
Figure 1.
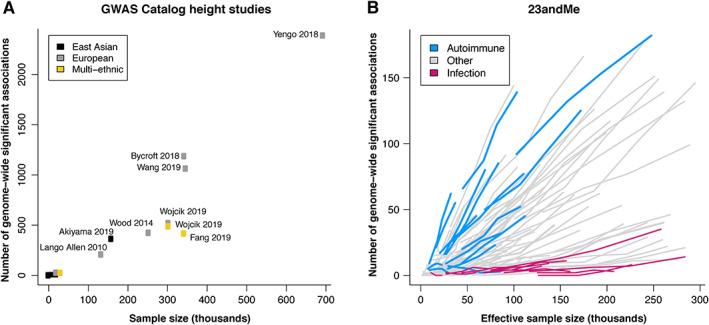
The number of genome‐wide significant loci discovered increases linearly as a function of sample size. (A) The number of genome‐wide significant loci discovered as a function of sample size for ‘body height’ GWAS recorded in the GWAS Catalog as of 1 November 2020 (see supplementary material, Table S1 for details of the studies used). The associated publication for each study was manually assessed, excluding (1) GWAS of traits other than adult height, (2) GWAS of individuals of European ancestry with fewer than 19 000 cases, and (3) GWAS conducted using whole‐genome or whole‐exome sequencing data. SNPs with p > 5 × 10−8 and SNPs that were only identified by conditional analysis were also excluded. The color of the points represents the ancestry of the individuals included in the study (black = East Asian; gray = European; gold = multi‐ethnic). (B) Trajectories for a selection of GWAS for 126 23andMe disease phenotypes conducted in individuals of European ancestry at four time points between October 2017 and August 2019. Effective sample size is defined as Neff = 4/(1/Ncases + 1/Ncontrols) for binary phenotypes and is equal to the sample size for continuous phenotypes. Trajectories for autoimmune diseases and infection phenotypes are highlighted in blue and pink, respectively.