

Abstract
Artificial temporal signaling systems, which mimic living out‐of‐equilibrium conditions, have made large progress. However, systems programmed by enzymatic reaction networks in multicomponent and unknown environments, and using biocompatible components remain a challenge. Herein, we demonstrate an approach to program temporal pH signals by enzymatic logic gates. They are realized by an enzymatic disaccharide‐to‐monosaccharide‐to‐sugar acid reaction cascade catalyzed by two metabolic chains: invertase‐glucose oxidase and β‐galactosidase‐glucose oxidase, respectively. Lifetimes of the transient pH signal can be programmed from less than 15 min to more than 1 day. We study enzymatic kinetics of the reaction cascades and reveal the underlying regulatory mechanisms. Operating with all‐food grade chemicals and coupling to self‐regulating hydrogel, our system is quite robust to work in a complicated medium with unknown components and in a biocompatible fashion.
Keywords: enzymatic logic gates, pH feedback, regulatory mechanisms, temporal signal, transient hydrogel
A tandem of a lag‐free YES gate and an enzymatic NAND gate with delay orchestrates a temporal pH feedback system: the YES gate lifts the system to a promoted state in seconds, and the NAND gate embedded with either invertase‐glucose oxidase or β‐galactosidase‐glucose oxidase chain drives it back to ground state with programmable lifetime. The system is very robust and able to use common foods as fuels.
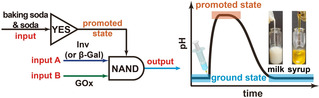
Introduction
Temporal signal production and transduction in living systems is an out‐of‐equilibrium process with high levels of complexity. It typically involves enzyme cascades (ECs) and enzymatic reaction networks (ERNs) to realize spatiotemporally controlled activation, inhibition, autocatalysis, and feedback and feedforward mechanisms. [1] Such ERNs allow for behavior [2] that is inaccessible by classical stimuli‐responsive artificial systems, which, in principle, use non‐processed external signals to switch between equilibrium states without temporal evolution. [3] The past decade has witnessed tremendous efforts in mimicking and applying the concept of natural out‐of‐equilibrium temporal signal systems into manmade self‐assembly and material system. [4] This has been achieved by temporal activation of building blocks, light stimulation, and chemical or enzymatic reaction networks coupled to molecules of interest. [5] Among them, ERNs are particularly attractive because of their selectivity, mild operation conditions and potential biocompatible nature.
Typically, such ERNs for transient generation of signals, self‐assemblies or material states use enzymes in an antagonistic fashion to activate and deactivate components on temporally pre‐designed pathways. Yet, the complexity of such artificial transient ERNs is still far away from concatenated ERNs in living systems. Therefore, a few reports have shifted focus to integrate ECs and more ERNs into temporal signal output system. [6] Apart from temporal signal output systems, ECs and ERNs have been extensively applied for Boolean logic gates for their potential ability in parallel computing by integrating numerous biomolecular units. [7] Although the construction of a real biocomputer based on enzymatic logic gates is far from realization, practical applications have been realized in biosensing, [8] where input signals are logically processed, generating a YES/NO output signal indicating the existence/absence of detected objects. This enzymatic signal processing system is essentially a signal‐switch device rather than inspired from transient out‐of‐equilibrium behavior.
Building on our and others’ work on pH‐feedback systems, [9] herein, we set out to bridge the gap between temporal signal output and enzymatic logic gates to construct a new type of temporal pH feedback system, in which the deactivating pathway is controlled by an enzymatic logic gate achieved through an EC. Here, we use the terminology of enzymatic logic gates to clearly express differences to earlier work on pH feedback systems by us and others, and to connect the reader to previous work on ECs. It should not be intended too closely as an approach for molecular computing that can be done on a much more sophisticated level, for instance by DNA computing. [10] We will first show the integration of an enzymatic disaccharide‐to‐monosaccharide‐to‐acid EC into a transient pH lifecycle system to program the lifetime of a transient alkaline pH output signal by regulating the EC kinetics. We will then investigate how enzymes involved in the logic gates interact and self‐regulate their activities and control the production of intermediates and output pH signal. Furthermore, we demonstrate robustness of the EC‐based logic gate‐programmed pH‐lifecycles by operating them with common multicomponent food products purchased in supermarket. We showcase robust, transient hydrogel system with programmable lifetimes adjusted by enzymatic logic gates. This translates the pH output signal to a macroscopically visible self‐assembly and material state, and critically underscores the robustness and (bio)compatibility of the developed networks.
Results and Discussion
Scheme 1 summarizes the concept of combining programmable pH lifecycles with tandem YES gate and enzymatic NAND gate. For the NAND gate, we build on two ECs, whereby Scheme 1 only shows the network for one system for clarity (see Scheme S1 in Supporting Information for the other network). Starting from the ground state (citric acid/sodium citrate (CA/Na3C) buffer containing the fuel, disaccharide), both the alkaline promoter (tris(hydroxymethyl)aminomethane (Tris) buffer) and enzymes (invertase (Inv) and glucose oxidase (GOx)) for the EC are injected. The Tris buffer quickly lifts the system to a promoted state, whereafter input A (Inv) catalyzes the hydrolysis of the chemical fuel sucrose to generate the intermediates fructose and glucose. Glucose is the substrate of input B (GOx). GOx catalyzes the conversion of glucose to gluconic acid δ‐lactone which spontaneously breaks down to gluconic acid (GLA). Meanwhile, GOx reduces oxygen dissolved in the system to hydrogen peroxide, that is regenerated to oxygen by a catalase (Cat). The output GLA lowers the pH and drives the system back to the ground state. This process realizes sucrose‐to‐glucose‐to‐GLA metabolic chain with a temporal pH lifecycle.
Scheme 1.
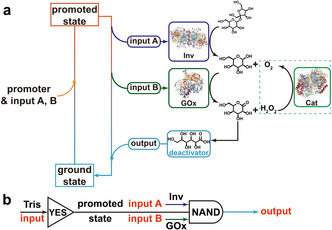
Transient alkaline pH profiles programmed by enzymatic logic gates. a) Adding a fast promoter lifts the system from ground state to promoted state. At promoted state, the dormant deactivator sucrose is catalytically hydrolyzed by Inv, generating intermediate products glucose and fructose. Glucose is further hydrolyzed to gluconic acid δ‐lactone by GOx in the presence of oxygen, which spontaneously hydrolyzes to GLA, driving the system back to ground state. b) The Inv‐GOx metabolic chain cooperates to form an enzymatic logic NAND gate, which together with Tris YES gate programs the temporal pH profile.
We define the ground state (acidic, pH <7) as 0 and promoted state as 1 (alkaline, pH >7), the addition of Tris is a lag‐free identity (YES) gate. We then define the presence of Inv and GOx in the system as input 1, the absence of which as input 0. The input signals Inv and GOx (1, 1) gradually convert the promoted state 1 to the ground state 0. In rest conditions without signals being present, the system stays at promoted state 1. Therefore, the EC works as a NAND gate, which is essentially the same as AND gate, [11] but with inverted output signal. Overall, the whole system is operated by a tandem of a lag‐free YES gate and a NAND gate with a delay to be programmed (Scheme 1 b). Figure 1 a and 1b demonstrate the proof of concept and the truth table of the temporal pH feedback system. The pH decreases from 8.5 to around 3.5 only in the presence of both Inv and GOx, namely, the output signal becomes 0 when the input signal (A, B) is (1, 1). Otherwise, the output signal remains at 1 and the pH only shows a minor decrease after extended (>500 min). The system stays in promoted state (Figure 1 a,b). In more detail, there is an obvious plateau where the alkaline pH almost does not change in the first tens of minutes in presence of the EC (1, 1). Afterwards, the pH dives down quickly to acidic regime. This delay is very different from the spontaneous hydrolysis of gluconic acid δ‐lactone (the intermediate produced by GOx) used as direct dormant deactivator in our previous report [9b] and is indicative of the EC‐based signal processing requiring the catalytic tandem conversion.
Figure 1.
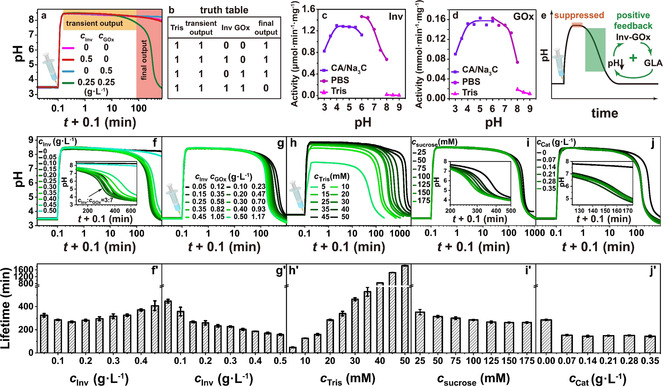
Programming the lifetime of transient alkaline states by Tris/Inv‐GOx logic gate. a) Proof of concept experiments: the pH profile only returns to acidic state in the presence of both Inv and GOx. Conditions: 100 mM sucrose, 1.5 mM CA/Na3C (pH 3.0; start), and 20 mM Tris (pH 8.8). b) Truth table generated in a). pH‐dependent activity of c) Inv and d) GOx. e) Positive feedback in the pH lifecycles programmed by Inv‐GOx EC. In the promoted state, the activities of both Inv and GOx are suppressed, however, not killed in high pH solution. As dormant deactivator (sucrose) is slowly transformed by Inv‐GOx, GLA gradually neutralizes the Tris buffer, driving the pH to the ground state. The decreasing pH enhances the activities of both enzymes and forms a positive feedback. f) Keeping c Inv+c GOx=0.5 g L−1, the t lf firstly decreases to a minimum when the c Inv is 0.15 g L−1 and then increases with c Inv. Conditions: 100 mM sucrose, 1.5 mM CA/Na3C (pH 3.0), and 20 mM Tris (pH 8.8). g) The t lf monotonically decreases when feeding more Inv and GOx. Conditions: c Inv/c GOx=3/7, 100 mM sucrose, 1.5 mM CA/Na3C (pH 3.0), and 20 mM Tris (pH 8.8). h) The t lf is effectively controlled by c Tris. Conditions: 0.15 g L−1 Inv, 0.35 g L−1 GOx, 100 mM sucrose, and 1.5 mM CA/Na3C (pH 3.0). i) Adding more chemical fuel can slightly shorten t lf. Conditions: 0.15 g L−1 Inv, 0.35 g L−1 GOx, 1.5 mM CA/Na3C (pH 3.0), and 20 mM Tris (pH 8.8). j) Adding Cat into the metabolic chain results in a shorter t lf. Conditions: Cat from 0 to 0.35 g L−1, 0.15 g L−1 Inv, 0.35 g L−1 GOx, 1.5 mM CA/Na3C (pH 3.0), and 20 mM Tris (pH 8.8). f′)–j′) The corresponding t lf of the transient alkaline states in (f)–(j). All data are an average of three measurements.
To better understand the EC, we measured the pH‐dependent activity of Inv and GOx (Figure 1 c, d; Supporting Information, Figures S1–S3). Different buffers need to be used to access different pH‐regimes. Both enzymes show very low activity at high pH (8–9), however, the activities rapidly increase to their maxima as the pH is decreased below 7, and keep constant until pH 4. At lower pH values, the activities drop to ca. 60 % of their maxima. Although measurements at the same pH points (pH 6.0 and 8.0) in different buffers show some buffer‐specific difference, overall, the activities of both enzymes show bell‐shaped curves. These bell‐shaped curves lead to a positive feedback loop and a self‐accelerating metabolic chain starting from alkaline regime (Figure 1 e).
After demonstrating the basic concept and positive feedback loop, we next elucidate the programming of the transient pH lifetimes (t lf) as a function of the ratio of Inv and GOx (Figure 1 f, f′). The t lf is defined from the midpoint values between the starting pH (state 0) and the highest pH of the transient promoted state 1. When keeping the total concentration of Inv and GOx constant (c Inv+c GOx=0.5 g L−1), Figure 1 f/1 f′ reveals that the t lf decreases from 325 to 268 min when the ratio c Inv/c GOx increases from 1/9 to 3/7, but then t lf increases to 407 min when inverting c Inv/c GOx up to 9/1. This basic screening reveals a non‐linear relationship, but more importantly it establishes a most suitable match of enzymes allowing a short t lf, by timely generation of enough glucose and conversion to GLA. While keeping this c Inv/c GOx ratio at 3/7, the t lf consistently decreases from 448 to 158 min when c Inv and c GOx are simultaneously increased from 0.05 and 0.12 g L−1 to 0.50 and 1.17 g L−1(Figure 1 g,g′). This is reasonable, because the transduction speed of sucrose is enhanced. An adjustment of the promoter concentration, c Tris, is the most effective way to program the t lf as the higher buffer capacity simply scavenges the acidic GLA product of the Inv‐GOx EC. At 0.15 Inv g L−1 and 0.50 g L−1 GOx, t lf is prolonged from 48 min to more than 30 h by a tenfold increase of c Tris from 5 to 50 mM (Figure 1 h,h′). As a comparison, increasing the feed amount of the chemical fuel sucrose from 25 to 175 mM only decreases the t lf from 352 to 263 min (Figure 1 i,i′). Considering that dissolved oxygen is limited (or has to diffuse from the vessel surface into the solution) and that the byproduct H2O2 is an inhibitor for GOx, [12] timely turning H2O2 back to oxygen should be favorable for a shorter t lf. Indeed, Figure 1 j and 1 j′ prove that adding a tiny amount of Cat can speed up the metabolic chain, but larger c Cat is not needed and the metabolic chain is also operational without Cat.
After having established a programmability of the pH lifecycles for the Inv‐GOx logic NAND, we now change to the second EC system that runs on a disaccharide found in animals, lactose. Therefore, we exchange Inv by β‐galactosidase (β‐Gal) to process the lactose into reaction products suitable for the downstream GOx. We firstly measured the pH‐dependent activity of β‐Gal (Figure 2 a; Supporting Information, Figure S4, S5), and found a narrower bell‐shaped activity curve. The behavior and programmability of the transient pH profiles programmed by the β‐Gal‐GOx EC follows similar consideration as for the Inv‐GOx one, as laid out in detail in Figure 2 b–d and the Supporting Information, Figure S6. In short, higher concentrations of β‐Gal at a constant c GOx lower the t lf due to faster conversion of the lactose. Similar to above, a symmetric increase of both enzymes decreases the t lf, whereas higher promoter concentrations c Tris drastically increase the t lf. A moderate influence of the fuel concentration, c lactose, is visible and the addition of Cat speeds up the lifecycle by reconversion of H2O2 into O2.
Figure 2.
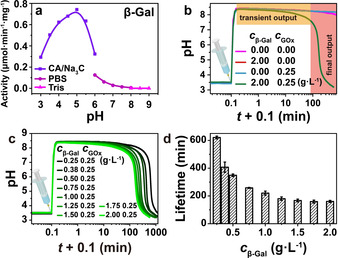
Programming the lifetime of transient alkaline states by Tris/β‐Gal‐GOx logic gate. a) pH‐dependent activity of β‐Gal; b) Proof of concept: the pH profile is only transient in presence of both β‐Gal and GOx. c) While keeping the c GOx as a constant of 0.25 g L−1, the t lf decreases as c β‐Gal is increased. d) The corresponding t lf of the transient alkaline states in (c). All data are an average of three measurements. Conditions: 100 mM lactose, 1.5 mM CA/Na3C (pH 3.0), and 20 mM Tris (pH 8.8). See also the Supporting Information, Figure S6.
So far we have established the concept of temporal programming of the pH feedback system by Inv‐GOx and β‐Gal‐GOx EC logic gates, discussed the positive feedback in the system, and programmed the lifetimes of the transient promoted state signal from less than 15 min to more than 1 day. Important remaining questions are, however, how the enzymatic kinetics of the EC within the pH lifecycle regulate the metabolic presence of chemical fuels, intermediates, and products, and how the dynamic pH changes affect the enzymatic kinetics.
To address these questions, we performed in‐depth high performance liquid chromatography (HPLC) analysis to quantify each component during the metabolic process (Supporting Information, Figures S7, S8). When starting from a plateau pH level around 8.5 (20 mM Tris, low activity regime of both enzymes), and when only in presence of Inv (0.5 g L−1) or β‐Gal (2.0 g L−1), the conversion of the substrates (sucrose and lactose, 100 mM) proceeds to a stable level of ca. 58 % sucrose and ca. 64 % lactose after 20 h, respectively. The time‐weighted derivatives of chemical fuels (d conversion/d time), that is, speed of conversion, gradually turn to flat as the substrate concentration decreases, and do not show any maxima or minima. However, the situation becomes critically different in presence of GOx that allows to further convert the intermediate (glucose) into GLA and concurrently lowers the pH (Figure 3; Supporting Information, Figure S9).
Figure 3.
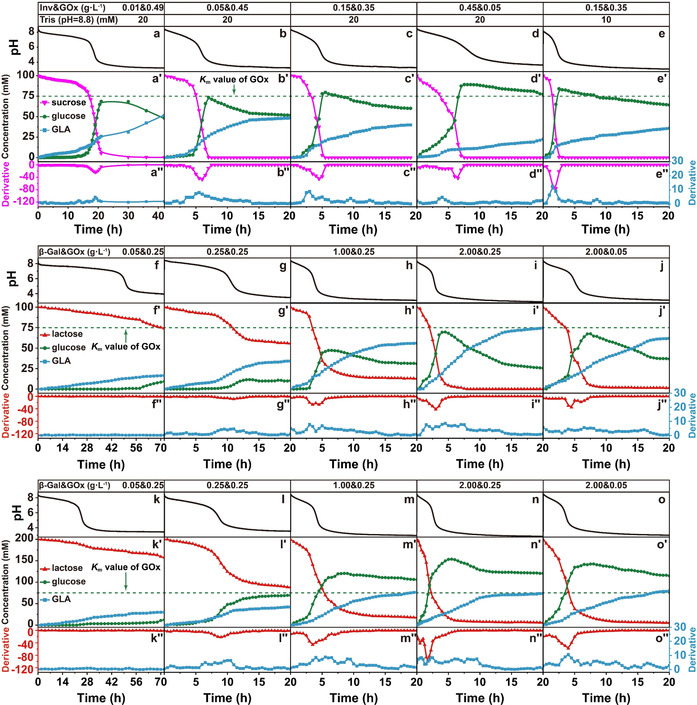
Temporal mapping of the major components of the metabolic chain during the temporal pH feedback system programmed by a)–e) Inv‐GOx and f)–o) β‐Gal‐GOx EC in the present of 1.5 mM CA/Na3C (pH 3.0) and varied Tris starting buffer. a)–a′′) Conditions: 0.01 g L−1 Inv, 0.49 g L−1 GOx, 100 mM sucrose, and 20 mM Tris (pH 8.8); b–b′′) Conditions: 0.05 g L−1 Inv, 0.45 g L−1 GOx, 100 mM sucrose, and 20 mM Tris (pH 8.8); c)–c′′) Conditions: 0.15 g L−1 Inv, 0.35 g L−1 GOx, 100 mM sucrose, and 20 mM Tris (pH 8.8); d)–d′′) Conditions: 0.45 g L−1 Inv, 0.05 g L−1 GOx, 100 mM sucrose, and 20 mM Tris (pH 8.8); e)–e′′) Conditions: 0.15 g L−1 Inv, 0.35 g L−1 GOx, 100 mM sucrose, and 10 mM Tris (pH 8.8); f–f′′) Conditions: 0.05 g L−1 β‐Gal, 0.25 g L−1 GOx, 100 mM lactose, and 20 mM Tris (pH 8.8); g)–g′′) Conditions: 0.25 g L−1 β‐Gal, 0.25 g L−1 GOx, 100 mM lactose, and 20 mM Tris (pH 8.8); h)–h′′) Conditions: 1.00 g L−1 β‐Gal, 0.25 g L−1 GOx, 100 mM lactose, and 20 mM Tris (pH 8.8); i)–i′′) Conditions: 2.00 g L−1 β‐Gal, 0.25 g L−1 GOx, 100 mM lactose, and 20 mM Tris (pH 8.8); j)–j′′) Conditions: 2.00 g L−1 β‐Gal, 0.05 g L−1 GOx, 100 mM lactose, and 20 mM Tris (pH 8.8); k)–k′′) Conditions: 0.05 g L−1 β‐Gal, 0.25 g L−1 GOx, 200 mM lactose, and 20 mM Tris (pH 8.8); l)–l′′) Conditions: 0.25 g L−1 β‐Gal, 0.25 g L−1 GOx, 200 mM lactose, and 20 mM Tris (pH 8.8); m)–m′′) Conditions: 1.00 g L−1 β‐Gal, 0.25 g L−1 GOx, 200 mM lactose, and 20 mM Tris (pH 8.8); n)–n′′) Conditions: 2.00 g L−1 β‐Gal, 0.25 g L−1 GOx, 200 mM lactose, and 20 mM Tris (pH 8.8); o)–o′′) Conditions: 2.00 g L−1 β‐Gal, 0.05 g L−1 GOx, 200 mM lactose, and 20 mM Tris (pH 8.8).
Looking first at the Inv‐GOx EC (at a constant total concentration of Inv and GOx, but varied c Inv/c GOx, Figure 3 a–e), the hydrolysis rates of both sucrose and glucose start to accelerate when the pH is decreased below 7, showing obvious minima and maxima in the derivatives of sucrose and GLA concentrations, respectively. This is a quantitative approximation for the existence of a positive feedback (see also Figure 1 f). The time point of this acceleration (location of maxima and minima) is brought forward from 17 to 2 h when the c Inv is increased from 0.01 to 0.15 g L−1 (Figure 3 a–c). Thereafter it is delayed to around 4 h by further increasing the c Inv to 0.45 g L−1 for the reason that insufficient GLA is produced because of less GOx present in the system and a concurrently slower pH decrease that would accelerate the sucrose conversion by Inv (Figure 3 d). In case less promoter c Tris is present (10 mM instead of 20 mM), the acceleration starts at 1 h, and thus even earlier because of the lower buffer capacity (Figure 3 e). It is worth noting that sucrose is 100 % hydrolyzed even in the case of 0.01 g L−1 Inv (Figure 3 a), which is due to the enhanced activity of Inv at lower pH and removal of glucose from the Inv equilibrium. During this wide range, sucrose is always totally converted to glucose, pushing the c glucose to a maximum, which can be higher than the Michaelis–Menten constant (K m) of GOx (which lies between 33 to 110 mM according to previous reports,[ 6a , 13 ] we take 75 mM as an average) when enough sucrose is fed (Figure 3 c–e; Supporting Information, Figure S10a,a′).
In comparison, the β‐Gal‐GOx EC shows a difference (Figure 3 f–f′′–3 o′–o′′): Not all lactose is hydrolyzed to glucose when the c β‐Gal is less than 2.00 g L−1. At 0.05 g L−1 β‐Gal (Figure 3 f–f′′), 74 % lactose is left after 72 h, and the highest c glucose is only 9.4 mM and thus far below the K m of GOx. No maximum or minimum is visible in the derivatives of c lactose and c GLA owing to low c β‐Gal. Hence there is no distinct metabolic chain‐induced positive feedback at this composition enabling an acceleration of the lactose conversion. The residual c lactose is lowered to ca. 56 % and 13 % after 20 h when the c β‐Gal is increased to 0.25 and 1.00 g L−1 (Figure 3 g,h). Thereupon, a distinct minimum and maximum start to appear in the derivatives of c lactose and c GLA after 10 h with 0.25 g L−1 β‐Gal, which appear earlier at around 4 and 3 h when the c β‐Gal is increased from 1.00 to 2.00 g L−1 (Figure 3 h,i). As anticipated, the appearance of the minima and maxima is delayed with decreasing c GOx (Figure 3 i–i′′ vs. Figure 3 j–j′′). When the c lactose is doubled to 200 mM (last row in Figure 3), we see a similar trend, that is, c glucose keeps below the K m and no acceleration is observed with low c β‐Gal (Figure 3 k–k′′), while the conversion of lactose gradually increases and positive feedback starts to emerge, pushing the c glucose to its maximum above the K m (Figure 3 l–l′′–3 o–o′′; Supporting Information, Figure S10b,b′).
Therefore, two kinds of regulatory mechanism exist in these EC systems (Figure 4): i) GOx operates far below K m because the chemical fuel (sucrose, lactose) cannot be converted fast enough by the first enzyme, and most glucose is timely hydrolyzed to GLA (Figure 4 a); ii) When enough chemical fuel is fed and largely converted to glucose, cglucose is pushed above K m, reaching to the maximum catalysis rate of GOx (v max, Figure 4 b). The Inv‐GOx EC largely works with the second regulatory mechanism owing to the wide pH range where Inv possesses its highest activity (Figure 1 c) and totally hydrolyzes sucrose to glucose, always pushing c glucose to levels near or above K m (Figure 4 c). By contrast, the β‐Gal‐GOx EC follows the first mode in the cases of low c β‐Gal because of the narrow pH range of maximum activity of β‐Gal (Figure 2 a). In a short time, the pH profile strides over this range to an acidic pH value where the activity of β‐Gal becomes low without generating additional glucose (Figure 3 f,g,k). Consequently, the initial fuel, lactose, remains to some extent in the system. However, given enough amount of β‐Gal and also increasing c lactose, all lactose can be converted to glucose in the narrow pH window, bringing the β‐Gal‐GOx chain to the second regulatory mechanism (Figure 4 d; Figure 3 m–o; Supporting Information, Figure S10b).
Figure 4.
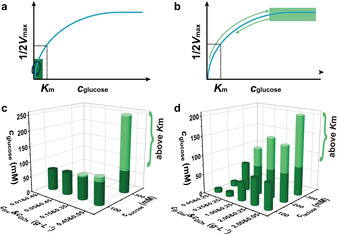
Two regulatory mechanisms operate within the EC metabolic chains: a) GOx operates far below K m, and b) c glucose is promoted above K m. Maximum c glucose intermediate in the temporal pH feedback systems programmed by c) Inv‐GOx and d) β‐Gal‐GOx ECs measured by HPLC (Supporting Information, Figures S9, S10). c) In Inv‐GOx EC, Inv always completely converts sucrose to glucose and pushes c glucose above K m of GOx (light green regime) when enough fuel is provided. d) Comparatively, in β‐Gal‐GOx EC, limited amount of glucose is produced in the case of low c β‐Gal, even though enough lactose is loaded, where the EC gate keeps working in a range below K m. The c glucose is only prompted above K m in the condition of high c β‐Gal and c lactose.
As stated earlier, natural temporal signal production and transduction takes place in an environment with high complexity such as cytoplasm, where multiple components are present. Hence, such ECs and ERNs need to precisely recognize input signal from noisy background. In contrast, current artificial temporal signal systems work in a very clean environment. We surmised that common foods containing the chemical fuels would in fact allow to produce such transient pH‐lifecycles using our established EC‐lifecycle systems. To this end, we replaced Tris promoter by supermarket‐grade soda and baking soda, sucrose by sugar beet syrup, and lactose by milk. The major “contaminant” components are listed in the Supporting Information, Figure S11.
Figure 5 a shows the pH profiles powered by different amounts of sugar beet syrup at c Inv=0.15 g L−1 and c GOx=0.35 g L−1 (promoter c(CO3 2−)=c(HCO3 −)=10 mM, whose pH is 9.9), where the c sucrose was calculated according to the nutrient information on package (Supporting Information, Figure S11). Comparing to the pH profiles fueled by sucrose (Figure 1), the profiles here have an additional plateau around pH 7.5, which is attributed to a buffering capacity of baking soda, soda, and, potentially, some unknown components in the syrup. Because of the additional buffer capacity, the pH profile supplied with 20 mM sucrose is not able to return to acidic state, but higher syrup concentrations successfully decrease the pH down to pH 3.7 (at 100 mM sucrose). The t lf of the transient pH signal decreases similarly from 468 to 362 min (Supporting Information, Figure S12a). The t lf can be further decreased to 106 min when the c Inv is increased from 0.15 to 0.75 g L−1 (Figure 5 b; Supporting Information, Figure S12b). Therefore, although running in a multicomponent solution with unknown components, our temporal pH feedback system operates rather well and exhibits an appealing robustness. Furthermore, the transient pH signal can be translated into macroscopic visible self‐assembly by introducing Fmoc‐ethylenediamine hydrochloride, a pH responsive gelator assembling into fibrils and forming hydrogels when pH >8.4. [14] Transient hydrogels are only accessible when both enzymes are present, and the lifetimes can be controlled following the pH lifecycles (Figure 5 c,d). Appropriate controls, that is, no enzymes present, or only one enzyme present, underscore that indeed the pH‐lifecycle is responsible for the behavior (Supporting Information, Figure S14a,b).
Figure 5.
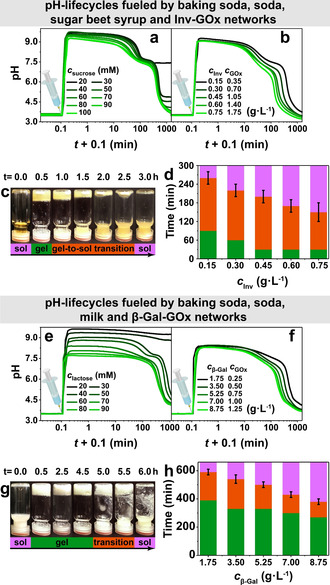
Programming the t lf with all‐food grade reagents. a) Transient pH profiles obtained by sugar beet syrup. Conditions: 0.15 g L−1 Inv, 0.35 g L−1 GOx, 1.5 mM CA/Na3C (pH 3.0, start), and c(CO3 2−)=c(HCO3 −)=10 mM. Other components as indicated. b) t lf decreases when for more Inv and GOx. Conditions: c Inv/c GOx=3:7, 100 mM sucrose, 1.5 mM CA/Na3C (pH 3.0), and c(CO3 2−)=c(HCO3 −)=10 mM. c) Autonomous sol‐to‐gel‐to‐sol transition of Fmoc‐ethylenediamine: Conditions: 0.60 g L−1 Inv, 1.40 g L−1 GOx, 100 mM sucrose, 1.5 mM CA/Na3C (pH 3.0), and c(CO3 2−)=c(HCO3 −)=10 mM. d) t lf of the transient gelation with different Inv and GOx concentrations. Conditions: c Inv/c GOx=3/7, 100 mM sucrose, 1 wt % Fmoc‐ethylenediamine hydrochloride, 1.5 mM CA/Na3C (pH 3.0), and c(CO3 2−)=c(HCO3 −)=10 mM. e) Transient pH profiles obtained by milk. Conditions: 1.75 g L−1 β‐Gal, 0.25 g L−1 GOx, 1.5 mM CA/Na3C (pH 3.0), and c(CO3 2−)=c(HCO3 −)=10 mM. Other components as indicated. f) Programmable t lf for increasing c β‐Gal and c GOx. Conditions: 60 mM lactose, 1.5 mM CA/Na3C (pH 3.0), and c(CO3 2−)=c(HCO3 −)=10 mM. g) Autonomous sol‐to‐gel‐to‐sol transition of Fmoc‐ethylenediamine: Conditions: 8.75 g L−1 β‐Gal, 1.25 g L−1 GOx, 60 mM lactose, 1.5 mM CA/Na3C (pH 3.0), and c(CO3 2−)=c(HCO3 −)=10 mM. h) t lf of the transient gelation with different β‐Gal and GOx concentrations. Conditions: c β‐Gal/c GOx=7/1, 60 mM lactose, 1 wt % Fmoc‐ethylenediamine hydrochloride, 1.5 mM CA/Na3C (pH 3.5), and c(CO3 2−)=c(HCO3 −)=10 mM. All the data are an average of three measurements.
We also used different amount of lactose‐containing cow milk to fuel the temporal pH feedback system programmed by β‐Gal‐GOx logic gate (Figure 5 e, 1.75 g L−1 β‐Gal, 0.25 g L−1 GOx, 1.5 mM CA/Na3C (pH 3.0), c(CO3 2−)=c(HCO3 −)=10 mM). Owing to more unknown components in the milk (Supporting Information, Figure S11b), higher concentrations are needed to drive the pH back to the acidic state (Figure 5 e), although this is on the expense of the height of the pH plateau. The accessible t lf decreases from ca. 792 to 376 min by increasing the feeding amount of milk (Supporting Information, Figure S13a), and t lf can be further decreased to 162 min by adding 8.75 g L−1 β‐Gal (Figure 5 f; Supporting Information, Figure S13b). A hydrogel system with programmable temporal self‐assembly is also accessible by coupling Fmoc‐ethylenediamine into the milk‐based pH lifecycles, and the t lf is programmable by variation of the enzyme concentration (Figure 5 g,h; Supporting Information, Figure S14c). Again, this underscores a good robustness of the pH‐lifecycle mechanisms and points to sufficient biocompatibility.
Conclusion
We have demonstrated temporal pH feedback system programmed by a tandem of a lag‐free Boolean YES gate and an enzymatic NAND gate with delay. The YES gate realized by adding Tris buffer quickly lifts the system from ground acidic state to a promoted alkaline state, resulting in a transient signal 1. Then the output signal 1 is driven back in an autonomous way to 0 by the NAND gate where an Inv‐GOx EC converts sucrose to GLA neutralizing Tris buffer and acidifying the system. We found an alternative β‐Gal‐GOx EC for the NAND gate, and showed that the lifetime of the transient promoted state 1 can be programmed from less than 15 min to more than 1 day by kinetically controlling both ECs. By measuring the pH‐dependent activity of Inv, β‐Gal and GOx, and by providing an in‐depth HPLC analysis of the metabolic components, it was possible to reveal detailed insights into the operation of a positive feedback arising from the metabolic chain. Different regulatory mechanisms of the pH feedback lifecycles operate with respect to the K m of the downstream GOx inside the ECs. The Inv‐GOx EC always tends to push intermediate glucose above the K m of GOx due its relatively wide pH‐dependent activity maximum. On the contrary, the β‐Gal‐GOx one leaves the glucose concentration below K m in the case of low c β‐Gal, and only pushes glucose above K m when enough β‐Gal and lactose are fed. Lactose can remain inside the system, which could be an interesting starting point to design new types of self‐oscillating reaction networks even outside continuously stirred tank reactors.
What is more, we demonstrated the robustness of our temporal pH feedback system programmed by enzymatic logic gates by using common foods to operate them. Even transient hydrogels can be obtained. This underscores compatibility with quite complex mixtures of ingredients and promises also robustness in biological application settings.
We foresee that the proposed concept provides new avenues for the development and application of temporal signal production and processing with enzymatic logic gates in environments with high levels of complexity (that is, multiple unknown components). In long perspective, the robust, artificial temporal signaling system tolerating unknown components may be a critical feature towards real‐life applications in materials systems with adaptive and autonomous behaviors.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge funding through the DFG WA‐3084/4‐2, the ERC Starting Grant “TimeProSAMat” (ID: 677960). X.F. acknowledges financial support from China Scholarship Council (CSC) and Deutscher Akademischer Austauschdienst (DAAD). We thank Wei Liu for partial snapshots. Open access funding enabled and organized by Projekt DEAL.
X. Fan, A. Walther, Angew. Chem. Int. Ed. 2021, 60, 11398.
References
- 1.
- 1a. Alam M. T., Olin-Sandoval V., Stincone A., Keller M. A., Zelezniak A., Luisi B. F., Ralser M., Nat. Commun. 2017, 8, 16018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Grüning N. M., Du D., Keller M. A., Luisi B. F., Ralser M., Open Biol. 2014, 4, 130232; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Vardi N., Chaturvedi S., Weinberger L. S., Proc. Natl. Acad. Sci. USA 2018, 115, E8803–E8810; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Page D. J., Thuret R., Venkatraman L., Takahashi T., Bentley K., Herbert S. P., Cell Rep. 2019, 27, 3139–3151; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Alon U., Nat. Rev. Genet. 2007, 8, 450–461; [DOI] [PubMed] [Google Scholar]
- 1f. Varusai T. M., Kolch W., Kholodenko B. N., Nguyen L. K., Mol. BioSyst. 2015, 11, 2750–2762; [DOI] [PubMed] [Google Scholar]
- 1g. Kochanowski K., Volkmer B., Gerosa L., van Rijsewijk B. R. H., Schmidt A., Heinemann M., Proc. Natl. Acad. Sci. USA 2013, 110, 1130–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Bisschop K., Mortier F., Etienne R. S., Bonte D., Proc. R. Soc. London Ser. B 2019, 286, 20190738; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Hao C., Gao Y., Wu D., Li S., Xu L., Wu X., Guo J., Sun M., Li X., Xu C., Adv. Mater. 2019, 31, 1903200. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Mellerup S. K., Wang S., Chem. Soc. Rev. 2019, 48, 3537–3549; [DOI] [PubMed] [Google Scholar]
- 3b. Merindol R., Walther A., Chem. Soc. Rev. 2017, 46, 5588–5619; [DOI] [PubMed] [Google Scholar]
- 3c. Walther A., Adv. Mater. 2020, 32, 1905111; [Google Scholar]
- 3d. Heinen L., Walther A., Soft Matter 2015, 11, 7857–7866. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Ragazzon G., Prins L. J., Nat. Nanotechnol. 2018, 13, 882–889; [DOI] [PubMed] [Google Scholar]
- 4b. Grzybowski B. A., Fitzner K., Paczesny J., Granick S., Chem. Soc. Rev. 2017, 46, 5647–5678; [DOI] [PubMed] [Google Scholar]
- 4c. Rieß B., Grötsch R. K., Boekhoven J., Chem 2020, 6, 552–578; [Google Scholar]
- 4d. Altay Y., Cao S., Che H., Abdelmohsen L. K., van Hest J. C., Biomacromolecules 2019, 20, 4053–4064; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4e. Deng J., Walther A., Adv. Mater. 2020, 32, 2002629. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Wang H., Wang Y., Shen B., Liu X., Lee M., J. Am. Chem. Soc. 2019, 141, 4182–4185; [DOI] [PubMed] [Google Scholar]
- 5b. della Sala F., Maiti S., Bonanni A., Scrimin P., Prins L. J., Angew. Chem. Int. Ed. 2018, 57, 1611–1615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1627–1631; [Google Scholar]
- 5c. Prins L. J., Cardona M. A., Chem. Sci. 2020, 11, 1518–1522; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Morrow S. M., Colomer I., Fletcher S. P., Nat. Commun. 2019, 10, 1011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Leira-Iglesias J., Tassoni A., Adachi T., Stich M., Hermans T. M., Nat. Nanotechnol. 2018, 13, 1021–1027; [DOI] [PubMed] [Google Scholar]
- 5f. Tamate R., Ueki T., Shibayama M., Yoshida R., Soft Matter 2017, 13, 4559–4568; [DOI] [PubMed] [Google Scholar]
- 5g. Semenov S. N., Wong A. S., Van Der Made R. M., Postma S. G., Groen J., Van Roekel H. W., De Greef T. F., Huck W. T., Nat. Chem. 2015, 7, 160–165; [DOI] [PubMed] [Google Scholar]
- 5h. Yucknovsky A., Mondal S., Burnstine-Townley A., Foqara M., Amdursky N., Nano Lett. 2019, 19, 3804–3810; [DOI] [PubMed] [Google Scholar]
- 5i. Singh D. P., Choudhury U., Fischer P., Mark A. G., Adv. Mater. 2017, 29, 1701328; [DOI] [PubMed] [Google Scholar]
- 5j. Kundu P. K., Samanta D., Leizrowice R., Margulis B., Zhao H., Börner M., Udayabhaskararao T., Manna D., Klajn R., Nat. Chem. 2015, 7, 646–652; [DOI] [PubMed] [Google Scholar]
- 5k. Ke H., Yang L. P., Xie M., Chen Z., Yao H., Jiang W., Nat. Chem. 2019, 11, 470–477; [DOI] [PubMed] [Google Scholar]
- 5l. Wang W., Giltinan J., Zakharchenko S., Sitti M., Sci. Adv. 2017, 3, e1602522; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5m. Pim W., Mater. Horiz. 2015, 2, 198–202; [Google Scholar]
- 5n. Bhangu S. K., Bocchinfuso G., Ashokkumar M., Cavalieri F., Nanoscale Horiz. 2020, 5, 553–563; [DOI] [PubMed] [Google Scholar]
- 5o. Heinen L., Walther A., Sci. Adv. 2019, 5, eaaw0590; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5p. Luo M., Xuan M., Huo S., Fan J., Chakraborty G., Wang Y., Zhao H., Herrmann A., Zheng L., Angew. Chem. Int. Ed. 2020, 59, 17250–17255; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5q. Che H., Zhu J., Song S., Mason A. F., Cao S., Pijpers I. A., Abdelmohsen L. K., van Hest J. C., Angew. Chem. Int. Ed. 2019, 58, 13113–13118; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13247–13252; [Google Scholar]
- 5r. Deng J., Walther A., J. Am. Chem. Soc. 2020, 142, 21102–21109; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5s. Deng J., Walther A., Chem 2020, 6, 3329–3343; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5t. Deng J., Walther A., Nat. Commun. 2020, 11, 3658; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5u. Deng J., Bezold D., Jessen H. J., Walther A., Angew. Chem. Int. Ed. 2020, 59, 12084–12092; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12182–12190; [Google Scholar]
- 5v. Kubota R., Makuta M., Suzuki R., Ichikawa M., Tanaka M., Hamachi I., Nat. Commun. 2020, 11, 3541; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5w. Dowari P., Das S., Pramanik B., Das D., Chem. Commun. 2019, 55, 14119–14122; [DOI] [PubMed] [Google Scholar]
- 5x. Vialetto J., Anyfantakis M., Rudiuk S., Morel M., Baigl D., Angew. Chem. Int. Ed. 2019, 58, 9145–9149; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9243–9247. [Google Scholar]
- 6.
- 6a. Nijemeisland M., Abdelmohsen L. K., Huck W. T., Wilson D. A., van Hest J. C., ACS Cent. Sci. 2016, 2, 843–849; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Helwig B., van Sluijs B., Pogodaev A. A., Postma S. G., Huck W. T., Angew. Chem. Int. Ed. 2018, 57, 14065–14069; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14261–14265; [Google Scholar]
- 6c. Bánsági T., Taylor A. F., Life 2019, 9, 63; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Pogodaev A. A., Lap T. T., Huck W. T., ChemSystemsChem 2020, 3, e2000033; [Google Scholar]
- 6e. Dubuc E., Pieters P. A., van der Linden A. J., van Hest J. C., Huck W. T., de Greef T. F., Curr. Opin. Biotechnol. 2019, 58, 72–80; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6f. Sorrenti A., Leira-Iglesias J., Markvoort A. J., de Greef T. F., Hermans T. M., Chem. Soc. Rev. 2017, 46, 5476–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Katz E., ChemPhysChem 2019, 20, 9–22. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Grattieri M., Minteer S. D., ACS Sens. 2018, 3, 44–53; [DOI] [PubMed] [Google Scholar]
- 8b. Amine A., Arduini F., Moscone D., Palleschi G., Biosens. Bioelectron. 2016, 76, 180–194. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Heuser T., Weyandt E., Walther A., Angew. Chem. Int. Ed. 2015, 54, 13258–13262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13456–13460; [Google Scholar]
- 9b. Heuser T., Steppert A. K., Molano Lopez C., Zhu B., Walther A., Nano Lett. 2015, 15, 2213–2219; [DOI] [PubMed] [Google Scholar]
- 9c. Heuser T., Merindol R., Loescher S., Klaus A., Walther A., Adv. Mater. 2017, 29, 1606842; [DOI] [PubMed] [Google Scholar]
- 9d. Heinen L., Walther A., Chem. Sci. 2017, 8, 4100–4107; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9e. Fan X., Walther A., Angew. Chem. Int. Ed. 2021, 60, 3619–3624; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 3663–3668; [Google Scholar]
- 9f. Heinen L., Heuser T., Steinschulte A., Walther A., Nano Lett. 2017, 17, 4989–4995; [DOI] [PubMed] [Google Scholar]
- 9g. Jee E., T. Bánsági Jr. , Taylor A. F., Pojman J. A., Angew. Chem. Int. Ed. 2016, 55, 2127–2131; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2167–2171; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9h. Bohner B., T. Bánsági Jr. , Tóth Á., Horváth D., Taylor A. F., Angew. Chem. Int. Ed. 2020, 59, 2823–2828; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 2845–2850; [Google Scholar]
- 9i. Man T., Ji W., Liu X., Zhang C., Li L., Pei H., Fan C., ACS Nano 2019, 13, 4826–4833; [DOI] [PubMed] [Google Scholar]
- 9j. Che H., Cao S., van Hest J. C., J. Am. Chem. Soc. 2018, 140, 5356–5359; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9k. Che H., Buddingh“ B. C., van Hest J. C., Angew. Chem. Int. Ed. 2017, 56, 12581–12585; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12755–12759. [Google Scholar]
- 10.
- 10a. Mailloux S., Gerasimova Y. V., Guz N., Kolpashchikov D. M., Katz E., Angew. Chem. Int. Ed. 2015, 54, 6562–6566; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6662–6666; [Google Scholar]
- 10b. Song T., Eshra A., Shah S., Bui H., Fu D., Yang M., Mokhtar R., Reif J., Nat. Nanotechnol. 2019, 14, 1075–1081. [DOI] [PubMed] [Google Scholar]
- 11. Motornov M., Zhou J., Pita M., Gopishetty V., Tokarev I., Katz E., Minko S., Nano Lett. 2008, 8, 2993–2997. [DOI] [PubMed] [Google Scholar]
- 12. Wong C. M., Wong K. H., Chen X. D., Appl. Microbiol. Biotechnol. 2008, 78, 927–938. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Zhang Y., Tsitkov S., Hess H., Nat. Catal. 2018, 1, 276–281; [Google Scholar]
- 13b. Gibson Q. H., Swoboda B. E., Massey V., J. Biol. Chem. 1964, 239, 3927–3934. [PubMed] [Google Scholar]
- 14. Panja S., Adams D. J., Chem. Commun. 2019, 55, 47–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary