and multilamellar bodies: Lysosomal dysfunction in GBA‐Parkinson disease. Autophagy 2018;14:717–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. García‐Sanz P, Orgaz L, Bueno‐Gil G, et al. N370S‐GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson's disease. Mov Disord 2017;32:1409–1422. [DOI] [PubMed] [Google Scholar]
- 60. Fernandes HJR, Hartfield EM, Christian HC, et al. ER stress and Autophagic perturbations Lead to elevated extracellular α‐Synuclein in GBA‐N370S Parkinson's iPSC‐derived dopamine neurons. Stem Cell Reports 2016;6:342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zunke F, Moise AC, Belur NR, et al. Reversible conformational conversion of α‐Synuclein into toxic assemblies by Glucosylceramide. Neuron 2018;97:92–107.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mazzulli JR, Xu Y‐H, Sun Y, et al. Gaucher disease glucocerebrosidase and α‐synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011;146:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. α‐Synuclein–induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci 2016;113:1931–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yun SP, Kim D, Kim S, et al. α‐Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP‐induced parkinsonism. Mol Neurodegener 2018;13:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rocha EM, Smith GA, Park E, et al. Progressive decline of glucocerebrosidase in aging and Parkinson's disease. Ann Clin Transl Neurol 2015;2:433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Balestrino R, Schapira AHV. Glucocerebrosidase and Parkinson disease: molecular, clinical, and therapeutic implications. Neurosci 2018;24:540–559. [DOI] [PubMed] [Google Scholar]
- 67. Blandini F, Cilia R, Cerri S, et al. Glucocerebrosidase mutations and synucleinopathies: toward a model of precision medicine. Mov Disord 2019;34:9–21. [DOI] [PubMed] [Google Scholar]
- 68. Yap TL, Gruschus JM, Velayati A, et al. α‐Synuclein interacts with Glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J Biol Chem 2011;286:28080–28088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huebecker M, Moloney EB, van der Spoel AC, et al. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson's disease. Mol Neurodegener 2019;14:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brekk OR, Moskites A, Isacson O, Hallett PJ. Lipid‐dependent deposition of alpha‐synuclein and tau on neuronal Secretogranin II‐positive vesicular membranes with age. Sci Rep 2018;8:15207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hallett PJ, Huebecker M, Brekk OR, et al. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol Aging 2018;67:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ikuno M, Yamakado H, Akiyama H, et al. GBA haploinsufficiency accelerates alpha‐synuclein pathology with altered lipid metabolism in a prodromal model of Parkinson's disease. Hum Mol Genet 2019;28:1894–1904. [DOI] [PubMed] [Google Scholar]
- 73. Henderson MX, Sedor S, McGeary I, et al. Glucocerebrosidase activity modulates neuronal susceptibility to pathological α‐synuclein insult. Neuron 2020;105:822–836.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim S, Yun SP, Lee S, et al. GBA1 deficiency negatively affects physiological α‐synuclein tetramers and related multimers. Proc Natl Acad Sci 2018;115:798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gegg ME, Verona G, Schapira AHV. Glucocerebrosidase deficiency promotes release of α‐synuclein fibrils from cultured neurons. Hum Mol Genet 2020;29:1716–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Burré J. The synaptic function of α‐synuclein. J Parkinsons Dis 2015;5:699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Xu W, Tan L, Yu J‐T. The link between the SNCA gene and parkinsonism. Neurobiol Aging 2015;36:1505–1518. [DOI] [PubMed] [Google Scholar]
- 78. Varkey J, Isas JM, Mizuno N, et al. Membrane curvature induction and Tubulation are common features of Synucleins and Apolipoproteins. J Biol Chem 2010;285:32486–32493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lykkebo S, Jensen PH. Alpha‐synuclein and presynaptic function: implications for Parkinson's disease. Neuromolecular Med 2002;2:115–129. [DOI] [PubMed] [Google Scholar]
- 80. Emanuele M, Chieregatti E. Mechanisms of alpha‐Synuclein action on neurotransmission: cell‐autonomous and non‐cell autonomous role. Biomolecules 2015;5:865–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci 2014;34:9364–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Huang M, Wang B, Li X, Fu C, Wang C, Kang X. Α‐Synuclein: a multifunctional player in exocytosis, endocytosis, and vesicle recycling. Front Neurosci 2019;13(28):eCollection. 10.3389/fnins.2019.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gibson Wood W, Igbavboa U, Müller WE, Eckert GP. Cholesterol asymmetry in synaptic plasma membranes. J Neurochem 2011;116:684–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lou H, Montoya SE, Alerte TNM, et al. Serine 129 phosphorylation reduces the ability of α‐Synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J Biol Chem 2010;285:17648–17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Peng XM. Synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J Cell Sci 2005;118:3523–3530. [DOI] [PubMed] [Google Scholar]
- 86. Gao N, Li Y‐H, Li X, Yu S, Fu G‐L, Chen B. Effect of α‐synuclein on the promoter activity of tyrosine hydroxylase gene. Neurosci Bull 2007;23:53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Van Maarschalkerweerd A, Vetri V, Vestergaard B. Cholesterol facilitates interactions between α‐synuclein oligomers and charge‐neutral membranes. FEBS Lett 2015;589(19):2661–2667. 10.1016/j.febslet.2015.08.013 [DOI] [PubMed] [Google Scholar]
- 88. Fantini J, Carlus D, Yahi N. The fusogenic tilted peptide (67–78) of α‐synuclein is a cholesterol binding domain. Biochim Biophys Acta ‐ Biomembr 2011;1808:2343–2351. [DOI] [PubMed] [Google Scholar]
- 89. Di Scala C, Yahi N, Boutemeur S, et al. Common molecular mechanism of amyloid pore formation by Alzheimer's β‐amyloid peptide and α‐synuclein. Sci Rep 2016;6:28781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mesa‐Herrera F, Taoro‐González L, Valdés‐Baizabal C, Diaz M, Marín R. Lipid and lipid raft alteration in aging and neurodegenerative diseases: a window for the development of new biomarkers. International Journal of Molecular Sciences 2019;20(15):3810. 10.3390/ijms20153810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Galvagnion C. The role of lipids interacting with α‐Synuclein in the pathogenesis of Parkinson's disease. J Parkinsons Dis 2017;7:433–450. [DOI] [PubMed] [Google Scholar]
- 92. Jakubec M, Bariås E, Furse S, Govasli ML, et al. Cholesterol is a strong promotor of an α‐Synuclein membrane binding mode that accelerates oligomerization. BioRxiv 2019;0(0):725–762. 10.1101/725762 [DOI] [Google Scholar]
- 93. Man WK, De Simone A, Barritt JD, Vendruscolo M, Dobson CM, Fusco G. A role of cholesterol in modulating the binding of α‐Synuclein to synaptic‐like vesicles. Front Neurosci 2020;14:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hsiao J‐H, Halliday G, Kim W. α‐Synuclein regulates neuronal cholesterol efflux. Molecules 2017;22:1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kim S, Kwon S‐H, Kam T‐I, et al. Transneuronal propagation of pathologic α‐Synuclein from the gut to the brain models Parkinson's disease. Neuron 2019;103:627–641.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Fujiwara H, Hasegawa M, Dohmae N, et al. α‐Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 2002;4:160–164. [DOI] [PubMed] [Google Scholar]
- 97. Ugalde CL, Lawson VA, Finkelstein DI, Hill AF. The role of lipids in α‐synuclein misfolding and neurotoxicity. J Biol Chem 2019;294:9016–9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Burré J, Sharma M, Südhof TC. Cell biology and pathophysiology of α‐synuclein. Cold Spring Harb Perspect Med 2018;8:a024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Alza NP, Iglesias González PA, Conde MA, Uranga RM, Salvador GA. Lipids at the crossroad of α‐Synuclein function and dysfunction: biological and pathological implications. Front Cell Neurosci 2019;13(175):eCollection. 10.3389/fncel.2019.00175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Davis AA, Inman CE, Wargel ZM, et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med 2020;12:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Nalls MA, Duran R, Lopez G, et al. A multicenter study of Glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 2013;70:727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cha S‐H, Choi YR, Heo C‐H, et al. Loss of parkin promotes lipid rafts‐dependent endocytosis through accumulating caveolin‐1: implications for Parkinson's disease. Mol Neurodegener 2015;10:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lim L, Jackson‐Lewis V, Wong LC, et al. Lanosterol induces mitochondrial uncoupling and protects dopaminergic neurons from cell death in a model for Parkinson's disease. Cell Death Differ 2012;19:416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Musanti R, Parati E, Lamperti E, Ghiselli G. Decreased cholesterol biosynthesis in fibroblasts from patients with Parkinson disease. Biochem Med Metab Biol 1993;49:133–142. [DOI] [PubMed] [Google Scholar]
- 105. Paul R, Dutta A, Phukan BC, et al. Accumulation of cholesterol and homocysteine in the nigrostriatal pathway of brain contributes to the dopaminergic neurodegeneration in mice. Neuroscience 2018;388:347–356. [DOI] [PubMed] [Google Scholar]
- 106. Paul R, Choudhury A, Kumar S, Giri A, Sandhir R, Borah A. Cholesterol contributes to dopamine‐neuronal loss in MPTP mouse model of Parkinson's disease: involvement of mitochondrial dysfunctions and oxidative stress. PLoS One 2017;12:e0171285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bousquet M, St‐Amour I, Vandal M, Julien P, Cicchetti F, Calon F. High‐fat diet exacerbates MPTP‐induced dopaminergic degeneration in mice. Neurobiol Dis 2012;45:529–538. [DOI] [PubMed] [Google Scholar]
- 108. Choi J‐Y, Jang E‐H, Park C‐S, Kang J‐H. Enhanced susceptibility to 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine neurotoxicity in high‐fat diet‐induced obesity. Free Radic Biol Med 2005;38:806–816. [DOI] [PubMed] [Google Scholar]
- 109. Hu G. Total cholesterol and the risk of Parkinson's disease: a review for some new findings. Parkinsons Dis 2010;2010:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Benn M, Nordestgaard BG, Frikke‐Schmidt R, Tybjærg‐Hansen A. Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer's disease and Parkinson's disease: Mendelian randomisation study. BMJ 2017;357:j1648. 10.1136/bmj.j1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hu G, Antikainen R, Jousilahti P, Kivipelto M, Tuomilehto J. Total cholesterol and the risk of Parkinson disease. Neurology 2008;70:1972–1979. [DOI] [PubMed] [Google Scholar]
- 112. Fang F, Zhan Y, Hammar N, et al. Lipids, apolipoproteins, and the risk of Parkinson disease. Circ Res 2019;125:643–652. [DOI] [PubMed] [Google Scholar]
- 113. Rozani V, Gurevich T, Giladi N, et al. Higher serum cholesterol and decreased Parkinson's disease risk: a statin‐free cohort study. Mov Disord 2018;33:1298–1305. [DOI] [PubMed] [Google Scholar]
- 114. de Lau LML, Koudstaal PJ, Hofman A, Breteler MMB. Serum cholesterol levels and the risk of Parkinson's disease. Am J Epidemiol 2006;164:998–1002. [DOI] [PubMed] [Google Scholar]
- 115. Huang X, Auinger P, Eberly S. et al; Parkinson Study Group DATATOP InvestigatorsSerum cholesterol and the progression of Parkinson's disease: results from DATATOP. PLoS One 2011;6:e22854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Huang X, Alonso A, Guo X, et al. Statins, plasma cholesterol, and risk of Parkinson's disease: a prospective study. Mov Disord 2015;30:552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wei Z, Li X, Li X, Liu Q, Cheng Y. Oxidative stress in Parkinson's disease: a systematic review and meta‐analysis. Front Mol Neurosci 2018;11(236):eCollection. 10.3389/fnmol.2018.0023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Guo X, Song W, Chen K, et al. The serum lipid profile of Parkinson's disease patients: a study from China. Int J Neurosci 2015;125:838–844. [DOI] [PubMed] [Google Scholar]
- 119. Zhang L, Wang X, Wang M, et al. Circulating cholesterol levels may link to the factors influencing Parkinson's risk. Front Neurol 2017;8(501):eCollection. 10.3389/fneur.2017.0050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Huang X, Chen H, Miller WC, et al. Lower low‐density lipoprotein cholesterol levels are associated with Parkinson's disease. Mov Disord 2007;22:377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Savica R, Grossardt BR, Ahlskog JE, Rocca WA. Metabolic markers or conditions preceding Parkinson's disease: a case‐control study. Mov Disord 2012;27(8):974–979. 10.1002/mds.25016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Sääksjärvi K, Knekt P, Männistö S, Lyytinen J, Heliövaara M. Prospective study on the components of metabolic syndrome and the incidence of Parkinson's disease. Parkinsonism Relat Disord 2015;21:1148–1155. [DOI] [PubMed] [Google Scholar]
- 123. Gudala K, Bansal D, Muthyala H. Role of serum cholesterol in Parkinson's disease: a meta‐analysis of evidence. J Parkinsons Dis 2013;3:363–370. [DOI] [PubMed] [Google Scholar]
- 124. Schommer J, Marwarha G, Schommer T, Flick T, Lund J, Ghribi O. 27‐Hydroxycholesterol increases α‐synuclein protein levels through proteasomal inhibition in human dopaminergic neurons. BMC Neurosci 2018;19:17. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 125. Marwarha G, Rhen T, Schommer T, Ghribi O. The oxysterol 27‐hydroxycholesterol regulates α‐synuclein and tyrosine hydroxylase expression levels in human neuroblastoma cells through modulation of liver X receptors and estrogen receptors‐relevance to Parkinson's disease. J Neurochem 2011;119:1119–1136. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 126. Marwarha G, Ghribi O. Does the oxysterol 27‐hydroxycholesterol underlie Alzheimer's disease–Parkinson's disease overlap? Exp Gerontol 2015;68:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hughes TM, Rosano C, Evans RW, Kuller LH. Brain cholesterol metabolism, Oxysterols, and dementia. J Alzheimer's Dis 2013;33:891–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Van Kampen JM, Robertson HA. The BSSG rat model of Parkinson's disease: progressing towards a valid, predictive model of disease. EPMA J 2017;8:261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Van Kampen JM, Baranowski DC, Robertson HA, Shaw CA, Kay DG. The progressive BSSG rat model of Parkinson's: recapitulating multiple key features of the human disease. PLoS One 2015;10:e0139694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gómez‐Suaga P, Bravo‐San Pedro JM, González‐Polo RA, Fuentes JM, et al. ER–mitochondria signaling in Parkinson's disease. Cell Death Dis 2018;9:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Panov A, Kubalik N, Brooks BR, Shaw CA. In vitro effects of cholesterol β‐d‐Glucoside, cholesterol and cycad Phytosterol Glucosides on respiration and reactive oxygen species generation in brain mitochondria. J Membr Biol 2010;237:71–77. [DOI] [PubMed] [Google Scholar]
- 132. Zambon F, Cherubini M, Fernandes HJR, et al. Cellular α‐synuclein pathology is associated with bioenergetic dysfunction in Parkinson's iPSC‐derived dopamine neurons. Hum Mol Genet 2019;28:2001–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Appelqvist H, Sandin L, Björnström K, et al. Sensitivity to lysosome‐dependent cell death is directly regulated by Lysosomal cholesterol content. PLoS One 2012;7:e50262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Appelqvist H, Nilsson C, Garner B, Brown AJ, Kågedal K, Öllinger K. Attenuation of the Lysosomal death pathway by Lysosomal cholesterol accumulation. Am J Pathol 2011;178:629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Aqul A, Liu B, Ramirez CM, et al. Unesterified cholesterol accumulation in late endosomes/lysosomes causes Neurodegeneration and is prevented by driving cholesterol export from this compartment. J Neurosci 2011;31:9404–9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Eriksson I, Nath S, Bornefall P, Giraldo AMV, Öllinger K. Impact of high cholesterol in a Parkinson's disease model: prevention of lysosomal leakage versus stimulation of α‐synuclein aggregation. Eur J Cell Biol 2017;96:99–109. [DOI] [PubMed] [Google Scholar]
- 137. Magalhaes J, Gegg ME, Migdalska‐Richards A, Doherty MK, et al. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum Mol Genet 2016;25:3432–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. García‐Sanz P, Moratalla R. The importance of cholesterol in Parkinson's disease. Mov Disord 2018;33:343–344. [DOI] [PubMed] [Google Scholar]
- 139. Martin S, Smolders S, Van den Haute C, et al. Mutated ATP10B increases Parkinson's disease risk by compromising lysosomal glucosylceramide export. Acta Neuropathol 2020;139:1001–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Neculai D, Schwake M, Ravichandran M, et al. Structure of LIMP‐2 provides functional insights with implications for SR‐BI and CD36. Nature 2013;504:172–176. [DOI] [PubMed] [Google Scholar]
- 141. Heybrock S, Kanerva K, Meng Y, et al. Lysosomal integral membrane protein‐2 (LIMP‐2/SCARB2) is involved in lysosomal cholesterol export. Nat Commun 2019;10:3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Castellano BM, Thelen AM, Moldavski O, et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9–Niemann‐pick C1 signaling complex. Science 2017;355:1306–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Eid W, Dauner K, Courtney KC, et al. mTORC1 activates SREBP‐2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc Natl Acad Sci 2017;114:7999–8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zaltieri M, Grigoletto J, Longhena F, et al. Alfa‐synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J Cell Sci 2015;128:2231–2243. [DOI] [PubMed] [Google Scholar]
- 145. Martínez‐Menárguez JÁ, Tomás M, Martínez‐Martínez N, Martínez‐Alonso E. Golgi fragmentation in neurodegenerative diseases: is there a common cause? Cells 2019;8:748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Salvioli R, Scarpa S, Ciaffoni F, et al. Glucosylceramidase mass and subcellular localization are modulated by cholesterol in Niemann‐pick disease type C. J Biol Chem 2004;279:17674–17680. [DOI] [PubMed] [Google Scholar]
- 147. Liou B, Zhang W, Fannin V, et al. Combination of acid β‐glucosidase mutation and Saposin C deficiency in mice reveals Gba1 mutation dependent and tissue‐specific disease phenotype. Sci Rep 2019;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Volpicelli‐Daley L, Brundin P. Prion‐like propagation of pathology in Parkinson disease. Handbook of Clinical Neurology. 153. Amsterdam, Netherlands: Elsevier; 2018:321–335. 10.1016/B978-0-444-63945-5.00017-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Li H, Ham A, Ma TC, et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 2019;15:113–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Roca‐Agujetas V, de Dios C, Lestón L, Morales A, Colell A. Recent insights into the mitochondrial role in autophagy and its regulation by oxidative stress. Oxid Med Cell Longev 2019;2019:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Desai R, Frazier AE, Durigon R, et al. ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 2017;140:1595–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. van Weely S, Brandsma M, Strijland A, Tager JM, Aerts JM. Demonstration of the existence of a second, non‐lysosomal glucocerebrosidase that is not deficient in Gaucher disease. Biochim Biophys Acta ‐ Mol Basis Dis 1993;1181:55–62. [DOI] [PubMed] [Google Scholar]
- 153. Wheeler S, Haberkant P, Bhardwaj M, et al. Cytosolic glucosylceramide regulates endolysosomal function in Niemann‐pick type C disease. Neurobiol Dis 2019;127:242–252. [DOI] [PubMed] [Google Scholar]
- 154. Körschen HG, Yildiz Y, Raju DN, et al. The non‐lysosomal β‐Glucosidase GBA2 is a non‐integral membrane‐associated protein at the endoplasmic reticulum (ER) and Golgi. J Biol Chem 2013;288:3381–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Aerts JMFG, Kuo C‐L, Lelieveld LT, et al. Glycosphingolipids and lysosomal storage disorders as illustrated by gaucher disease. Curr Opin Chem Biol 2019;53:204–215. [DOI] [PubMed] [Google Scholar]
- 156. Marques ARA, Mirzaian M, Akiyama H, et al. Glucosylated cholesterol in mammalian cells and tissues: formation and degradation by multiple cellular β‐glucosidases. J Lipid Res 2016;57:451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Boer DEC, Mirzaian M, Ferraz MJ, et al. Glucosylated cholesterol in skin: synthetic role of extracellular glucocerebrosidase. Clin Chim Acta 2020;510:707–710. [DOI] [PubMed] [Google Scholar]
- 158. Marques ARA, Aten J, Ottenhoff R, et al. Reducing GBA2 activity ameliorates neuropathology in Niemann‐pick type C mice. PLoS One 2015;10:e0135889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Akiyama H, Ide M, Nagatsuka Y, et al. Glucocerebrosidases catalyze a transgalactosylation reaction that yields a newly‐identified brain sterol metabolite, galactosylated cholesterol. J Biol Chem 2020;295:5257–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Burke DG, Rahim AA, Waddington SN, et al. Increased glucocerebrosidase (GBA) 2 activity in GBA1 deficient mice brains and in Gaucher leucocytes. J Inherit Metab Dis 2013;36:869–872. [DOI] [PubMed] [Google Scholar]
- 161. Lelieveld LT, Mirzaian M, Kuo C‐L, et al. Role of β‐glucosidase 2 in aberrant glycosphingolipid metabolism: model of glucocerebrosidase deficiency in zebrafish. J Lipid Res 2019;60:1851–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Schöndorf DC, Aureli M, McAllister FE, et al. iPSC‐derived neurons from GBA1‐associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun 2014;5:4028. [DOI] [PubMed] [Google Scholar]
- 163. Akiyama H, Hirabayashi Y. A novel function for glucocerebrosidase as a regulator of sterylglucoside metabolism. Biochim Biophys Acta ‐ Gen Subj 2017;1861:2507–2514. [DOI] [PubMed] [Google Scholar]
- 164. Franco R, Sánchez‐Arias JA, Navarro G, Lanciego JL. Glucocerebrosidase mutations and Synucleinopathies. Potential role of Sterylglucosides and relevance of studying both GBA1 and GBA2 genes. Front Neuroanat 2018;12(52):eCollection. 10.3389/fnana.2018.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Boer DEC, van Smeden J, Bouwstra JA, Aerts JMF. Glucocerebrosidase: functions in and beyond the lysosome. J Clin Med 2020;9:736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia 2013;61:71–90. [DOI] [PubMed] [Google Scholar]
- 167. Sarkar S, Dammer EB, Malovic E, et al. Molecular signatures of Neuroinflammation induced by αSynuclein aggregates in microglial cells. Front Immunol 2020;11(33):eCollection. 10.3389/fimmu.2020.00033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kam T‐I, Hinkle JT, Dawson TM, Dawson VL. Microglia and astrocyte dysfunction in parkinson's disease. Neurobiol Dis 2020;144:105028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017;541:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Pajares M, Rojo AI, Manda G, Boscá L, Cuadrado A. Inflammation in Parkinson's disease: mechanisms and therapeutic implications. Cells 2020;9:1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Gustafsson G, Lindström V, Rostami J, et al. Alpha‐synuclein oligomer‐selective antibodies reduce intracellular accumulation and mitochondrial impairment in alpha‐synuclein exposed astrocytes. J Neuroinflammation 2017;14(1):241. 10.1186/s12974-017-1018-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Rocha EM, Smith GA, Park E, et al. Sustained systemic Glucocerebrosidase inhibition induces brain α‐Synuclein aggregation, microglia and complement C1q activation in mice. Antioxidants Redox Signal 2015;23(6):550–564. 10.1089/ars.2015.6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Soria FN, Engeln M, Martinez‐Vicente M, et al. Glucocerebrosidase deficiency in dopaminergic neurons induces microglial activation without neurodegeneration. Hum Mol Genet 2017;26:2603–2615. [DOI] [PubMed] [Google Scholar]
- 174. Pandey MK, Burrow TA, Rani R, et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017;543:108–112. [DOI] [PubMed] [Google Scholar]
- 175. Aerts JMFG, Hollak CEM. 4 plasma and metabolic abnormalities in Gaucher's disease. Baillieres Clin Haematol 1997;10:691–709. [DOI] [PubMed] [Google Scholar]
- 176. Chahine LM, Qiang J, Ashbridge E, et al. Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol 2013;70:852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Aflaki E, Moaven N, Borger DK, et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 2016;15(1):77–88. 10.1111/acel.12409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Lee E, Hwang I, Park S, et al. MPTP‐driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ 2019;26:213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. González‐Guevara E, Cárdenas G, Pérez‐Severiano F, Martínez‐Lazcano JC. Dysregulated brain cholesterol metabolism is linked to neuroinflammation in Huntington's disease. Mov Disord 2020;35:1113–1127. [DOI] [PubMed] [Google Scholar]
- 180. Shin SD, Shin A, Mayagoitia K, Wilson CG, Bellinger DL, Soriano S. Interferon downstream signaling is activated early in pre‐symptomatic Niemann‐pick disease type C. Neurosci Lett 2019;706:43–50. [DOI] [PubMed] [Google Scholar]
- 181. Mayagoitia K, Shin SD, Rubini M, et al. Short‐term exposure to dietary cholesterol is associated with downregulation of interleukin‐15, reduced thigmotaxis and memory impairment in mice. Behav Brain Res 2020;393:112779. [DOI] [PubMed] [Google Scholar]
- 182. Mutemberezi V, Buisseret B, Masquelier J, Guillemot‐Legris O, Alhouayek M, Muccioli GG. Oxysterol levels and metabolism in the course of neuroinflammation: insights from in vitro and in vivo models. J Neuroinflammation 2018;15:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Mouzat K, Chudinova A, Polge A, et al. Regulation of brain cholesterol: what role Do liver X receptors play in neurodegenerative diseases? Int J Mol Sci 2019;20:3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Miller YI, Navia‐Pelaez JM, Corr M, Yaksh TL. Lipid rafts in glial cells: role in neuroinflammation and pain processing. J Lipid Res 2020;61:655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Low H, Mukhamedova N, Capettini L dos SA, et al. Cholesterol efflux‐independent modification of lipid rafts by AIBP (Apolipoprotein A‐I binding protein). Arterioscler Thromb Vasc Biol 2020;40:2346–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Lynch‐Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson's disease. Cold Spring Harb Perspect Med 2012;2:a009357–a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Shibuya Y, Chang CCY, Huang L‐H, Bryleva EY, Chang T‐Y. Inhibiting ACAT1/SOAT1 in microglia stimulates autophagy‐mediated Lysosomal proteolysis and increases a 1‐42 clearance. J Neurosci 2014;34:14484–14501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Urano Y, Ho Vo D‐K, Hirofumi A, Noguchi N. 24(S)‐Hydroxycholesterol induces ER dysfunction‐mediated unconventional cell death. Cell Death Discov 2019;5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Shahmoradian SH, Lewis AJ, Genoud C, et al. Lewy pathology in Parkinson's disease consists of crowded organelles and lipid membranes. Nat Neurosci 2019;22:1099–1109. [DOI] [PubMed] [Google Scholar]
- 190. Velayati A, Yu WH, Sidransky E. The role of Glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr Neurol Neurosci Rep 2010;10:190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Guerreiro R, Ross OA, Kun‐Rodrigues C, et al. Investigating the genetic architecture of dementia with Lewy bodies: a two‐stage genome‐wide association study. Lancet Neurol 2018;17:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Cenedella RJ. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids 2009;44:477–487. [DOI] [PubMed] [Google Scholar]
- 193. Elgner F, Ren H, Medvedev R, et al. The intracellular cholesterol transport inhibitor U18666A inhibits the exosome‐dependent release of mature hepatitis C virus. J Virol 2016;90:11181–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Breiden B, Sandhoff K. Emerging mechanisms of drug‐induced phospholipidosis. Biol Chem 2019;401:31–46. [DOI] [PubMed] [Google Scholar]
- 195. Smolders S, Van Broeckhoven C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson's disease pathogenesis. Acta Neuropathol Commun 2020;8:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Ferraz MJ, Marques ARA, Appelman MD, et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett 2016;590:716–725. [DOI] [PubMed] [Google Scholar]
- 197. Taguchi YV, Liu J, Ruan J, et al. Glucosylsphingosine promotes α‐synuclein pathology in mutant GBA‐associated Parkinson's disease. J Neurosci 2017;37:9617–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Gegg ME, Sweet L, Wang BH, Shihabuddin LS, Sardi SP, Schapira AHV. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov Disord 2015;30:1085–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Hallett PJ, Engelender S, Isacson O. Lipid and immune abnormalities causing age‐dependent neurodegeneration and Parkinson's disease. J Neuroinflammation 2019;16:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Luzio JP, Hackmann Y, Dieckmann NMG, Griffiths GM. The biogenesis of lysosomes and lysosome‐related organelles. Cold Spring Harb Perspect Biol 2014;6(9):a016840. 10.1101/cshperspect.a016840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Swinn L, Schrag A, Viswanathan R, Bloem BR, Lees A, Quinn N. Sweating dysfunction in Parkinson's disease. Mov Disord 2003;18:1459–1463. [DOI] [PubMed] [Google Scholar]
- 202. Jucevičiūtė N, Banaitytė I, Vaitkus A, Balnytė R. Preclinical signs of Parkinson's disease: a possible association of Parkinson's disease with skin and hair features. Med Hypotheses 2019;127:100–104. [DOI] [PubMed] [Google Scholar]
- 203. Ridsdale R, Na CL, Xu Y, Greis KD, Weaver T. Comparative proteomic analysis of lung lamellar bodies and lysosome‐related organelles. PLoS One 2011;6(1):e16482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Akiyama M. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest 2005;115:1777–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Bhagavan NV, Ha C‐E. Lipids II. Essentials of Medical Biochemistry. Amsterdam, Netherlands: Elsevier; 2015:299–320. 10.1016/B978-0-12-416687-5.00017-8 [DOI] [Google Scholar]
- 206. Höppner S, Kinting S, Torrano AA, et al. Quantification of volume and lipid filling of intracellular vesicles carrying the ABCA3 transporter. Biochim Biophys Acta ‐ Mol Cell Res 2017;1864:2330–2335. [DOI] [PubMed] [Google Scholar]
- 207. Piehler AP, Özcurumez M, Kaminski WE. A‐subclass ATP‐binding cassette proteins in brain lipid homeostasis and neurodegeneration. Front Psychiatry 2012;3:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Wenzel JJ. ABC A‐subclass proteins: gatekeepers of cellular phospho‐ and sphingolipid transport. Front Biosci 2007;12:3177. [DOI] [PubMed] [Google Scholar]
- 209. Liu J, Zhang JP, Shi M, et al. Rab11a and HSP90 regulate recycling of extracellular α‐synuclein. J Neurosci 2009;29:1480–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Breda C, Nugent ML, Estranero JG, et al. Rab11 modulates α‐synuclein‐mediated defects in synaptic transmission and behaviour. Hum Mol Genet 2015;24:1077–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Martello A, Platt FM, Eden ER. Staying in touch with the endocytic network: the importance of contacts for cholesterol transport. Traffic 2020;21:354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Pfeffer SR. NPC intracellular cholesterol transporter 1 (NPC1)‐mediated cholesterol export from lysosomes. J Biol Chem 2019;294:1706–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Parkinson‐Lawrence EJ, Shandala T, Prodoehl M, Plew R, Borlace GN, Brooks DA. Lysosomal storage disease: revealing lysosomal function and physiology. Physiology 2010;25:102–115. [DOI] [PubMed] [Google Scholar]
- 214. Johnson PH, Weinreb NJ, Cloyd JC, Tuite PJ, Kartha RV. GBA1 mutations: prospects for exosomal biomarkers in α‐synuclein pathologies. Mol Genet Metab 2020;129:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Kong SMY, Chan BKK, Park J‐S, et al. Parkinson's disease‐linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α‐Synuclein externalization via exosomes. Hum Mol Genet 2014;23:2816–2833. [DOI] [PubMed] [Google Scholar]
- 216. Grey M, Dunning CJ, Gaspar R, et al. Acceleration of α‐synuclein aggregation by exosomes. J Biol Chem 2015;290:2969–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Mielke MM, Maetzler W, Haughey NJ, et al. Plasma Ceramide and Glucosylceramide metabolism is altered in sporadic Parkinson's disease and associated with cognitive impairment: a pilot study. PLoS One 2013;8:e73094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Bellomo G, Paciotti S, Gatticchi L, Parnetti L. The vicious cycle between α ‐Synuclein aggregation and Autophagic‐Lysosomal dysfunction. Mov Disord 2020;35:34–44. [DOI] [PubMed] [Google Scholar]
- 219. Yang S, Gegg M, Chau D, Schapira A. Glucocerebrosidase activity, cathepsin D and monomeric α‐synuclein interactions in a stem cell derived neuronal model of a PD associated GBA1 mutation. Neurobiol Dis 2020;134:104620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Papadopoulos VE, Nikolopoulou G, Antoniadou I, et al. Modulation of β‐glucocerebrosidase increases α‐synuclein secretion and exosome release in mouse models of Parkinson's disease. Hum Mol Genet 2018;27(10):1696–1710. 10.1093/hmg/ddy075 [DOI] [PubMed] [Google Scholar]
- 221. Thomas RE, Vincow ES, Merrihew GE, MacCoss MJ, Davis MY, Pallanck LJ. Glucocerebrosidase deficiency promotes protein aggregation through dysregulation of extracellular vesicles. PLOS Genet 2018;14:e1007694 [DOI] [PMC free article] [PubMed] [Google Scholar]