

Abstract
Spinal muscular atrophy (SMA) is caused by bi‐allelic loss or pathogenic variants in the SMN1 gene. SMN2, the highly homologous copy of SMN1, is considered the major phenotypic modifier of the disease. Determination of SMN2 copy number is essential to establish robust genotype–phenotype correlations and predict disease evolution, to stratify patients for clinical trials, as well as to define those eligible for treatment. Discordant genotype–phenotype correlations are not uncommon in SMA, some of which are due to intragenic SMN2 variants that may influence the amount of complete SMN transcripts and, therefore, of full‐length SMN protein. Detection of these variants is crucial to predict SMA phenotypes in the present scenario of therapeutic advances and with the perspective of SMA neonatal screening and early diagnosis to start treatments. Here, we present a novel, affordable, and versatile method for complete sequencing of the SMN2 gene based on long‐range polymerase chain reaction and next‐generation sequencing. The method was validated by analyzing samples from 53 SMA patients who lack SMN1, allowing to characterize paralogous, rare variants, and single‐nucleotide polymorphisms of SMN2 as well as SMN2–SMN1 hybrid genes. The method identifies partial deletions and can be adapted to determine rare pathogenic variants in patients with at least one SMN1 copy.
Keywords: next‐generation sequencing, paralogous variants, phenotype–genotype correlations, SMN2 copies, spinal muscular atrophy
We developed a new affordable and versatile method that, by means of long PCR and NGS, allows sequencing the entire SMN2 genes of spinal muscular atrophy (SMA) patients detecting point variants, copy number variants and hybrid SMN1‐SMN2 genes. This complete characterization of the SMN2 structure in each patient will improve our knowledge of the genotype‐phenotype correlation, the phenotype prediction in the context of newborn screening and pre‐symptomatic diagnosis and also will potentially allow the identification of new phenotype modifier variants.
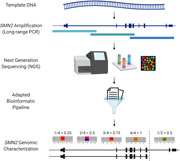
1. INTRODUCTION
Spinal muscular atrophy (SMA) is the second most common recessive genetic disease of infancy and early childhood, with an incidence of 1 in 5000–10,000 live births and a worldwide carrier frequency of 1:51 (Sugarman et al., 2012). SMA patients are classified into different clinical groups based on the age of onset, clinical severity, and achieved motor milestones. In the most severe form, type I SMA, patients are never able to sit and generally die of respiratory failure before the age of 2 years. Intermediate type II SMA patients are able to sit but never walk, thus being confined to wheel‐chair. Type III patients walk unassisted but may lose this ability during infancy or adolescence (Wang et al., 2007; Zerres & Rudnik‐Schöneborn, 1995).
Bi‐allelic absence or pathogenic variants of the Survival of Motor Neuron 1 (SMN1) gene cause SMA (Lefebvre et al., 1995). A centromeric and nearly identical paralog, SMN2, encodes in principle the same protein as SMN1 (Lefebvre et al., 1995; Monani et al., 1999; Rochette et al., 2001). However, a silent transition within exon 7 of the SMN2 gene causes exon skipping and results in a truncated, nonfunctional variant (SMN‐Δ7) (Lorson et al., 1999). It has been estimated that each SMN2 copy can produce only around 10% to 15% of functional SMN protein, depending on the cells and tissues studied (Boza‐Morán et al., 2015; Soler‐Botija et al., 2005; Wirth et al., 2013).
The number of SMN2 copies and the presence of intragenic SMN2 variants are known modifiers of SMA disease severity (Bernal et al., 2010; Prior et al., 2009; Ruhno et al., 2019). Indeed, numerous studies show that the higher the SMN2 copy number, producing larger amount of full‐length SMN protein, the milder the associated SMA phenotype and vice versa. However, this inverse correlation is not absolute (Calucho et al., 2018). Whereas the determination of SMN2 copy number is widely implemented to study SMA patients, the actual structures and genomic sequences of SMN2 copies are usually not included in the characterization of SMA patients.
The current scenario of SMA therapy is rapidly evolving due to the approval in the last years of nusinersen/Spinraza, an antisense‐tailored therapy (Finkel et al., 2017), AVXS101/Zolgensma, an adeno‐associated viral‐based gene therapy (Mendell et al., 2017), as well as the recent approval of the first oral drug to treat SMA, risdiplam/Evrysdi (http://www.fda.gov). However, these disease‐modifying therapies are expensive treatments, and their efficacy needs to be periodically assessed. Although responses to treatment vary in SMA patients, it is not yet known whether specific features of SMN2 are correlated with these responses (Cuscó et al., 2020). Thus, it becomes crucial to investigate genomic SMN2 data to better characterize SMA patients and accurately predict disease evolution.
Here, we report a novel method for sequencing the whole SMN2 gene based on long‐range polymerase chain reaction (PCR) and next‐generation sequencing (NGS). The method allows determining all variants described so far as disease modifiers in SMA patients without SMN1 as well as to identify new variants and structural changes. Furthermore, the technique can be adapted to determine rare pathogenic variants in heterozygous patients with at least one SMN1 copy. Inclusion of this technique in the routine diagnosis of SMA patients is expected to improve individual genotype–phenotype correlations and, therefore, to help predict more accurately the evolution of the disease.
2. MATERIALS AND METHODS
2.1. Patients
We studied 53 genetically confirmed SMA patients with homozygous absence of SMN1. The vast majority of studied patients had three SMN2 gene copies (n = 51), but the cohort included one patient each with two and four SMN2 copies, respectively. In addition, samples from three patients who carry one SMN1 copy were studied to assess the versatility of the method to detect SMN1‐specific pathogenic variants. These patients were: patient SMA54, who had a heterozygous deletion of SMN1, the pathogenic variant c.399_402del (p.(Glu134Serfs*14)) in the other SMN1 allele, and three SMN2 copies; patient SMA55, who had a heterozygous deletion of SMN1, the pathogenic variant c.815A>G (p.(Tyr272Cys)) in the other SMN1 allele, one complete SMN2 copy, and two partial SMN copies comprising exons 1 to 6 (also known as SMN1/2Δ7‐8 deletion, Arkblad et al., 2006), and SMA55F, father of SMA55 harboring 2 SMN1 copies(one with the variant c.815A>G), and two SMN2 copies.
Genetic confirmation of SMA by bi‐allelic defects in SMN1 (Alías et al., 2009) as well as SMN2 copy number determination by multiplex ligation‐dependent probe amplification (MLPA) were carried out as previously described (Alías et al., 2011). Patients were classified as I, II, or III according to their severity and motor milestone achievements. One patient presenting with type 0 (congenital) SMA was also studied. All patients were unrelated with the exception of the SMA55/55F pair mentioned above (a child with type 0 SMA and his father) and two pairs of siblings, SMA17/SMA18 (both type II) and SMA27/SMA51 (types II and III, respectively). DNA samples were obtained from peripheral blood. All participants or their legal guardians signed written informed consent. The study was approved by the Ethical Committee of our Hospital (PR(AG)229/2018).
2.2. PCR design and library preparation
We studied the complete genomic SMN2 sequence, including promoter, 5′‐UTR and 3′‐UTR regions. To this end, we designed three overlapping PCRs (~12‐kb each) to amplify a target region of approximately 31.5 Kb (chr5:69,342,511‐69,374,064). These long‐range PCRs were successfully set up using TaKaRa LA Taq® DNA polymerase (#RR002A; Takara Bio). Given the high homology between SMN1 and SMN2 sequences, primers were not specific to SMN2, but the 53 patients analyzed have zero SMN1 copies, allowing in principle an SMN2 exclusive analysis. In addition, samples from three patients with at least one SMN1 copy were analyzed. Primer sequences and PCR conditions are given in Table S1.
After amplification, the concentration of the three PCR products was measured using Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and mixed equimolarly. One thousand nanogram of the obtained mixture was fragmented with NEBNext® dsDNA Fragmentase® (New England Biolabs) to generate DNA fragments of ~200 bp. Then, the NEBNext Ultra DNA library prep kit for Illumina and NEBNext® MultiplexOligos for Illumina® Dual Index Primers Set 1 (New England Biolabs) were used to generate the libraries. The necessary purifications and size selections were performed using AMPure XP beads (Beckman Coulter).
The quality and size of the libraries were assessed using QIAxcel (Qiagen) and were quantified with Qubit. Finally, the libraries of all patients were equimolarly mixed and sequenced using a 500‐cycle MiSeq reagent kit v2 with a paired‐end run of 2 × 251 bp reads in a MiSeq instrument (Illumina). The number of patients included in each run was calculated to ensure a minimum coverage of ×400. All procedures were performed following the manufacturer's instructions.
In samples harboring at least one copy of SMN1, pathogenic variants were ascribed to SMN1 using a long‐range PCR and Sanger sequencing of SMN1 as previously described (Kubo et al., 2015), with slight modifications.
2.3. Bioinformatics analysis
The data analysis pipeline included the quality trimming of Illumina sequences using Trimmomatic (Bolger et al., 2014). Sequences were mapped to an artificial genome reference that contains only the SMN2 coordinates, based on the reference genome (UCSC hg 19 version, build 37.1). This strategy avoids arbitrarily reporting only one of the possible alignments produced by most mapping algorithms and therefore avoids dispersion of the read‐depth signal from all SMN genes. This approach increases the power to detect small changes of coverage and variants with lower AB ratios, which are characteristic of heterozygous variants in multicopy regions. Results show a single location per each read that corresponds to any of the SMN genes. Mapping was performed using burrows‐wheeler aligner (BWA)‐align and BWA‐sample with default parameters. Variant calling was performed with Genome Analysis Toolkit (GATK) Unified Genotyper and Haplotype Caller (McKenna et al., 2010), and variant annotation with ANNOVAR (Codina‐Solà et al., 2016; Wang et al., 2010).
2.4. Genetic variant types
We discriminated the genetic variants identified in three categories: (1) paralogous sequence variants (PSVs), which are positions differing between duplicated genes (in this context, variants differing between SMN1 and SMN2); (2) rare single‐nucleotide variants (SNVs), which are those present in less than 1% of the population or never described; and (3) single‐nucleotide polymorphisms (SNPs), which are variations in a single position present in over 1% of the population. Determining whether the variants found are SNPs, SNVs, or PSVs allowed us to evaluate the specificity of the method in the detection of SMN1 versus SMN2 and to identify hybrid structures in the samples. This classification is not related to a possible effect of variants on SMN2 expression and phenotype, a topic out of the scope of this article.
PSV ratios were computed as quotients of the number of reads belonging to a given functional copy and the total coverage.
Mutation nomenclature refers to GenBank NC_000005.9, RefSeq NM_000344.3 for SMN1, and RefSeq NM_017411.4 for SMN2.
3. RESULTS
In all analyzed samples, a mean depth of coverage of 1720× (435×–7478×) was obtained. This high coverage allows to accurately determine the allelic frequency of the detected variants in each sample (AB ratio). Given that the copy number of SMN2 genes had been already studied by MLPA, we could use the AB ratio to calculate the number of copies in which the detected variant was present.
To assess the specificity of our method to identify genetic variants in the 5q13 complex region, we first analyzed PSVs that are specific for the SMN2 gene. To this end, a list of SMN1 and SMN2 PSVs was created based initially on a literature review (Monani et al., 1999), and expanded with the information obtained from a basic local alignment search tool‐like alignment tool (BLAT) between the two genes using the reference genome Hg19. The detailed information obtained from sequencing the SMN2 gene in samples from 53 patients was used to determine and eventually confirm which positions should be considered real PSVs or SNPs or SNVs. Based on previous reports and BLAT data, we initially considered 22 PSVs (Table 1). Four of these variants exhibited high variability between samples, indicating that they should not be considered genuine PSVs (by definition). Two other variants, including the candidate position g.69371981A/C previously described as a PSV (Monani et al., 1999) and g.69367553G/A were not found neither in SMN1 nor SMN2 genes from our samples and were considered as very infrequent variants. The remaining 16 changes are genuine PSVs between SMN1 and SMN2 genes, being c.835‐1606C/T in intron 6 considered here for the first time as a PSV (Table 1). These PSVs were present in all the reads and in more than 95% of the samples, confirming that all these positions were specific for SMN2. However, in two patients (SMA04 and SMA39) we observed some discrepancies in the PSV ratio compatible with the presence of hybrid genes. Both patients presented six SMN1 PSVs all located in intron 6 (Chr5: 69370451‐ 69370895). The PSV ratio indicates that in SMA04, two of the three SMN copies are SMN2–SMN1 hybrids, while in SMA39, only one of the three copies is an SMN2–SMN1 hybrid (Figure 1).
Table 1.
The 22 candidate positions for paralogous sequence variants (PSVs) between SMN1 and SMN2 are shown
| SMN1 position | SMN2 position | Gene location | Ref SMN1 | Ref SMN2 | Categorization | Conversion nomenclature SMN2>SMN1 | Source of information |
|---|---|---|---|---|---|---|---|
| 70231509 | 69356085 | Intron 1 | G | A | SNPa | c.82‐3157A>G | BLAT |
| 70240028 | 69364605 | Intron 4 | G | A | SNPa | c.628‐457A>G | BLAT |
| 70242435 | 69367010 | Intron 6 | T | C | SNPa | c.834+432C>T | BLAT |
| 70242978 | 69367553 | Intron 6 | A | G | SNVb | c.834+975G>A | BLAT |
| 70244142 | 69368717 | Intron 6 | A | G | SNPa | c.834+2139G>A | Monani et al. /BLAT |
| 70245876 | 69370451 | Intron 6 | T | C | PSV | c.835‐1897C>T | Monani et al. /BLAT |
| 70246016 | 69370591 | Intron 6 | G | A | PSV | c.835‐1757A>G | Monani et al. /BLAT |
| 70246019 | 69370594 | Intron 6 | T | C | PSV | c.835‐1754C>T | Monani et al. /BLAT |
| 70246156 | 69370731 | Intron 6 | G | A | PSV | c.835‐1617A>G | Monani et al. /BLAT |
| 70246167 | 69370742 | Intron 6 | T | C | PSV | c.835‐1606C>T | BLAT |
| 70246320 | 69370895 | Intron 6 | G | A | PSV | c.835‐1453A>G | Monani et al. /BLAT |
| 70246793 | 69371368 | Intron 6 | G | A | PSV c | c.835‐980A>G | Monani et al. /BLAT |
| 70246872 | 69371448 | Intron 6 | ‐ | AGGCA | PSV c | c.835‐900_835‐896del | Monani et al. /BLAT |
| 70246919 | 69371499 | Intron 6 | A | C | PSV c | c.835‐849C>A | Monani et al. /BLAT |
| 70247219 | 69371799 | Intron 6 | G | A | PSV c | c.835‐549A>G | Monani et al. /BLAT |
| 70247290 | 69371870 | Intron 6 | T | C | PSV | c.835‐478C>T | Monani et al. /BLAT |
| 70247401 | 69371981 | Intron 6 | C | A | SNVb, c | c.835‐367A>C | Monani et al. /BLAT |
| 70247724 | 69372304 | Intron 6 | G | A | PSV c | c.835‐44A>G | Monani et al. /BLAT |
| 70247773 | 69372353 | Exon 7 | C | T | PSV c | c.840T>C | Monani et al. /BLAT |
| 70247921 | 69372501 | Intron 7 | A | G | PSV c | c.*3+100G>A | Monani et al. /BLAT |
| 70248036 | 69372616 | Intron 7 | A | G | PSV c | c.*3+215G>A | Monani et al. /BLAT |
| 70248501 | 69373081 | Exon 8 | G | A | PSV c | c.*239A>G | Monani et al. /BLAT |
Note: These positions were obtained from a previous bibliographic compilation (Monani et al., 1999) and were complemented with a BLAT between the two genes, as deposited in the reference genome Hg19. Repetitive regions (polyA, polyT, and polyGT) were discarded. The candidate positions were genotyped in the patients studied in this study (n = 53) and in samples with at least one SMN1 (n = 3) to check for consistency. From the 22 candidates, six were discarded as PSVs, four of them are listed as SNPs instead, and the remaining two as rare SNV. Therefore, a total of 16 nucleotides (in bold in the Table) differentiate SMN1 and SMN2 genes, 10 of which had been previously described and validated, 5 had been described but not validated, and 1 is considered here as a PSV for the first time.
Abbreviations: BLAT, BLAST‐like alignment tool; SNP, single‐nucleotide polymorphism; SNV, single‐nucleotide variant.
These positions show high variability between samples and were therefore classified as SNPs (including position c.835‐367C/A previously validated by Monani et al., 1999).
In these positions, the same nucleotide has always been found in both SMN1 and SMN2. Thus, G>A and A>C exchanges appears to be very rare SNV found in the reference genome.
These 11 positions were previously classified as bona fide PSVs, after being tested in a control population of 15 individuals (Monani et al., 1999).
Figure 1.
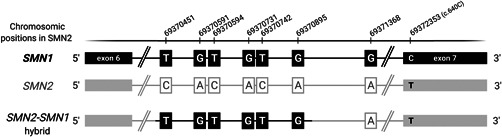
Structure of the SMN2–SMN1 hybrid detected in patients SMA4 and SMA39. PSVs were genotyped in all patients. In SMA4 and SMA39, six SMN1 PSVs located in intron 6 (Chr5: 69370451‐69370895) were detected, which indicates the presence of hybrid genes. The AB ratio indicates that in SMA04, two of the three copies are SMN2–SMN1 hybrids (SMN1 PSVs in 66%) while in SMA39, only one of the three copies is an SMN2–SMN1 hybrid (SMN1 PSVs in 33%). PSV, paralogous sequence variant
We also detected variants previously described as beneficial for SMN2 function (Bernal et al., 2010; Prior et al., 2009; Ruhno et al., 2019; Wu et al., 2017). Using the AB ratio data, we were able to confirm the presence of the c.859G>C (p.(Gly287Arg)) variant in patient SMA52 with two SMN2 copies in the heterozygous state and of c.835‐44A>G (g.69372304A>G, commonly known as A‐44G) in one of the three SMN2 copies of patient SMA21 (Figure 2 and Table S2). Furthermore, in two sisters with different phenotypes (SMA27, type II and SMA51, type III) and three SMN2 copies, we found the PSV conversion c.835‐1897C>T (g.69370451C>T) in one of their three alleles. In our series, we did not identify any of the other more recently published variants considered as candidates to be modifiers (Ruhno et al., 2019; Wadman et al., 2020).
Figure 2.
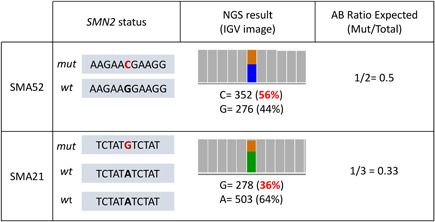
Utility of AB ratios to calculate the number of the copies in which variants are present. Patient SMA52 has two SMN2 copies (determined by MLPA) and the variant NM_017411.4:c.859G>C (p.(Gly287Arg)) was detected with a frequency of 56%, in agreement with the AB ratio expected for the variant in one over two alleles. Patient SMA21 has three SMN2 copies (determined by MLPA), and the variant c.835‐44A>G (NC_000005.9: g.69372304A>G) was detected with a frequency of 36%, in agreement with the AB ratio expected for the variant in one over three alleles. MLPA, multiplex ligation‐dependent probe amplification
Finally, to verify whether our technique could also be extended to identify SMN1 variants, we studied samples from three SMA patients harboring at least one SMN1 copy. The PSV genotype in all three cases was in agreement with the copy numbers previously determined by MLPA. SMA54 (1_SMN1/3_SMN2 MLPA genotype) showed the SMN1 PSVs at a frequency of ~25% (one of the four SMN copies of the patient). The SMN1 PSV frequency in SMA55 and SMA55F was ~50% in line with their genotypes (1_SMN1/1_SMN2 and 2_SMN1/2_SMN2, respectively). A similar consistency was obtained with SMN1 pathogenic variants. Thus, c.399_402del (p.(Glu134Serfs*14)) in SMA54 showed a frequency of ~25%, while c.815A>G (p.(Tyr272Cys)) showed a frequency of ~50% in SMA55 and of 25% in SMA55F. SMN1‐specific Sanger sequencing (Kubo et al., 2015) revealed that patients SMA54 and SMA55 present this variant in hemizygous state, whereas in SMA55F it was detected in heterozygous state (Figure 3).
Figure 3.
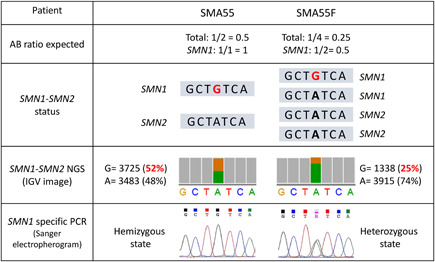
Detection of the pathogenic variant NM_017411.4:c.815A>G in samples SMA55 and SMA55F. Patient SMA55 has one SMN1 copy (with the variant c.815A>G) and one SMN2 copy, while his father (SMA55F) has two SMN1 (one copy with the variant c.815A>G) and two SMN2 copies (determined by MLPA and Sanger). The pathogenic variant c.815A>G was detected in SMA55 and SMA55F through NGS with a frequency of 52% and 25%, respectively. The SMN1‐specific PCR performed confirms that the pathogenic variant c.815A>G is present in SMN1 since we observed the variant in hemizygous status in SMA55 and in heterozygous status in SMA55F. MLPA, multiplex ligation‐dependent probe amplification; NGS, next‐generation sequencing; PCR, polymerase chain reaction
In addition, in patient SMA55, it was also possible to corroborate the presence of the common partial SMN1/2Δ7‐8 deletion of the 3′ region previously detected by MLPA (Figure 4). Indeed, analysis of the AB ratios of all SNVs revealed a discrepancy between the 5′ and 3′ SMN region, showing ratios compatibles with four copies (25%–75%) in the 5′ region (from promoter to exon 6), but compatible with two copies (50%–100%) in the 3′ region confirming the presence of the two alleles with the SMN1/2Δ7‐8 deletion (further explanation in Figure 4).
Figure 4.
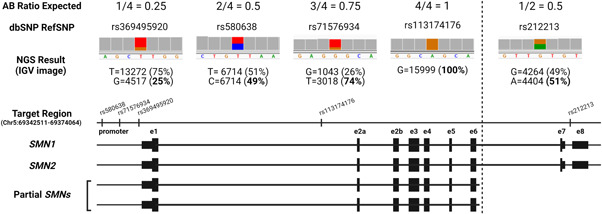
Description of the utility of AB ratios to determine the presence of two partial SMN genes (SMN1/2Δ7/8). The patient (SMA55) has one SMN1 (with a pathogenic variant in exon 6*), one SMN2, and two partial SMNs from promoter to exon 6 (determined by MLPA). In the 5′ region (promoter‐ex6), the patient has a total of four SMN copies; consequently, we detected different SNPs with an allelic frequency of 25%, 50%, 75%, and 100%. By contrast, in the 3′ region (in6‐ex8), the patient has two SMN copies, and only variants with an allelic frequency of 50% are detected. Note that PSVs are located in the 3′ region; therefore, it is not possible to determine whether these partial genes are derived from SMN1 or SMN2. MLPA, multiplex ligation‐dependent probe amplification; PSV, paralogous sequence variant; SNP, single‐nucleotide polymorphisms
4. DISCUSSION
We have developed a novel, practical method for the genomic analysis of both SMN1 and SMN2 regions. The technique focuses on the genomic characterization of each SMN2 copy in SMA patients regardless of the gene copy number and can also be applied to detect pathogenic variants in heterozygous SMA patients with at least one SMN1 copy.
Currently published methods to study the complete sequence of the SMN2 gene include whole genome sequencing (WGS) and multiplexed direct genomic selection (MDiGS) sequencing (Chen et al., 2020; Ruhno et al., 2019). WGS is a rather expensive and laborious technique and usually needs a complex bioinformatics analysis. In MDiGS, whole DNA is prepared in libraries, target regions are captured by bacterial artificial chromosomes (BAC) probes of SMN (not specific for SMN1 or SMN2), as well as of CFTR and PLS3, for quantitative comparison. Thus, the hybridization step is rather large and complex. By contrast, the new method described here based on long‐range PCR and NGS can be easily implemented in any genetics laboratory performing NGS applications.
The main advantages of our method, specifically designed to deal with the complexity of the SMA region, include accessible cost, relative simplicity, and speedy results (usually obtained in around 3 days). This practical protocol can easily genotype both PSVs and rare variants already described in SMA patients, but it might also help to identify new variants and SMN2–SMN1 hybrid genes as part of more investigative and personalized approaches. The high coverage obtained at the nucleotide level with the new method allows the determination of the number of copies in which a specific variant is present, which, in turn, is essential to characterize the genomic architecture of each SMN2 copy. Indeed, although the method does not directly quantify SMN2 copy numbers, calculation of the number of copies and the presence of partial SMN genes is straightforward using the allelic frequencies of the variants and might be used to, for example, confirm results reported by other methods, such as MLPA.
Employing this method to samples from SMA patients, we elaborated an updated list of 16 PSVs between SMN1 and SMN2, including one previously never described. PSV genotyping would help to detect hybrid genes and to discover new, potentially relevant conversions that might act as disease modifiers (Ruhno et al., 2019; Wu et al., 2017). Our method is highly specific given that it allows detection of positive modifiers, such as c.859G>C, c.835‐44A>G (g.69372304A>G), and c.835‐1897C>T (g.69370451C>T). The method also determines the number of SMN2 copies carrying modifier variants through analysis of allelic frequencies (Figure 2).
Based on the analysis of the entire genomic SMN2 region and according to the PSV genotyping, we also identified specific SMN2–SMN1 hybrid structures previously undetected by MLPA. The presence of hybrid SMN genes has been previously described based mainly on the analysis of exons 7 and 8 (Cuscó et al., 2001; Hahnen et al., 1996). This phenomenon occurs because the complex 5q13 region contains segmental duplications prone to nonallelic homologous recombinations, deletions, duplications, and gene conversion events. Usually, hybrid genes are detected because of the homozygous absence of SMN1 exon 7 coupled to the presence of SMN1 exon 8. Thus, hybrids upstream exon 7 cannot be detected by common methods of diagnosis, including MLPA. In contrast, with our long PCR‐based approach, we were able to detect novel hybrid genes consisting of a SMN2 gene with a fragment of intron 6 derived from SMN1 (Figure 1). Although additional studies are needed to characterize the function of these two hybrid genes, their detection might provide clues about possible functional differences between SMN2 genes.
The method described here also allows a complete genomic SMN1 analysis. In fact, we were able to detect the pathogenic variants c.399_402del (p.(Glu134Serfs*14)) and c.815A>G (p.(Tyr272Cys)) in patients and carriers, and to determine their frequencies (e.g., homozygous vs. heterozygous cases). Further confirmation that a certain variant is located in SMN1 can be achieved with a specific PCR of the gene (Kubo et al., 2015). Given that in our method, we analyze the whole SMN1, including promoter, 5′ and 3′ regions as well as all introns, it emerges as potentially useful to study complex SMA cases in which only one allele alteration (deletion or point mutation) has been detected using conventional techniques (Alías et al., 2009).
Finally, we were able to confirm the presence of partial SMN copies (SMN1/2Δ7‐8) using the AB ratios. The SMN1/2Δ7‐8 deletion has been widely described in the literature (Arkblad et al., 2006; Calucho et al., 2018; Chen et al., 2020; Ruhno et al., 2019; Vijzelaar et al., 2019) as a variant relatively common in general population (Europeans 15.7%, Vijzelaar et al., 2019). In almost all cases, the breakpoints are consistent (3643 bp before exon 7 and 1587 bp after exon 8 according to Ruhno et al., 2019), so it is considered that this deletion is the result of a single deletion event (Vijzelaar et al., 2019).
The number of SMN2 copies and specific variants of the gene have been established as the main disease modifiers of the SMA phenotype. However, to date, studies in discordant siblings do not support the hypothesis that the intra‐familiar variability is due to variants in the SMN2 locus (Calucho et al., 2018; Cuscó et al., 2006; Ruhno et al., 2019) as is the case of our sisters sharing the modifier variant c.835‐1897C>T (see Table S2). Although SMN2 is the target for splicing modifiers in the current therapeutic scenario, thorough sequencing of the gene is almost never performed in genetic diagnostic laboratories. Application of our method and thus the availability of detailed SMN2 sequences of SMA patients would help to solve discrepancies in genotype–phenotype correlations, as well as deepen the study of intra‐familiar variability, in a prospective manner, for the analysis of presymptomatic cases detected in newborn screening. Indeed, being the method so specific and fast, it is envisaged that it could be accommodated within the time frame for therapeutic decisions in SMA newborns. It is also essential to unveil possible linkages between specific SMN2 variants, factors involved in SMN2 splicing, and responses to therapies. SMA treatments are very expensive, and proof of their efficacy is periodically assessed in SMA patients. In particular, nusinersen is an 18‐mer oligonucleotide that binds the ISNN1 region of the intron 7, and to date, none of the modifier variants described is located in this region (Ruhno et al., 2019; Wadman et al., 2020; this study). However, in addition to the known modifier variants, other features of their SMN2 genes may be correlated with the level of responsiveness and effectiveness, an issue that warrants further investigation. Thus, the discovery and validation of positive and negative SMN2 variants in each patient remain a crucial issue in SMA diagnosis and research. An additional benefit of implementing the new method besides the characterization of SMN2 sequences in patients with a homozygous deletion of SMN1 is its application to study SMN1 in SMA patients retaining at least one SMN1 copy. Therefore, the versatile method described here is a useful tool to approach SMN1 and SMN2 deep sequencing, which can be easily implemented in most SMA diagnostic laboratories.
CONFLICT OF INTERESTS
A patent is in preparation for this methodology and pipeline (I.C. and E. F. T.). E.F.T has received grant support to conduct clinical trials on SMA from Ionis/Biogen and serves as a consultant to AveXis, Novartis, Biogen, Biologix, Cytokinetics, Roche. The remaining authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
The enclosed manuscript has been seen and approved by all the authors, and they have taken care to ensure the integrity of the work.
WEB RESOURCES
The following web sources have been used to carry out this study:
https://genome-euro.ucsc.edu/,
http://www.hgmd.cf.ac.uk/ac/index.php,
Supporting information
Supporting information.
ACKNOWLEDGMENTS
This study was supported by Grants from Biogen ESP‐SMG‐17‐11256 and Galicia AME (to E. F. T. supporting L. B.‐P.), “Fundación Daniel Bravo Andreu” (to E. F. T. and P. F.‐P.), SMA Europe (to E.F.T and P.F.‐P.), and Spanish Instituto de Salud Carlos III, Fondo de Investigaciones Sanitarias and cofunded with ERDF funds (Grant No. FIS PI18/000687) (to E. F. T. and L. A.).
Blasco‐Pérez, L. , Paramonov, I. , Leno, J. , Bernal, S. , Alias, L. , Fuentes‐Prior, P. , Cuscó, I. , & Tizzano, E. F. (2021). Beyond copy number: A new, rapid, and versatile method for sequencing the entire SMN2 gene in SMA patients. Human Mutation, 42, 787–795. 10.1002/humu.24200
Contributor Information
Ivon Cuscó, Email: icusco@vhebron.net.
Eduardo F. Tizzano, Email: etizzano@vhebron.net.
DATA AVAILABILITY STATEMENT
All data and scripts used to generate the analyses of this paper are available upon request unless that the type of request compromises ethical standards or legal requirements. All variants reported in this article are now listed in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/).
REFERENCES
- Alías, L. , Bernal, S. , Barceló, M. J. , Also‐Rallo, E. , Martínez‐Hernández, R. , Rodríguez‐Alvarez, F. J. , Hernández‐Chico, C. , Baiget, M. , & Tizzano, E. F. (2011). Accuracy of marker analysis, quantitative real‐time polymerase chain reaction, and multiple ligation‐dependent probe amplification to determine SMN2 copy number in patients with spinal muscular atrophy. Genetic Testing and Molecular Biomarkers, 15(9), 587–594. 10.1089/gtmb.2010.0253 [DOI] [PubMed] [Google Scholar]
- Alías, L. , Bernal, S. , Fuentes‐Prior, P. , Barceló, M. J. , Also, E. , Martínez‐Hernández, R. , Rodríguez‐Alvarez, F. J. , Martín, Y. , Aller, E. , Grau, E. , Peciña, A. , Antiñolo, G. , Galán, E. , Rosa, A. L. , Fernández‐Burriel, M. , Borrego, S. , Millán, J. M. , Hernández‐Chico, C. , Baiget, M. , & Tizzano, E. F. (2009). Mutation update of spinal muscular atrophy in Spain: Molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Human Genetics, 125(1), 29–39. 10.1007/s00439-008-0598-1 [DOI] [PubMed] [Google Scholar]
- Arkblad, E. L. , Darin, N. , Berg, K. , Kimber, E. , Brandberg, G. , Lindberg, C. , Holmberg, E. , Tulinius, M. , & Nordling, M. (2006). Multiplex ligation‐dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscular Disorders, 16(12), 830–838. 10.1016/j.nmd.2006.08.011 [DOI] [PubMed] [Google Scholar]
- Bernal, S. , Alías, L. , Barceló, M. J. , Also‐Rallo, E. , Martínez‐Hernández, R. , Gámez, J. , Guillén‐Navarro, E. , Rosell, J. , Hernando, I. , Rodriguez‐Alvarez, F. J. , Borrego, S. , Millán, J. M. , Hernández‐Chico, C. , Baiget, M. , Fuentes‐Prior, P. , & Tizzano, E. F. (2010). The c.859>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. Journal of Medical Genetics, 47(9), 640–642. [DOI] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boza‐Morán, M. G. , Martínez‐Hernández, R. , Bernal, S. , Wanisch, K. , Also‐Rallo, E. , Le Heron, A. , Alías, L. , Denis, C. , Girard, M. , Yee, J.‐K. , Tizzano, E. F. , & Yáñez‐Muñoz, R. J. (2015). Decay in survival motor neuron and plastin 3 levels during differentiation of iPSC‐derived human motor neurons. Scientific Reports, 5, 11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calucho, M. , Bernal, S. , Alías, L. , March, F. , Venceslá, A. , Rodríguez‐Álvarez, F. J. , Aller, E. , Fernández, R. M. , Borrego, S. , Millán, J. M. , Hernández‐Chico, C. , Cuscó, I. , Fuentes‐Prior, P. , & Tizzano, E. F. (2018). Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscular Disorders, 28(3), 208–215. 10.1016/j.nmd.2018.01.003 [DOI] [PubMed] [Google Scholar]
- Chen, X. , Sanchis‐Juan, A. , French, C. E. , Connell, A. J. , Delon, I. , Kingsbury, Z. , Chawla, A. , Halpern, A. L. , Taft, R. J. , Bentley, D. R. , Butchbach, M. E. R. , Raymond, F. L. , & Eberle, M. A. (2020). Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genetics in Medicine, 22(5), 945–953. 10.1038/s41436-020-0754-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codina‐Solà, M. , Flores, R. , Homs, A. , Aguado, C. , Pérez‐Jurado, L. , & Cuscó, I. (2016). Genomic and genetic variation at complex segmental duplications in Autism Spectrum disorders [Conference presentation abstract]. European Journal of Human Genetics, 24, 181. [Google Scholar]
- Cuscó, I. , Barceló, M. J. , Del Rio, E. , Martín, Y. , Hernández‐Chico, C. , Bussaglia, E. , Baiget, M. , & Tizzano, E. F. (2001). Characterisation of SMN hybrid genes in Spanish SMA patients: De novo, homozygous and compound heterozygous cases. Human Genetics, 108(3), 222–229. 10.1007/s004390000452 [DOI] [PubMed] [Google Scholar]
- Cuscó, I. , Barceló, M. J. , Rojas‐García, R. , Illa, I. , Gámez, J. , Cervera, C. , Pou, A. , Izquierdo, G. , Baiget, M. , & Tizzano, E. F. (2006). SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. Journal of Neurology, 253(1), 21–25. 10.1007/s00415-005-0912-y [DOI] [PubMed] [Google Scholar]
- Cuscó, I. , Bernal, S. , Blasco‐Pérez, L. , Calucho, M. , Alias, L. , Fuentes‐Prior, P. , & Tizzano, E. F. (2020). Practical guidelines to manage discordant situations of SMN2 copy number in patients with spinal muscular atrophy. Neurology: Genetics, 6(6), e530. 10.1212/nxg.0000000000000530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel, R. S. , Mercuri, E. , Darras, B. T. , Connolly, A. M. , Kuntz, N. L. , Kirschner, J. , Chiriboga, C. A. , Saito, K. , Servais, L. , Tizzano, E. , Topaloglu, H. , Tulinius, M. , Montes, J. , Glanzman, A. M. , Bishop, K. , Zhong, Z. J. , Gheuens, S. , Bennett, C. F. , Schneider, E. , … De Vivo, D. C. (2017). Nusinersen versus sham control in infantile‐onset spinal muscular atrophy. New England Journal of Medicine, 377(18), 1723–1732. 10.1056/NEJMoa1702752 [DOI] [PubMed] [Google Scholar]
- Hahnen, E. , Schönling, J. , Rudnik‐Schöneborn, S. , Zerres, K. , & Wirth, B. (1996). Hybrid survival motor neuron genes in patients with autosomal recessive spinal muscular atrophy: New insights into molecular mechanisms responsible for the disease. American Journal of Human Genetics, 59(5), 1057–1065. [PMC free article] [PubMed] [Google Scholar]
- Kubo, Y. , Nishio, H. , & Saito, K. (2015). A new method for SMN1 and hybrid SMN gene analysis in spinal muscular atrophy using long‐range PCR followed by sequencing. Journal of Human Genetics, 60(5), 233–239. 10.1038/jhg.2015.16 [DOI] [PubMed] [Google Scholar]
- Lefebvre, S. , Bürglen, L. , Reboullet, S. , Clermont, O. , Burlet, P. , Viollet, L. , Benichou, B. , Cruaud, C. , Millasseau, P. , Zeviani, M. , Le Paslier, D. , Frézal, J. , Cohen, D. , Weissenbach, J. , Munnich, A. , & Melki, J. (1995). Identification and characterization of a spinal muscular atrophy‐determining gene. Cell, 80(1), 155–165. [DOI] [PubMed] [Google Scholar]
- Lorson, C. L. , Hahnen, E. , Androphy, E. J. , & Wirth, B. (1999). A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proceedings of the National Academy of Sciences of the United States of America, 96(11), 6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The genome analysis toolkit: A map reduce framework for analyzing next‐generation DNA sequencing data. Genome Res, 20(9), 1297–303. 10.1101/gr.107524.110.20 [DOI] [PMC free article] [PubMed]
- Mendell, J. R. , Al‐Zaidy, S. , Shell, R. , Arnold, W. D. , Rodino‐Klapac, L. R. , Prior, T. W. , Lowes, L. , Alfano, L. , Berry, K. , Church, K. , Kissel, J. T. , Nagendran, S. , L'Italien, J. , Sproule, D. M. , Wells, C. , Cardenas, J. A. , Heitzer, M. D. , Kaspar, A. , Corcoran, S. , … Kaspar, B. K. (2017). Single‐dose gene‐replacement therapy for spinal muscular atrophy. New England Journal of Medicine, 377(18), 1713–1722. 10.1056/NEJMoa1706198 [DOI] [PubMed] [Google Scholar]
- Monani, U. R. , Lorson, C. L. , Parsons, D. W. , Prior, T. W. , Androphy, E. J. , Burghes, A. H. M. , & McPherson, J. D. (1999). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Human Molecular Genetics, 8(7), 1177–1183. [DOI] [PubMed] [Google Scholar]
- Prior, T. W. , Krainer, A. R. , Hua, Y. , Swoboda, K. J. , Snyder, P. C. , Bridgeman, S. J. , Burghes, A. H. M. , & Kissel, J. T. (2009). A positive modifier of spinal muscular atrophy in the SMN2 gene. American Journal of Human Genetics, 85(3), 408–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochette, C. F. , Gilbert, N. , & Simard, L. R. (2001). SMN gene duplications and the emergence of the SMN2 gene ocurred in distinct hominids: SMN2 is unique to Homo sapiens. Human Genetics, 108(3), 255–266. [DOI] [PubMed] [Google Scholar]
- Ruhno, C. , McGovern, V. L. , Avenarius, M. R. , Snyder, P. J. , Prior, T. W. , Nery, F. C. , Muhtaseb, A. , Roggenbuck, J. S. , Kissel, J. T. , Sansone, V. A. , Siranosian, J. J. , Johnstone, A. J. , Nwe, P. H. , Zhang, R. Z. , Swoboda, K. J. , & Burghes, A. H. M. (2019). Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Human Genetics, 138, 241–256. 10.1007/s00439-019-01983-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler‐Botija, C. , Cuscó, I. , Caselles, L. , López, E. , Baiget, M. , & Tizzano, E. F. (2005). Implication of fetal SMN2 expression in type I SMA pathogenesis: Protection or pathological gain of function? Journal of Neuropathology and Experimental Neurology, 64(3), 215–223. [DOI] [PubMed] [Google Scholar]
- Sugarman, E. A. , Nagan, N. , Zhu, H. , Akmaev, V. R. , Zhou, Z. , Rohlfs, E. M. , Flynn, K. , Hendrickson, B. C. , Scholl, T. , Sirko‐Osadsa, D. A. , & Allitto, B. A. (2012). Pan‐ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72 400 specimens. European Journal of Human Genetics, 20(1), 27–32. 10.1038/ejhg.2011.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijzelaar, R. , Snetselaar, R. , Clausen, M. , Mason, A. G. , Rinsma, M. , Zegers, M. , Molleman, N. , Boschloo, R. , Yilmaz, R. , Kuilboer, R. , Lens, S. , Sulchan, S. , & Schouten, J. (2019). The frequency of SMN gene variants lacking exon 7 and 8 is highly population dependent. PLOS One, 14(7), 1–12. 10.1371/journal.pone.0220211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadman, R. I. , Jansen, M. D. , Stam, M. , Wijngaarde, C. A. , Curial, C. A. D. , Medic, J. , Sodaar, P. , Schouten, J. , Vijzelaar, R. , Lemmink, H. H. , van den Berg, L. H. , Groen, E. J. N. , & van der Pol, W. L. (2020). Intragenic and structural variation in the SMN locus and clinical variability in spinal muscular atrophy. Brain Communications, 2(2), 1–13. 10.1093/braincomms/fcaa075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. H. , Finkel, R. S. , Bertini, E. S. , Schroth, M. , Simonds, A. , Wong, B. , Aloysius, A. , Morrison, L. , Main, M. , Crawford, T. O. , & Trela, A. (2007). Consensus statement for standard of care in spinal muscular atrophy. Journal of Child Neurology, 22(8), 1027–1049. [DOI] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Research, 38(16), 1–7. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth, B. , Garbes, L. , & Riessland, M. (2013). How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Current Opinion in Genetics and Development, 23(3), 330–338. [DOI] [PubMed] [Google Scholar]
- Wu, X. , Wang, S. H. , Sun, J. , Krainer, A. R. , Hua, Y. , & Prior, T. W. (2017). A‐44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Human Molecular Genetics, 26(14), 2768–2780. 10.1093/HMG/DDX166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerres, K. , & Rudnik‐Schöneborn, S. (1995). Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Archives of Neurology, 52(5), 518–523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
All data and scripts used to generate the analyses of this paper are available upon request unless that the type of request compromises ethical standards or legal requirements. All variants reported in this article are now listed in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/).