

Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease associated with mitochondrial oxidative stress. Mitochondrial reactive oxygen species (mtROS) are important for cell homeostasis by regulating mitochondrial dynamics. Here, we show that IPF BAL cells exhibited increased mitochondrial biogenesis that is, in part, due to increased nuclear expression of peroxisome proliferator‐activated receptor‐ɣ (PPARɣ) coactivator (PGC)‐1α. Increased PPARGC1A mRNA expression directly correlated with reduced pulmonary function in IPF subjects. Oxidant‐mediated activation of the p38 MAPK via Akt1 regulated PGC‐1α activation to increase mitochondrial biogenesis in monocyte‐derived macrophages. Demonstrating the importance of PGC‐1α in fibrotic repair, mice harboring a conditional deletion of Ppargc1a in monocyte‐derived macrophages or mice administered a chemical inhibitor of mitochondrial division had reduced biogenesis and increased apoptosis, and the mice were protected from pulmonary fibrosis. These observations suggest that Akt1‐mediated regulation of PGC‐1α maintains mitochondrial homeostasis in monocyte‐derived macrophages to induce apoptosis resistance, which contributes to the pathogenesis of pulmonary fibrosis.
Keywords: mitochondrial biogenesis, monocyte‐derived macrophages, PGC‐1α, pulmonary fibrosis
Abbreviations
- BAL
bronchoalveolar lavage
- CS
citrate synthase
- MDM
monocyte‐derived macrophage
- mtDNA
mitochondrial DNA
- mtROS
mitochondrial reactive oxygen species
- nDNA
nuclear DNA
- RAM
resident alveolar macrophage
- TFAM
mitochondrial transcription factor A
1. INTRODUCTION
Pulmonary fibrosis affects millions of people worldwide and has a prevalence that is increasing to over 80 per 100 000 persons. 1 , 2 The disease is devastating with an average life expectancy of 3‐5 years after diagnosis for certain forms of pulmonary fibrosis, such as idiopathic pulmonary fibrosis (IPF), and only 20% of patients survive 5 years after diagnosis. 3 The incidence and mortality from IPF is increasing worldwide, with an estimated 40 000 lives lost in the United States each year, 1 , 4 which is similar to the number of deaths from breast cancer. 5 Approved therapies for IPF have limited efficacy in that either drug is associated with improvements in quality of life or mortality. 6 , 7 Thus, understanding the basic molecular mechanisms that regulate fibrosis may uncover novel therapeutic modalities.
In fibrotic disorders of the lung, including IPF, lung macrophages have a decisive role in fibrotic repair of the injured lung. Lung macrophages play an integral function in the normal resolution of organ injury, but also contribute to the pathogenesis of pulmonary fibrosis by initiating an innate immune response and by generating reactive oxygen species (ROS), particularly mitochondrial ROS. 8 , 9 Considerable heterogeneity exists in the population of lung macrophages during fibrosis; however, monocyte‐derived macrophages have recently been identified to be the critical myeloid cell in lung fibrosis. 10 , 11 , 12 , 13 Lung remodeling during pulmonary fibrosis is poorly understood, but changes in mitochondrial ROS (mtROS) and mitochondrial dynamics in monocyte‐derived macrophages are emerging as critical determinants of fibrotic repair.
Our previous work showed that IPF BAL cells have increased mtROS and increased mitophagy. 14 Mitochondria are dynamic organelles that undergo fission and fusion. Mitochondrial fission is required for mitophagy to occur, as mitophagy is attenuated in cells with decreased mitochondrial fission. 15 Additionally, mtROS, including H2O2 generation, play an important role in cell signaling by regulating mitochondrial biogenesis. 16 Mitophagy, which can be induced by mtROS, 14 is important for the quality control of the mitochondrial population suggesting the interdependence of mitophagy and mitochondrial biogenesis is critical for cell homeostasis. 17 The removal of mitochondria by mitophagy is typically followed by biogenesis, especially during cellular stress when biogenesis increases to meet the energy demand of the cell. 18 This cycle of mitophagy and mitochondrial biogenesis modulates cell fate in models of sepsis and cardiomyopathy. 19 , 20
The peroxisome proliferator‐activating receptor‐ɣ co‐activator (PGC)‐1α, a master transcriptional regulator of mitochondrial biogenesis, regulates the expression of nuclear respiratory factors (NRFs) and mitochondrial transcription factor A (Tfam) to induce the expression of many nuclear and mitochondrial genes. 21 Additionally, PGC‐1α induces the production of antioxidant enzymes to reduce ROS levels; however, PGC‐1α expression is increased in response to ROS and bioenergetic demands. 22 Here, we show that Akt1‐mediated mtROS generation increased the nuclear localization and activation of PGC‐1α in fibrotic monocyte‐derived macrophages thereby promoting mitochondrial biogenesis. Mice harboring a conditional deletion of Ppargc1a in monocyte‐derived macrophages or mice administered mdivi‐1, a chemical inhibitor of mitochondrial fission, were protected from bleomycin‐induced pulmonary fibrosis by diminishing mitochondrial biogenesis and inducing macrophage apoptosis. This is translationally relevant as lung macrophage Ppargc1a gene expression in humans with IPF is directly associated with a reduction in lung function. These observations suggest that PGC‐1α is critical for mitochondrial homeostasis in monocyte‐derived macrophages and is directly associated to the pathogenesis of pulmonary fibrosis.
2. MATERIALS AND METHODS
2.1. Human subjects
We obtained human BAL cells as previously described 23 from normal subjects and IPF patients under an approved protocols (300001124 and 01670) by the Human Subjects Institutional Review Boards of UAB and the Birmingham VAMC. Human BAL specimens were used for research only. All subjects provided prior written consent to participate in the study. Normal volunteers had to meet the following criteria: (i) age between 18 and 65 years; (ii) no history of cardiopulmonary disease or other chronic disease; (iii) no prescription or nonprescription medication except oral contraceptives; (ⅳ) no recent or current evidence of infection; and (v) lifetime nonsmoker. Idiopathic pulmonary fibrosis (IPF) subjects had to meet the following criteria: (i) FVC (forced vital capacity) at least 50% predicted; (ii) current nonsmoker; (iii) no recent or current evidence of infection; (iv) evidence of restrictive physiology on pulmonary function tests; and (v) usual interstitial pneumonia on high resolution chest computed tomography. Fiberoptic bronchoscopy with bronchoalveolar lavage (BAL) was performed after subjects received local anesthesia. Three sub‐segments of the lung were lavaged with five 20‐mL aliquots of normal saline, and the first aliquot in each was discarded. The percentage of macrophages was determined by Wright‐Giemsa stain and varied from 90% to 98%.
2.2. Mice
Animal experiments were approved by UAB Institutional Animal Care and Use Committee under protocols 21076 and 21149 and were performed in accordance with NIH guidelines. WT C57BL/6J mice were purchased from JAX Labs, and Akt1−/−Lyz2‐cre, Akt2−/−Lyz2‐cre, and Akt1/2−/−Lyz2‐cre mice were generated by selective disruption of Akt1, Akt2, or both genes in the cells of the granulocyte/monocyte lineage as previously described. 14 Akt1fl/fl, Akt2 fl/fl, and Akt1/2 fl/fl mice were used as controls unless otherwise noted. Ppargc1a−/−Csf1rMeriCreMer mice were generated by crossing Ppargc1afl/fl mice (B6N.129(FVB)‐PPargc1atm2.1Brsp/J, Jackson Laboratory) with tamoxifen inducible Mer‐iCre‐Mer driven by the Csf1r promoter (FVB‐Tg(Csf1r‐cre/Esr1*)1Jwp/J, stock number 019098, Jackson Laboratory). Ppargc1a was selectively and conditionally deleted from the monocytes/macrophages following administration of tamoxifen chow for 14 days prior to bleomycin exposure and continued until euthanization. Ppargc1afl/fl mice were used as controls. From 8‐ to 12‐week‐old male and female mice were intratracheally administered 1.75 U/kg of bleomycin or saline, as a negative control, after being anesthetized with 3% isoflurane using a precision Fortec vaporizer. Alveolar epithelial cells (AECs) were isolated as previously described. 14
2.3. Administration of mdivi‐1
WT mice were administered the small molecule Drp1 inhibitor, Mdivi‐1 (50 mg/kg body weight/day, Cayman Chemical) 10 days after saline or bleomycin exposure by intraperitoneal injection and continued daily for 11 days.
2.4. Cell culture
Human monocyte (THP‐1) and mouse alveolar macrophage (MH‐S) cell lines were obtained from American Type Culture Collection. Macrophages were maintained in RPMI 1640 media with 10% fetal bovine serum and penicillin/streptomycin supplements. All experiments were conducted in RPMI containing 0.5% serum.
2.5. Quantitative real time PCR
Total RNA was isolated, reverse transcribe, and quantitative real‐time PCR was performed as described previously. 14 Data were calculated by the cycle threshold (ΔΔCT) method, normalized to β‐actin or HPRT, and expressed in arbitrary units. The following primer sets were used: human 12s ribosomal RNA: 5′‐CGA TGG TGC AGC CGC TAT TA‐3′ and 5′‐AGA TGT GGC GGG TTT TAG GG‐3′; human citrate synthase, 5′‐CTC AGG ACG GGT TGT TCC AG‐3′ and 5′‐CCA GTA CAC CCA ATG CTC GT‐3′; human COX IV, 5′‐GGA AGA CGA GGG ATG CAC AG‐3′ and 5′‐AAA TAC GTA GAC CCG CTG CC‐3′; human PGC‐1α, 5′‐GAG TGT GTG CTC TGT GTC ACT and 5′‐CAG CAC ACT CGA TGT CAC TCC AT; human Tfam, 5′‐GAA CAA CTA CCC ATA TTT AAA GCT CA‐3′ and 5′‐GAA TCA GGA AGT TCC CTC CA‐3′; mouse citrate synthase, 5′‐CTG CTC CAG TAC TAT GGC ATG A‐3′ and 5′‐TTA AAG GCC CCT GAA ACA AAA CA‐3′; mouse COX IV, 5′‐CCG TCT TGG TCT TCC GGT TG‐3′ and 5′‐TGG AAG CCA ACA TTC TGC CA‐3′; mouse NRF1, 5′‐CCA TGG CCC TCA ACA GTG AA ‐3′ and 5′‐GCT GTC CGA TAT CCT GGT GG‐3′; mouse Nrf2, 5′‐AGA TGA CCA TGA GTC GCT TGC‐3′ and 5′‐CCA GCG AGG AGA TCG ATG AG‐3′; mouse PGC‐1α, 5′‐GGC AGT AGA TCC TCT TCA AGA TC and 5′‐TCA CAC GGC GCT CTT CAA TTG; mouse Tfam, 5′‐CCA AAA AGA CCT CGT TCA GC‐3′ and 5′‐CCA TCT GCT CTT CCC AAG AC‐3′.
2.6. Plasmids, transfections, small interfering RNA (siRNA), and luciferase assays
The pcDNA‐MKK6(Glu), pCMV‐Flag‐p38DN (generous gifts from Dr. Roger Davis, University of Massachusetts), and pUSE‐Akt1CA plasmids have been previously described. 24 , 25 Constitutively active Akt2 (Akt2CA) plasmid was a gift from Dr. William Sellers, purchased from Addgene #9016. Human PGC‐1α gene expression was evaluated using a luciferase reporter plasmid a gift from Dr. Bruce Spiegelman purchased from Addgene #8887. 26 pCDNA4‐PGC‐1α (PGC‐1αWT) plasmid was a gift from Dr. Toren Finkel, purchased from Addgene# 10974. 27 A Kozak sequence was introduced using Q5 Site‐Directed Mutagenesis Kit (BioLabs# E0552S). Four different site mutations of PGC‐1α (T262A, S265A, T298A, and S570A) were performed using the Q5 Site‐Directed Mutagenesis Kit. The correct reading frame and sequence were verified by the Heflin Center Genomics Core at UAB. Cells were transfected using X‐treme GENE 9 Transfection Reagent (Roche Applied Scientific) according to the manufacturer’s protocol. Cells were transfected with 100 nM scramble, human Akt1, human or mouse PGC‐1α, human Parkin, or human Tfam siRNA duplex (IDT) utilizing Dharmafect 2 (Thermo Scientific) according to manufacturer’s protocol. Eight hours after transfection, media was replaced, and cells were allowed to recover for 24‐72 hours. Renilla and firefly luciferase activity was determined in cell lysates using the Dual Luciferase reporter assay kit (Promega) and normalized to control (firefly).
2.7. Quantification of mitochondrial DNA
Purification of DNA was performed using DNeasy Blood & Tissue kit (QIAGEN). The ratio of mitochondrial to nuclear DNA was assessed using the Human Mitochondrial DNA Monitoring Primer Set Ratio kit (Takara Bio) and the mtDNA/nDNA ratio in mice was assessed by amplifying Nd2 gene of the mitochondrial genome and Gapdh gene of the nuclear genome. Primer sequences were as follows: Gapdh: 5′‐CCT GCA CCA CCA ACT GCT TAG‐3′and 5′‐GTG GAT GCA GGG ATG ATG TTC; Nd2: 5′‐CCC ATT CCA CTT CTG ATT ACC‐3′ and 5′‐ATG ATA GTA GAG TTG AGT AGC G‐3′.
2.8. Flow cytometry
Macrophages were stained with MitoTracker green (50 nM) for mitochondrial localization. Data were collected with a Becton Dickinson LSR‐II flow cytometer using FACS Diva software (BD Biosciences) and further analyzed with FlowJo software (version 8.5.2; TreeStar). BAL cells were blocked with 1% BSA containing TruStain fcX (anti‐mouse CD16/32) antibody (101319; BioLegend), followed by staining with antibodies. Antibodies used: Rat anti‐mouse CD45‐PE (12‐0451‐82; eBiosciences), LIVE Dead‐eflour506 (65‐0866; Invitrogen), Rat anti‐mouse CD11b‐APC‐Cy7 (101225; BioLegend), anti‐mouse CD64‐PE‐Cy7 (139313; BioLegend), Rat anti‐mouse Ly6G‐AF700 (561236; BD), Rat anti‐mouse Siglec F‐APC (155507; BioLegend), Rat anti‐mouse Ly6C: eflour450 (48‐5932‐82; Invitrogen), Rat anti‐mouse MHC II‐PerCP‐Cy5.5 (562363; BD), and Dead Cell Apoptosis Kit with Annexin V FITC and PI (V13242, Molecular Probes). Hierarchical gating strategy was used to represent the resident alveolar macrophages as CD45+CD11b+/−Ly6G−CD64+Ly6c−Siglec Fhi and monocyte‐derived macrophages as CD45+CD11b+/−Ly6G−CD64+Ly6c−Siglec Flow. Data were acquired on FACSAria II or LSR II (BD Biosciences) using BD FACS DIVA software (version 8.0.1). Data were analyzed using FlowJo (FlowJo LLC) software (Version 10.5.0).
2.9. Transmission electron microscopy
BAL cells were fixed in 2.5% paraformaldehyde and 2.5% glutaraldehyde in Sorenson’s phosphate buffer as previously described. 14 Cells were processed and sectioned with a diamond knife (Diatome, Electron Microscopy Sciences) at 70‐80 nm and sections were placed on copper mesh grids. Sections were stained with uranyl acetate and lead citrate for contrast and viewed on a Tecnai Twin 120 kv TEM (FEI).
2.10. Confocal imaging
Macrophages were stained with MitoTracker green for mitochondrial determination. BAL cells were fixed with 4% paraformaldehyde in PBS for 45 minutes at room temperature, followed by permeabilization for 3 minutes and incubated with PBS containing 5% BSA for 45 minutes. BAL cells were incubated with hamster anti‐CD11c‐FITC (117305; BioLegend) or rat anti‐CD11b (101201; BioLegend) and goat anti‐rat‐IgG‐FITC (3030‐02; Southern Biotech) and counterstained with DAPI. Nikon A1 Confocal was utilized for imaging.
2.11. Oxygen consumption rate determination (OCR) and proton leak
OCR was determined using a Seahorse XF24 bioanalyzer (Seahorse Bioscience) as previously described. 28 Briefly BAL cells were subjected to OCR measurement with sequential additions of: oligomycin (0.25 μg/mL), carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (0.5 μM), and antimycin A/rotenone (4/1 μM). Proton leak was determined by determining the difference of minimum rate measurement after oligomycin injection from non‐mitochondrial respiration.
2.12. Determination of H2O2 generation
H2O2 production was determined fluorometrically. Mitochondria were isolated as described below and incubated in phenol‐red free Hanks’ balanced salt solution supplemented with 6.5 mM glucose, 1 mM HEPES, 6 mM sodium bicarbonate, 1.6 mM pHPA, and 0.95 μg/mL HRP. Fluorescence of pHPA‐dimer was measured using a spectrofluorometer at excitation of 320 nm and emission of 400 nm. 14
2.13. Isolation of nucleus
Nuclear isolation was performed by resuspending cells in a lysis buffer (10 mM HEPES, 10 mM KCL, 2 mM MgCl2, and 2 mM EDTA) for 15 minutes on ice. Nonidet P‐40 (10%) was added to lyse the cells, and the cells were centrifuged at 4°C at 14 000 rpm. The nuclear pellet was resuspended in an extraction buffer (50 mM HEPES, 50 mM KCL, 300 mM NaCl, 0.1 mM EDTA, and 10% glycerol) for 20 minutes on ice. After centrifuging at 4°C at 14 000 rpm, the supernatant was collected as nuclear extract. 29
2.14. Isolation of mitochondria and cytoplasm
Mitochondria were isolated by lysing the cells in mitochondria buffer containing 10 mM Tris, pH 7.8, 0.2 mM EDTA, 320 mM sucrose, and protease inhibitors. Lysates were homogenized using a Kontes Pellet Pestle Motor and centrifuged at 2000 ×g for 8 minutes at 4°C. The supernatant was removed and incubated at 4°C and the pellet was lysed, homogenized, and centrifuged again. The two supernatants were pooled and centrifuged at 12 000 ×g for 15 minutes at 4°C. The pellet was washed in the mitochondrial buffer twice and then resuspended in mitochondria buffer without sucrose. 23
2.15. Immunoblot analysis
Primary antibodies used: Akt1 (2938), Akt2 (5239), Akt1/2 (9272), GSK‐3α/β (5676), LC3 (4108S), Lamin A/C (2032), NFR1 (12381), PGC‐1α (2178), phospho‐p38 MAPK (9215), phospho‐Akt1 (9018), phospho‐Akt2 (8599), phospho‐DRP1 (Ser616) (4494), phospho‐GSK‐3α/β (9331), Parkin (2132S), Tfam (7495S), VDAC (4866) (Cell Signaling); β‐actin (A5441) (Sigma); PINK1 (ab23707), Mfn‐2 (ab124773) (Abcam); Drp1 (611113) (BD Biosciences); phospho‐PGC‐1α (S571) (AF6650) (R&D Systems); p38 (sc‐7972) and Tom20 (sc‐11415) (Santa Cruz); SPC (AB3786) (Millipore).
2.16. ELISA
TGF‐β1 and TNF‐α expression were determined in BALF using ELISA kits (R&D Systems) according to the manufacturer’s instructions.
2.17. ATP production
Mitochondrial and total ATP was quantitated by Cell Titer‐Glo Luminescent Cell Viability Assay kit (Promega) according to manufacturer’s protocol.
2.18. TUNEL assay
Detection of apoptosis was determined in macrophages and in BAL cells by TUNEL analysis In Situ Cell Death Detection Kit, TMR red (Sigma) according to the manufacturer’s instructions and as previously described. 25
2.19. Caspase‐3 activity analysis
Caspase‐3 activity was measured using EnzChek Caspase‐3 Assay Kit Number 2 (Molecular Probes) according to the manufacturer’s protocol. Cells were lysed in 1× lysis buffer, subjected to a freeze‐thaw cycle, centrifuged to remove cellular debris, and loaded into individual microplate wells. The 2× reaction buffer with substrate was immediately added to the samples, and fluorescence was measured (excitation/emission 496/520 nm). A supplied inhibitor was used as a negative control in all experiments. 25
2.20. Hydroxyproline analysis
Lung tissues were dried to a stable weight and acid hydrolyzed with 6N HCl for 24 hours at 110°C. Samples were resuspended in 1.5 mL phosphate‐buffered saline followed by incubation at 60°C for 1 hours. Samples were centrifuged at 13 000 rpm, and the supernatant was taken for hydroxyproline analysis by using chloramine‐T. Hydroxyproline concentration was normalized to the dry weight of the tissue. 14
2.21. Materials
Bleomycin was obtained from the University of Alabama at Birmingham Animal Resources Program. α‐Ketogluterate, p‐Hydroxyphenyl acetic acid (pHPA), HRP, LY294002, and MG‐132 were purchased from Sigma Chemical Company. Mdivi‐1 was purchased from Cayman Chemical. MitoTempo was purchased from Enzo.
2.22. Statistics
Statistical comparisons were performed using a Student’s t test when only two groups of data are presented, or one‐way ANOVA with a Tukey’s post hoc test or and two‐way ANOVA followed by Bonferroni post‐test when multiple data groups are presents. All statistical analysis was expressed as ±S.E.M. and P < .05 was considered to be significant. GraphPad Prism statistical software was used for all analysis.
3. RESULTS
3.1. IPF BAL cells have increased mitochondrial biogenesis
Because dysfunctional mitochondria in BAL cells from IPF subjects are removed by mitophagy, 14 we questioned if mitochondria maintain functionality through biogenesis and rapid turnover. As PGC‐1α is a critical regulator of mitochondrial biogenesis, we found that the nuclear localization of PGC‐1α was significantly increased in IPF subjects (Figure 1A,B), and IPF BAL cells expressed nearly fivefold more PPARGC1A mRNA than normal subjects (Figure 1C). Additionally, IPF subjects had a significant increase in TFAM, Citrate Synthase (CS), and greater than 200‐fold more 12s rRNA compared to normal subjects (Figure 1C). Furthermore, the ratio of mtDNA to nuclear DNA was increased 10‐fold in IPF BAL cells (Figure 1D). Although mitochondrial fusion is another form of biogenesis, Mfn‐2 was significantly reduced in IPF subjects (Figure S1A). The impact of the increased mitochondrial turnover resulted in a marked increase in mitochondrial ATP content (Figure S1B). These data suggest that IPF BAL cells have increased PGC‐1α to maintain mitochondrial and cellular homeostasis.
FIGURE 1.
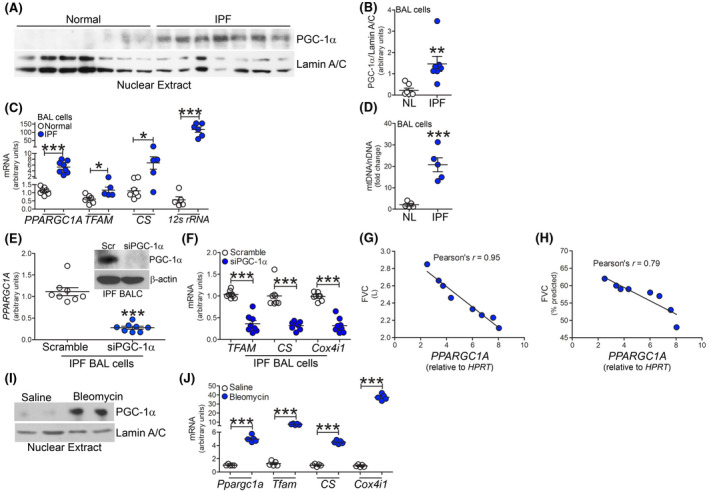
IPF BAL cells have increased mitochondrial biogenesis. A, Nuclear immunoblot analysis and B, quantification of PGC‐1α in BAL cells from normal and IPF subjects (n = 7). mRNA expression in BAL cells from normal (n = 5‐7) or IPF subjects (n = 5‐8) for C, PPARGC1A, TFAM, CS, and 12s rRNA. D, Ratio of mtDNA to nuclear DNA in normal and IPF BAL cells (n = 5). E, mRNA analysis of PPARGC1A in IPF BALC with silencing of PGC‐1α (n = 8). Inset, PGC‐1α immunoblot analysis. F, TFAM, CS, and COX4I1 expression (n = 8) in IPF BALC with silencing of PGC‐1α. Pearson’s correlation of PPARGC1A mRNA in BAL cells and G, FVC or H, FVC % predicted in IPF subjects (n = 5). I, Nuclear immunoblot of PGC‐1α in BAL cells from saline or bleomycin‐exposed mice at 21 days. J, mRNA analysis Ppargc1a, Tfam, Cs, and Cox4i1 mRNA in BAL cells from saline or bleomycin‐exposed mice at 21 days (n = 5). *P < .05; **P < .001; ***P < .0001. Values shown as mean ± S.E.M. Two‐tailed t‐test statistical analysis was utilized for B‐F, J. Pearson’s coefficient was used for G, H
Demonstrating the importance of PGC‐1α mediated mitochondrial biogenesis, we silenced PGC‐1α in IPF BAL cells ex vivo (Figure 1E). Mitochondrial biogenesis was abrogated in IPF BAL cells with PGC‐1α silenced (Figure 1F). Moreover, lower forced vital capacity (FVC) and FVC % predicted were highly correlated with increased PPARGC1A mRNA expression in BAL cells (Figure 1G,H). These data suggest that PPARGC1A expression in macrophages is associated with fibrosis progression in IPF.
To determine the biological significance of the observations in human subjects, we investigated mitochondrial biogenesis in mice with pulmonary fibrosis. BAL cells isolated from bleomycin‐injured mice showed nuclear localization of PGC‐1α and a five‐fold increase in Ppargc1a mRNA (Figure 1I,J). BAL cells isolated from bleomycin‐injured mice showed a significant increase in Tfam, Cs, and greater than 40‐fold more Cox4i1 mRNA (Figure 1J). These results suggest PGC‐1α is critical for a fibrotic phenotype.
3.2. Akt1 promotes mitochondrial biogenesis
Because macrophage Akt1 activation modulates mitophagy and pulmonary fibrosis, we asked if mitochondrial biogenesis was also regulated by Akt1. Macrophages expressing constitutively active Akt1 (Akt1CA) had greater mitochondrial fluorescence (Figure 2A), and these observations were confirmed by flow cytometry (Figure 2B). Silencing Akt1 or treating macrophages with LY294002, an inhibitor of PI3K, the primary activator of Akt, had the opposite effect (Figure S2A,B).
FIGURE 2.
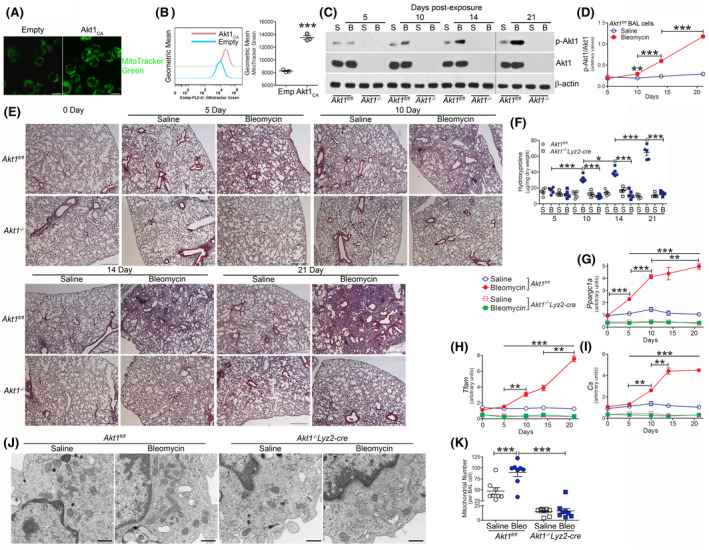
Akt1 promotes mitochondrial biogenesis. A, MitoTracker green staining of MH‐S cells transfected with empty (Emp) (n = 4) or Akt1CA (n = 4). Scale bars represent 10 μm. B, Flow cytometry of transfected THP‐1 cells (n = 3). BAL cells were isolated at indicated days after saline (S) (n = 5/time point) or bleomycin (B, bleo) exposure (n = 5/time point) from Akt1fl/fl or Akt1−/−Lyz2‐cre mice (Akt1−/−). C, Immunoblot analysis and D, quantification of p‐Akt1 (n = 5). E, Representative histology of lung sections with Masson’s trichome staining (n = 5). Scale bar = 500 μm. F, Hydroxyproline analysis of lung homogenates (n = 5). mRNA analysis of G, Ppargc1a (n = 5), H, Tfam (n = 5), and I, Cs (n = 5). J, TEM analysis of BAL cells (n = 8) from Akt1fl/fl and Akt1−/−Lyz2‐cre mice. Scale bar represents 1 μm. K, Total number of mitochondria per BAL cell (n = 8). *P < .05; **P < .001; ***P < .0001. Values shown as mean ± S.E.M. Two‐tailed t test statistical analysis was utilized for B. One‐way ANOVA followed by Tukey’s multiple comparison test was utilized for D, F, K. Two‐way ANOVA followed by Bonferroni post‐test was utilized for G‐I. ***with open line in G‐I refer to Akt1fl/fl bleomycin vs Akt1fl/fl saline at 5, 10, 14, and 21 days and Akt1fl/fl bleomycin vs Akt1−/−Lyz2‐cre bleomycin at 5, 10, 14, and 21 days. Bracketed lines in G‐I is comparing Akt1fl/fl bleomycin at indicated timepoints
Because Akt1 activation regulated mitochondrial dynamics, we asked if this effect was fundamental to the pathogenesis of pulmonary fibrosis. Bleomycin‐induced injury resulted in an increase in the total number of BAL cells in Akt1fl/fl and mice harboring a conditional deletion of Akt1 in cells of monocyte lineage (Akt1−/−Lyz2‐cre) (Figure S2C). Monocytic cells were the primary cell type in the BAL from both strains (Figure S2D‐F). BAL cells from Akt1fl/fl mice showed significantly increased Akt1 activation 10 days after bleomycin, and Akt1 activation further increased in a time‐dependent manner (Figure 2C,D). The deletion of Akt1 was not present in neutrophils (Figure S2G). Activation of Akt1 correlated with an increase in collagen deposition in lungs from Akt1fl/fl mice. Masson’s trichrome staining of lung sections showed fibrosis was evident 10 days after bleomycin exposure and increased in a time‐dependent manner (Figure 2E). In contrast, Akt1−/−Lyz2‐cre mice were protected from bleomycin‐induced fibrosis at all timepoints. Hydroxyproline content in the lungs of Akt1fl/fl mice showed increased levels 10 days after bleomycin‐induced injury that increased in a time‐dependent manner through 21 days (Figure 2F). Akt1−/−Lyz2‐cre mice showed similar hydroxyproline levels as saline controls. The increase in Akt1 activation promoted an increase in genes associated with mitochondrial biogenesis. Ppargc1a expression was significantly increased 5 days after bleomycin, while Tfam and Cs (Figure 2G‐I) were greater at 10 days post‐exposure. Ppargc1a, Tfam, and Cs continued to increase in a time‐dependent manner. In contrast, Akt1−/−Lyz2‐cre BAL cells showed reduced expression of these genes below the Akt1fl/fl saline controls regardless of bleomycin‐induced injury. These results suggest that Akt1 contributes to the PGC‐1α‐mediated increase in mitochondria biogenesis.
To determine the effect of Akt1 expression visually, we used transmission electron microscopy to examine the abundance of mitochondria. Bleomycin‐induced injury increased the number of mitochondria in BAL cells isolated from Akt1fl/fl mice compared to macrophages isolated from saline controls (Figure 2J). Conversely, BAL cells isolated from Akt1−/−Lyz2‐cre mice had a significant reduction in the number of mitochondria below the Akt1fl/fl saline control (Figure 2K). These findings provide further evidence that Akt1 mediates mitochondrial biogenesis in macrophages in a PGC‐1α‐dependent manner.
Macrophage mitochondrial redox responses that utilize oxidative phosphorylation are crucial for pulmonary fibrosis development. 14 , 30 Because oxygen consumption is correlated to bioenergetics, we determined that the oxygen consumption rate (OCR) in BAL cells from bleomycin‐injured Akt1fl/fl mice was significantly increased compared to saline‐exposed mice (Figure S2H,I). OCR in bleomycin‐injured Akt1−/−Lyz2‐cre mice was no different than the saline‐exposed Akt1fl/fl mice. This difference in OCR was associated with a significant reduction in mitochondrial ATP content in Akt1−/−Lyz2‐cre macrophage mitochondria (Figure S2J) suggesting that fibrotic macrophages optimize energy production during fibrosis.
3.3. Monocyte‐derived macrophages are resistant to apoptosis
Macrophages in chronic disease exhibit apoptosis resistance, and their prolonged survival is generally associated with disease progression. 14 We determined if modulating PGC‐1α reversed the apoptosis resistance seen in these cells. Silencing PGC‐1α in IPF BAL cells ex vivo significantly increased caspase‐3 activity (Figure 3A). Moreover, Akt1fl/fl BAL cells displayed reduced TUNEL staining 10 days after bleomycin injury, and the resistance to apoptosis remained consistent through 21 days (Figure 3B,C). Akt1−/−Lyz2‐cre BAL cells had a significant increase in TUNEL staining compared to Akt1fl/fl saline controls, while no difference in TUNEL staining was observed at day 0 between the strains (Figure S3A). These observations suggest that the fate of macrophages in pulmonary fibrosis may require sufficient mitochondrial turnover and mitochondrial biogenesis.
FIGURE 3.
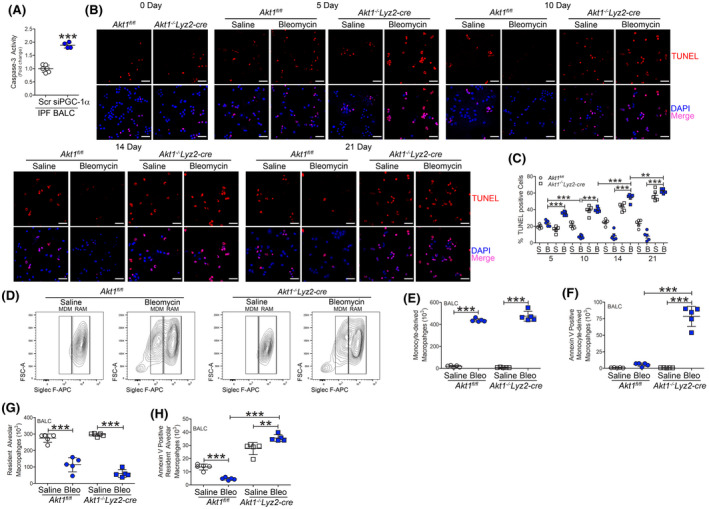
Monocyte‐derived macrophages are resistant to apoptosis. A, Caspase‐3 activity (n = 4‐6) in IPF BALC with silencing of PGC‐1α. BAL cells were isolated at indicated days after saline (n = 5/time point) or bleomycin exposure (n = 5/time point) from Akt1fl/fl or Akt1−/−Lyz2‐cre mice. B, TUNEL staining (n = 5) and C, quantification (n = 5). Scale bar represents 40 μm. BAL cells were isolated 21 days after exposure. D, Representative flow cytometry plots of monocyte‐derived macrophages (MDM, CD45+CD11b+/−Ly6G−CD64+Ly6c−Siglec Flow) and resident alveolar macrophages (RAM, CD45+CD11b+/−Ly6G−CD64+Ly6c−Siglec Fhi) from saline or bleomycin‐exposed Akt1fl/fl and Akt1−/−Lyz2‐cre mice. Total cell number of (E) MDM (n = 5), F, annexin V positive MDM (n = 5), (G) RAM (n = 5), and H, annexin V positive RAM from Akt1fl/fl and Akt1−/−Lyz2‐cre BAL (n = 5). **P < .001; ***P < .0001. Values shown as mean ± S.E.M. Two‐tailed t‐test statistical analysis was utilized for A. One‐way ANOVA followed by Tukey’s multiple comparison test was utilized for C, E‐H
Because monocyte‐derived macrophages have been implicated in promoting pulmonary fibrosis, 10 , 12 , 13 we asked which monocyte/macrophage cell subset displayed apoptosis. Bleomycin‐induced injury significantly increased monocyte‐derived macrophages in Akt1fl/fl and Akt1−/−Lyz2‐cre mice (Figures 3D,E and S3B). Recruited monocyte‐derived macrophages from bleomycin‐injured Akt1−/−Lyz2‐cre mice showed 8‐fold increase in annexin V staining compared to bleomycin‐injured Akt1fl/fl mice (Figure 3F). These results were analyzed visually by confocal analysis in CD11b expressing cells with TUNEL staining (Figure S3C). Mice receiving bleomycin showed a significant reduction in the number of resident alveolar macrophages (Figure 3D,G). Resident alveolar macrophages from bleomycin‐injured mice displayed apoptosis resistance compared to saline‐exposed, while Akt1−/−Lyz2‐cre mice showed significantly greater annexin V staining than saline‐exposed Akt1fl/fl mice (Figure 3H). Taken together, these data show that bleomycin‐injured mice show enhanced recruitment of monocyte‐derived macrophages that are resistant to apoptosis, suggesting this may have a critical role in fibrotic repair.
Active TGF‐β1 in BALF from bleomycin‐injured Akt1fl/fl mice was significantly increased at 10 days, and it continued to increase in a time‐dependent manner (Figure S3D). The reverse was seen with the anti‐fibrotic cytokine TNF‐α, as it was increased at 10 days in BALF from Akt1−/−Lyz2‐cre mice (Figure S3E). These data suggest that Akt1 activation in monocyte‐derived macrophages is critical for a fibrotic phenotype in mice, emerging as early as 10 days after bleomycin injury.
3.4. Pulmonary fibrosis does not require Akt2
Because the Akt isoform, Akt2, was shown to inhibit PGC‐1α activity 31 and to determine if our findings were specific for the Akt1 isoform, we generated mice with a conditional deletion of another prominent Akt isoform, Akt2 (Akt2−/−Lyz2‐cre). BAL cells isolated from Akt2−/−Lyz2‐cre mice had a complete absence of p‐Akt2 and Akt2 expression, whereas Akt1 activation was present in the Akt2−/−Lyz2‐cre mice similar to that in Akt2fl/fl mice after bleomycin injury (Figure S4A). The recombination occurred only in monocytic cells. Type II AECs isolated from Akt2fl/fl and Akt2−/−Lyz2‐cre mice had similar expression of p‐Akt2 and Akt2 (Figure S4B). Akt2fl/fl and Akt2−/−Lyz2‐cre mice had similar numbers of BAL cells after bleomycin exposure, and monocytic cells were the predominant cell type in the BAL from both strains (Figure S4C,D).
Because mice with a conditional deletion of Akt1 showed increased apoptosis, we determined if this also occurred in Akt2‐deficient mice. BAL cells from bleomycin‐injured Akt2−/−Lyz2‐cre mice showed a significant reduction in TUNEL staining compared to saline‐exposed, and the percent of TUNEL positive cells was similar to bleomycin‐injured Akt2fl/fl mice (Figure 4A,B). These results were confirmed by determining caspase‐3 activity (Figure 4C). Ppargc1a gene expression (Figure 4D) was essentially equivalent in Akt2fl/fl and Akt2−/−Lyz2‐cre mice as were mitochondrial biogenesis genes with increases in Tfam, Cs, and Cox4i1 mRNA expression after bleomycin injury (Figure 4E‐G). Furthermore, mitochondrial ATP production in BAL cells from Akt2−/−Lyz2‐cre mice was similar to Akt2fl/fl mice (Figure S4E).
FIGURE 4.
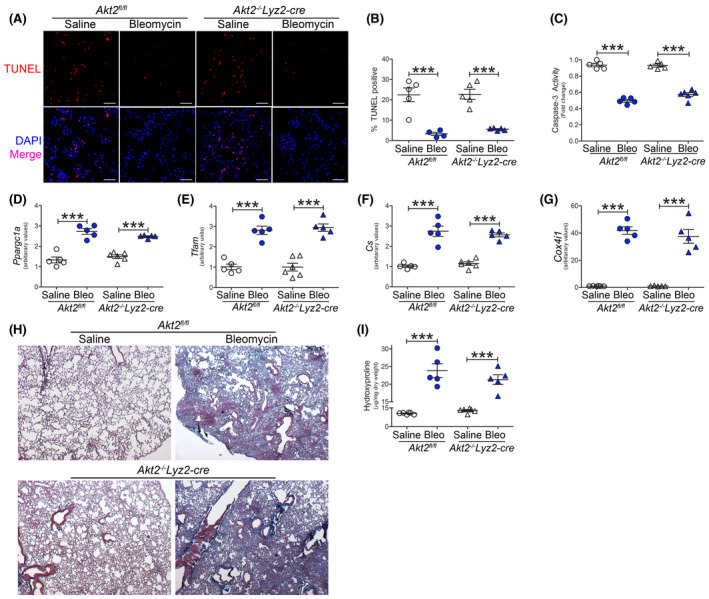
Pulmonary fibrosis does not require Akt2. Akt2fl/fl or Akt2−/−Lyz2‐cre mice were exposed to saline or bleomycin (Bleo). BAL cells were isolated 21 days later. A, TUNEL staining (n = 5), B, TUNEL quantification (n = 4‐5), and C, caspase‐3 activity (n = 5). Scale bar represent 50 μm. mRNA analysis of D, Ppargc1a (n = 5‐6), E, Tfam (n = 5‐6), F, Cs (n = 5‐6), and G, Cox4i1 expression (n = 5‐6) in BAL cells. H, Histology of lung sections with Masson’s trichrome staining (n = 5) and I, hydroxyproline analysis of homogenized lung (n = 5‐6). ***P < .0001. Values shown as mean ± S.E.M. One‐way ANOVA followed by Tukey’s multiple comparison test was utilized
The conditional deletion of Akt2 had no effect on the lung parenchyma in saline‐exposed mice; however, the Akt2−/−Lyz2‐cre mice developed extensive bleomycin‐induced fibrosis as seen in Akt2fl/fl mice (Figure 4H). The histological findings were validated biochemically showing that Akt2fl/fl and Akt2−/−Lyz2‐cre mice had similar lung collagen content (Figure 4I). These data suggest that Akt1, rather than Akt2, regulates PGC‐1α during fibrotic repair. Moreover, these observations indicate that Akt2 activation in monocyte‐derived macrophages is not required for bleomycin‐induced pulmonary fibrosis.
Because mice harboring a conditional deletion of Akt1 are protected from fibrosis and mice with a deletion of Akt2 are not protected, we generated mice with a conditional deletion of both Akt isoforms (Akt1/2−/−Lyz2‐cre) to determine the effect on fibrosis. BAL cells isolated from Akt1/2−/−Lyz2‐cre mice had no expression of Akt1 and Akt2 (Figure S4F). There was a similar increase in the number of BAL cells in bleomycin‐exposed Akt1/2fl/fl and Akt1/2−/−Lyz2‐cre mice (Figure S4G), and monocytic cells were the predominant cell type in BALF from both strains (Figure S4H). AECs isolated from Akt1/2fl/fl and Akt1/2−/−Lyz2‐cre mice had similar expression of Akt1 and Akt2, confirming recombination occurred in monocytes/macrophages (Figure S4I).
Gene expression of Ppargc1a in isolated BAL cells from bleomycin‐injured Akt1/2−/−Lyz2‐cre mice was significantly reduced compared to Akt1/2fl/flmice (Figure S4J). The reduced Ppargc1a expression correlated with significantly lower Tfam, Cs, Cox4i1, and mtDNA levels (Figure S4K‐N) in BAL cells. Masson’s trichrome staining of lung sections revealed that Akt1/2−/−Lyz2‐cre mice were protected from bleomycin‐induced fibrosis (Figure S4O). These results were confirmed by hydroxyproline assay (Figure S4P). These data suggest Akt2 is not activated in monocyte‐derived macrophages during fibrosis; however, these observations also indicate that Akt1‐mediated Ppargc1a expression enhances mitochondrial homeostasis to mediate the pathogenesis of pulmonary fibrosis.
3.5. Mitochondrial ROS activates PGC‐1α via phosphorylation of p38 MAPK
Akt2 phosphorylates PGC‐1α at serine 570 leading to PGC‐1α inhibition 31 ; however, Akt1 has not been shown to regulate activation or inhibition of PGC‐1α. Overexpression of Akt1CA resulted in increased nuclear localization of PGC‐1α (Figure 5A). PPARGC1A promoter activity was nearly fourfold higher in cells transfected with Akt1CA (Figure 5B), whereas macrophages expressing Akt2CA showed a significant reduction in PPARGC1A promoter activity below the empty control. We confirmed that Akt2CA expressing macrophages increased phosphorylation of PGC‐1α (S570) and reduced nuclear localization of PGC‐1α, while this response was the opposite in Akt1CA expressing macrophages (Figures 5C and S5A). Akt1CA increased activation of p38 (Figure 5C), which was associated with PGC‐1α activation, as well as gene activation inducing mitochondrial biogenesis. These findings were absent in cells expressing Akt2CA.
FIGURE 5.
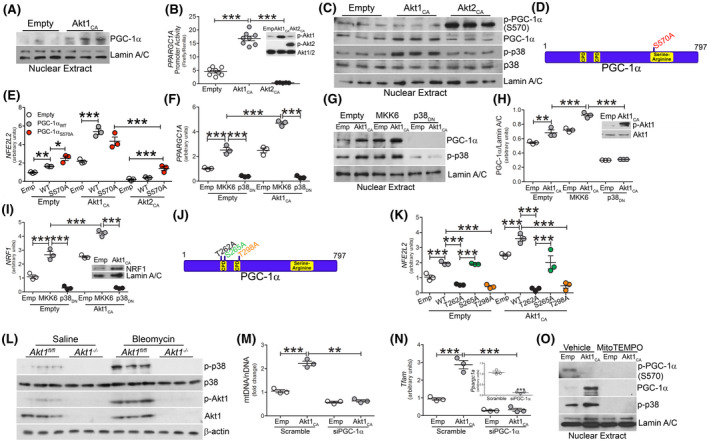
Akt1‐mediated mtROS activates PGC‐1α via phosphorylation of p38 MAPK. A, Nuclear immunoblot analysis for p‐PGC‐1α (S570) and PGC‐1α in transfected macrophages. B, PPARGC1A promoter activity in transfected macrophages (n = 8). Inset, p‐Akt1 and p‐Akt2 immunoblot. C, Nuclear immunoblot analysis in transfected macrophages. D, Schematic of Akt2 phosphorylation and mutation sites on PGC‐1α. E, NFE2L2 mRNA analysis in macrophages expressing empty (emp), Akt1C(A) or Akt2CA together with PGC‐1αWT (WT) or PGC‐1αS570A (S570A) (n = 3). Macrophages were transfected with empty or Akt1CA together with MKK6(Glu) or p38DN. F, PPARGC1A mRNA expression (n = 3), G, nuclear immunoblot analysis, H, quantification of PGC‐1α expression (n = 3); inset, Akt1 immunoblot analysis, I, NRF1 mRNA expression (n = 3). J, Schematic of p38MAPK phosphorylation and mutation sites on PGC‐1α. K, NFE2L2 mRNA analysis of macrophages expressing empty or Akt1CA together with PGC‐1αWT (WT), PGC‐1αT262A (T262A), PGC‐1αS265A (S265A), PGC‐1αT298A (T298A) (n = 3). L, Immunoblot analysis of BAL cells from saline or bleomycin‐exposed Akt1fl/fl and Akt1−/−Lyz2‐cre mice. Macrophages were transfected with scramble or PGC‐1α siRNA and empty or Akt1CA. M, Mitochondrial DNA content (n = 3) and N, Tfam mRNA expression (n = 3). Inset, PGC‐1α mRNA expression. O, Nuclear immunoblot analysis in macrophages expressing empty or Akt1CA and treated with vehicle or mitoTEMPO (10 µM, overnight). **P < .001; ***, P < .0001. Values shown as mean ± S.E.M. One‐way ANOVA followed by Tukey’s multiple comparison test was utilized. CPD = Cdc4 phosphodegron motifs
Validating that Akt1 does not regulate the phosphorylation of PGC‐1α at Ser570, we mutated serine 570 to alanine (PGC‐1αS570A) (Figure 5D). No expression of p‐PGC‐1α (S570) was detected in macrophages expressing Akt1CA with either WT or the S570A mutant (Figure S5B); however, macrophages expressing Akt2CA had increased PGC‐1α phosphorylation at Ser570 that was absent in cells expressing the S570A mutant.
PGC‐1α is known to augment NRF1 and Nrf2 expression to mediate transcription of mtDNA genes, including Tfam. 32 This effect was relevant to PGC‐1α‐dependent genes. Macrophages expressing Akt2CA and PGC‐1αS570A showed a partial increase in Nrf2 gene expression, whereas PGC‐1αS570A did not alter Nrf2 in macrophages expressing Akt1CA (Figure 5E). Nrf2 expression was similar with Akt1CA and PGC‐1αWT, and when Akt1CA and PGC‐1αWT were expressed together there was greater than five‐fold increase over control levels. Phosphorylation at Ser570 by Akt2 was linked to PGC‐1α degradation as treatment with MG‐132, a proteasome inhibitor, resulted in PGC‐1α accumulation in the nucleus of Akt2CA‐expressing macrophages (Figure S5C). The results suggest Akt1 and Akt2 have divergent effects in stabilizing and activating PGC‐1α.
Because the phosphorylation of PGC‐1α by the p38 MAPK enhances its activity, 33 we questioned if Akt1 regulation of PGC‐1α was p38‐dependent. Macrophages expressing constitutively active MKK6(Glu), the upstream activator of p38, increased PGC‐1α gene expression to a similar level as Akt1CA (Figure 5F). The nuclear localization of PGC‐1α was increased by constitutively active Akt1 and MKK6(Glu) alone, and the nuclear content was significantly increased when they were expressed together (Figure 5G,H). Both gene expression and nuclear localization were abrogated in macrophages expressing a dominant negative p38 (p38DN). Similar results were seen with expression of NRF1, TFAM, CS, and COX4I1 (Figures 5I and S5D‐F).
Mutation of the p38 MAPK phosphorylation sites on PGC‐1α (T262→A262, S265→A265, and T298→A298) (Figure 5J) confirmed that Akt1 regulated the expression and activation of PGC‐1α by activating p38. The T262A and T298A mutants significantly reduced NFE2L2 gene expression in macrophages expressing Akt1CA, whereas there was no alteration in NFE2L2 expression in cells expressing T265A (Figure 5K). The p38‐mediated increase of PGC‐1α activation by Akt1 also resulted in increased phosphorylation of dynamin‐related protein (p‐Drp) 1 (S616) (Figure S5G). The activation of p38 MAPK was biologically relevant, as p38 was activated in monocyte‐derived macrophages isolated from bleomycin‐injured Akt1fl/fl mice, while its expression was absent in Akt1−/−Lyz2‐cre mice (Figure 5L). The p38 MAPK was activated in Akt2−/−Lyz2‐cre mice similar to Akt2fl/fl mice (Figure S5H).
Because Akt1 regulated PGC‐1α, we questioned if Akt1‐mediated mitochondrial biogenesis required PGC‐1α. Silencing PGC‐1α in macrophages expressing Akt1CA significantly reduced mitochondrial DNA content, as well as Tfam (Figure 5M,N), Nrf1, Nfe2l2, Cs, and Cox4i1 (Figure S5I‐L). These results suggest that the Akt1‐mediated regulation of PGC‐1α required p38 MAPK activation and phosphorylation of PGC‐1α at Thr262 and Thr298.
Because mitochondrial biogenesis can be regulated by mtROS, we determined that Akt1 promoted mtROS generation via regulating complex III of the mitochondrial electron transport chain. By silencing NDUFB8 (complex I), Rieske (complex III), or cytochrome c, macrophages expressing Akt1CA showed increased proton leak by OCR, while silencing complex III abrogated this response (Figure S5M). Because PGC‐1α is known to suppress mtROS by increasing antioxidant enzymes, 22 we determined if PGC‐1α regulated mtROS in our model. Macrophages expressing PGC‐1α had a significant reduction in mtROS generation in control cells, whereas the Akt1CA‐mediated mtROS generation was not altered by overexpression of PGC‐1α (Figure S5N). To determine the effect of Akt1‐mediated mtROS on PGC‐1α, we quenched mtROS with MitoTEMPO (Figure S5O), which abolished the nuclear localization of PGC‐1α (Figure 5O). MitoTEMPO also inhibited Akt1‐mediated p38 MAPK activation. These data provided evidence that nuclear localization and activation of PGC‐1α is redox‐dependent.
3.6. Inhibition of mitochondrial fission protects against pulmonary fibrosis
Because PGC‐1α increases mitochondrial homeostasis by inducing biogenesis, we questioned if inhibition of biogenesis alone would have an effect on fibrotic repair. The increase in biogenesis in BAL cell mitochondria from IPF subjects was secondary to fission as significantly more p‐Drp1 was present in IPF mitochondria compared to normal subjects (Figure 6A,B). Targeting mitochondrial fission therapeutically, BAL cells were isolated from IPF subjects and treated with mdivi‐1, a Drp1 inhibitor. TUNEL staining was significantly increased in IPF BAL cells treated with mdivi‐1 (Figure 6C,D).
FIGURE 6.
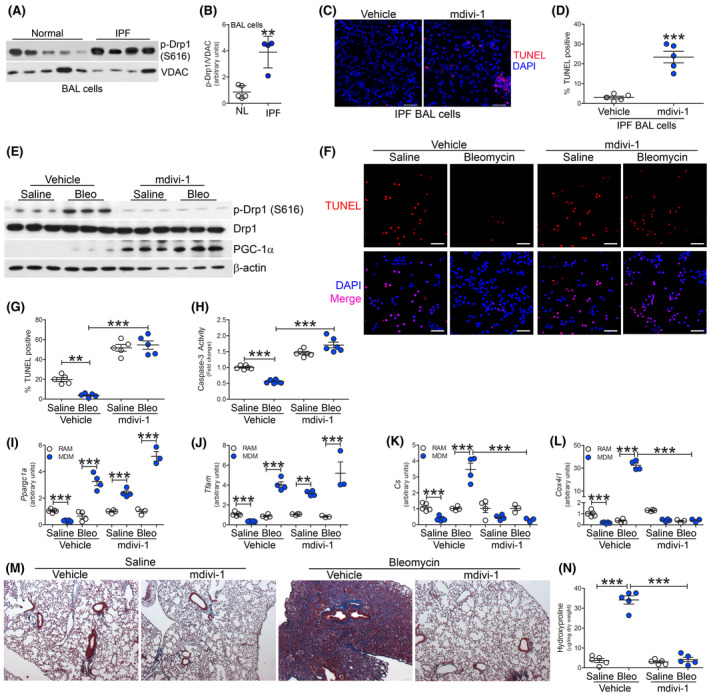
Mitochondrial division inhibitor prevents bleomycin‐induced pulmonary fibrosis. A, Immunoblot analysis and B, quantification of p‐Drp1 (S616) expression in BAL cells from normal (n = 5) or IPF subjects (n = 4). C, TUNEL staining (n = 5) and D, TUNEL quantification (n = 5) of IPF BAL cells treated with vehicle or mdivi‐1 (20 µM, overnight). Scale bar represents 50 μm. C57BL/6J WT mice were exposed to saline or bleomycin (bleo), 10 days after exposure mice were administered daily ip injections of vehicle or mdivi‐1. BAL cells were isolated 21 days later. E, Immunoblot analysis in BAL cells. F, TUNEL staining (n = 5), G, TUNEL quantification (n = 5), and H, Caspase‐3 activity (n = 6), Scale bar represent 50 μm. mRNA analysis of I, Ppargc1a (n = 5), J, Tfam (n = 5), K, Cs (n = 5), and L, Cox4i1 expression (n = 5). M, Masson’s trichrome staining of lung sections (n = 5) and N, hydroxyproline analysis (n = 5). **P < .001; ***P < .0001. Values shown as mean ± S.E.M. Two‐tailed t‐test statistical analysis was utilized for B and D. One‐way ANOVA followed by Tukey’s multiple comparison test was utilized for G‐L, N
To determine the biological relevance of mitochondrial fission in fibrosis, we exposed mice to bleomycin and administered mdivi‐1 daily starting at day 10 after bleomycin injury. Bleomycin exposure resulted in an increase in BAL cells from vehicle or mdivi‐1 treated mice (Figure S6A), and monocytic cells were the predominant cell type in the BALF (Figure S6B). Mdivi‐1 treatment resulted in a marked reduction in p‐Drp1 (S616) expression in BAL cells, while there was increased PGC‐1α expression and Akt1 activation (Figures 6E and S6C). Akt2 activation did not occur under any condition, including bleomycin injury or mdivi‐1 treatment.
Inhibition of Drp1 increased TUNEL staining in BAL cells (Figure 6F). Mdivi‐1‐treated mice showed 20‐fold greater TUNEL‐positive BAL cells after bleomycin injury (Figure 6G). Similarly, mdivi‐1‐treated mice showed increased caspase‐3 activity in BAL cells, whereas macrophages from bleomycin‐injured WT mice treated with vehicle showed a significant reduction in caspase‐3 activity (Figure 6H).
To determine the macrophage subset responsible for the enhanced PGC‐1α expression, tissue resident and monocyte‐derived macrophages were FACS‐sorted. The number of monocyte‐derived macrophages increased in mice treated with mdivi‐1, which was further increased in bleomycin‐exposed mice treated with mdivi‐1 (Figure S6D). The expression of Ppargc1a was significantly increased in monocyte‐derived macrophages from mdivi‐1‐treated mice and further increased after bleomycin‐induced injury (Figure 6I). A similar trend was seen with Tfam gene expression (Figure 6J). Cs and Cox4i1 gene expression were significantly increased in monocyte‐derived macrophages from bleomycin‐injured mice treated with vehicle, whereas mdivi‐1 treatment significantly reduced expression in bleomycin‐injured mice below the saline control (Figure 6K,L). These results suggest that abrogation of apoptosis resistance in BAL cells occurred by targeting mitochondrial biogenesis downstream of PGC‐1α.
Because dysfunctional IPF BAL cell mitochondria are removed by mitophagy 14 and show increased mitochondrial biogenesis, we determined if these processes were co‐dependent. BAL cells isolated from mice treated with mdivi‐1 showed an absence of mitochondrial PINK1 and Parkin (Figure S6E) and reduced LC3 expression after bleomycin‐induced injury (Figure S6F). Silencing PGC‐1α reduced the mitochondrial localization of PINK1 and Parkin (Figure S6G) as well as LC3 expression (Figure S6H). Similar results were seen when Tfam was silenced (Figure S6I,J). Moreover, silencing Parkin in macrophages led to a significant reduction in mtDNA genes (Figure S6K‐M).
Systemic administration of mdivi‐1 did not alter caspase‐3 activity, mtDNA genes, or mitophagy/autophagy in isolated lung fibroblasts compared to vehicle‐treated mice (Figure S6N‐R); however, mdivi‐1‐treated mice showed a sixfold reduction in lung fibroblast Col1a1 mRNA (Figure S6S). Bleomycin‐injured mice administered mdivi‐1 showed a significant reduction in caspase‐3 activity in isolated AECs (Figure S6T), while no difference was detected in mtDNA genes with mdivi‐1 treatment (Figure S6U,W). Potentially explaining the reduction of caspase‐3 activity, isolated AECs from bleomycin‐exposed mice treated with mdivi‐1 showed increased LC3 expression suggesting increased mitophagy/autophagy (Figure S6X), which is known to be dysregulated AECs in pulmonary fibrosis. 34 These data suggest the interdependence of mitophagy and mitochondrial biogenesis is cell type‐specific.
To validate the biologic importance of mitochondrial biogenesis, mdivi‐1‐treated mice were protected from bleomycin‐induced pulmonary fibrosis (Figure 6M). These results were confirmed biochemically (Figure 6N). These observations signify the critical role of mitochondrial biogenesis and apoptosis resistance in monocyte‐derived macrophages in the pathogenesis and progression of pulmonary fibrosis.
3.7. Deletion of PGC‐1α in monocyte‐derived macrophages protects against pulmonary fibrosis
Due to the significant role of PGC‐1α in mitochondrial and macrophage homeostasis and human lung function, we determined the biological relevance of PGC‐1α in fibrotic repair by generating mice harboring a conditional deletion of Ppargc1a in monocyte‐derived macrophages using a tamoxifen inducible Cre driven by the colony‐stimulating factor 1 receptor (Csf1r) promoter (Ppargc1a−/−Csf1rMeriCreMer). The Csf1rMeriCreMer mice have been used to investigate the role of recruited monocyte‐derived macrophages in multiple diseases. 35 , 36 , 37 Bleomycin‐injured Ppargc1afl/fl mice showed a significant increase in Ppargc1a gene expression, while tamoxifen induced the deletion of Ppargc1a lung macrophages from Ppargc1a−/−Csf1rMeriCreMer mice (Figure 7A). FACS‐sorted BAL cells confirmed that the tamoxifen‐induced Ppargc1a deletion occurred in monocyte‐derived macrophages (Figure S7A). Bleomycin injury led to an increase in BAL cells (Figure S7B), and the majority of cells were monocytic regardless of strain or diet (Figure S7C).
FIGURE 7.
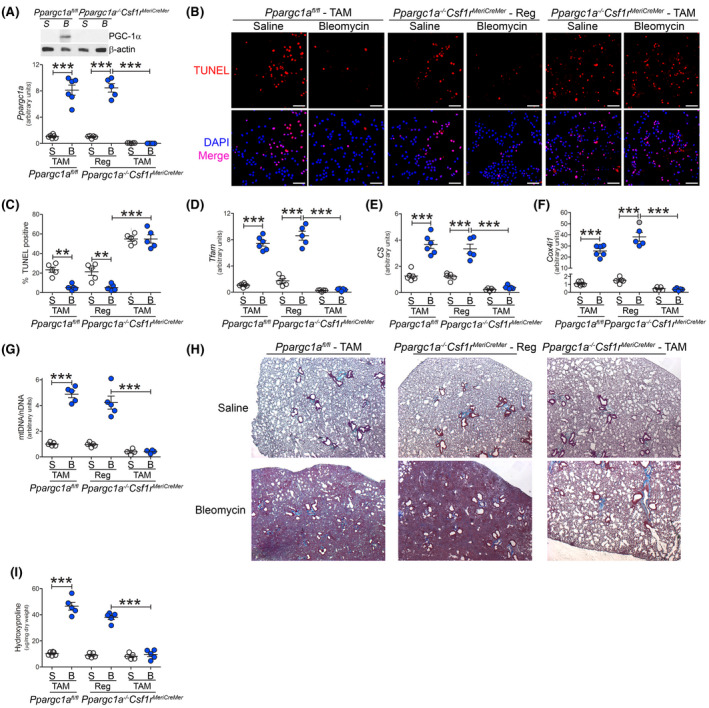
Deletion of PGC‐1α in monocyte‐derived macrophages protects against pulmonary fibrosis. Ppargc1afl/fl or Ppargc1a−/−Csf1rMeriCreMer mice were administered tamoxifen (TAM) or regular (Reg) chow and exposed to saline or bleomycin (bleo). BAL cells were isolated 21 days later. A, mRNA analysis of PGC‐1α in BAL cells (n = 5‐6). B, TUNEL staining (n = 5‐6), and C, TUNEL quantification (n = 5‐6). Scale bar represents 50 μm. D, Tfam (n = 5), E, Cs (n = 5‐6), and F, Cox4i1 mRNA expression (n = 5‐6). G, mtDNA/nDNA in BAL cells (n = 4‐5). H, Masson’s trichrome staining of lung sections (n = 5‐6) and I, hydroxyproline analysis (n = 5). BAL cells were isolated from IPF patients by BAL. **P < .001; ***P < .0001. Values shown as mean ± S.E.M. One‐way ANOVA followed by Tukey’s multiple comparison test was utilized
Tamoxifen‐induced deletion of PGC‐1α resulted in increased TUNEL staining in monocyte‐derived macrophages regardless of exposure (Figure 7B). Lung macrophages from bleomycin‐injured Ppargc1afl/fl mice and Ppargc1a−/−Csf1rMeriCreMer mice fed regular chow showed a significant reduction in TUNEL staining compared to their saline‐exposed counterpart (Figure 7C). To further validate PGC‐1α in apoptosis resistance, silencing PGC‐1α increased caspase‐3 activity, and over expression of Akt1CA had no effect on caspase‐3 activity with PGC‐1α silenced (Figure S7D). These results suggest that PGC‐1α expression is critical for the resistance to apoptosis in monocyte‐derived macrophages during fibrosis.
Confirming that PGC‐1α regulates mitochondrial biogenesis, tamoxifen‐induced deletion of Ppargc1a resulted in greater than six‐fold reduction in Tfam and significantly reduced Cs and Cox4i1 gene expression (Figure 7D‐F). Bleomycin‐injured Ppargc1afl/fl mice and Ppargc1a−/−Csf1rMeriCreMer mice fed regular chow showed a fourfold increase in the ratio of mtDNA to nuclear DNA in lung macrophages compared to saline–exposed mice (Figure 7G). The deletion of Ppargc1a led to a significant reduction in mtDNA. The biological significance of PGC‐1α in lung macrophages was demonstrated by complete abrogation of bleomycin‐induced fibrosis in mice harboring the conditional deletion of Ppargc1a (Figure 7H). The histological observations were verified by hydroxyproline analysis (Figure 7I). Taken together, these results suggest that PGC‐1α in monocyte‐derived macrophages is a novel therapeutic target to attenuate and/or halt lung fibrosis.
4. DISCUSSION
Lung remodeling during pulmonary fibrosis is poorly understood; however, modulation of mitochondrial dynamics in lung macrophages is emerging as a critical determinant of fibrotic repair. Mitochondrial dynamics are induced by mtROS and can contribute to apoptosis resistance during cellular stress. 14 , 16 , 20 The generation of new and the removal of damaged or dysfunctional mitochondria are highly regulated processes that must be precisely coordinated for the maintenance of mitochondrial and cellular homeostasis. Mitophagy and mitochondrial biogenesis are typically co‐dependent. This is particularly true during cellular stress when biogenesis is needed to meet the energy demand of the cell. 38 We have previously shown that IPF BAL cells have increased mtROS and mitophagy. 14 Here, we show that monocyte‐derived macrophages have increased PCC‐1α‐mediated mitochondrial biogenesis to maintain apoptosis resistance during pulmonary fibrosis (Figure 8). Importantly, the increased expression of PPARGC1A in IPF lung macrophages correlated with reduced pulmonary function in IPF subjects. Biological evidence to support this is that the deletion of Ppargc1a in monocyte‐derived macrophages in mice prevented pulmonary fibrosis after bleomycin injury.
FIGURE 8.
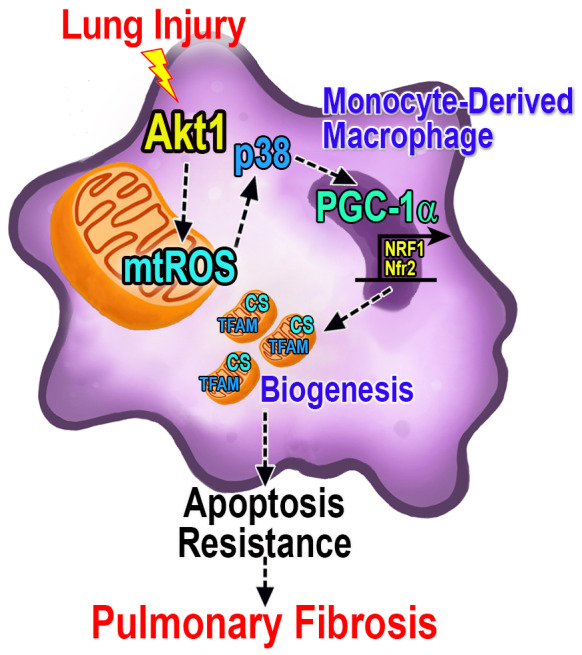
PGC‐1α regulates fibrotic progression in monocyte‐derived macrophages. Lung injury promotes Akt1 activation and the generation of the mitochondrial ROS (mtROS), which induces p38 MAPK. The p38‐mediated increase of PGC‐1α activation occurs via Akt1 and augments the transcription of mitochondrial DNA genes, including TFAM and CS (citrate synthase) to induce apoptosis resistance in monocyte‐derived macrophages and fibrotic progression
Mitochondrial dynamics have an important role in mediating apoptosis resistance. We have shown that BAL cells from IPF subjects are resistant to apoptosis compared with normal subjects. 14 Here, we have extended these findings to suggest that infiltrating monocyte‐derived macrophages are responsible for fibrosis development. A prior study suggests that the increase in the number of infiltrating monocyte‐derived macrophages compared to resident macrophages during fibrosis development persisted even after resolution of the injury. 10 Monocytes typically undergo spontaneous apoptosis unless exposed to particular survival signals or are involved in innate immune activities, 39 such as seen in dysregulated fibrotic repair. Our data indicate that apoptosis resistance is necessary for recruited monocyte‐derived macrophages to mediate fibrotic repair.
Controversy exists on the role of macrophage apoptosis in disease development. Studies show that disease occurs in association with apoptosis, as seen in heart and liver disease, ulcerative colitis, and non‐small cell lung cancer. 40 , 41 , 42 , 43 , 44 In contrast, resistance to apoptosis is associated with progression of Crohn’s disease, breast cancer, rheumatoid arthritis, and atherosclerosis. 45 , 46 , 47 , 48 , 49 We recently showed that increased macrophage apoptosis resistance was present in IPF subjects and in fibrotic mice. 14 Moreover, macrophage apoptosis has been shown to attenuate fibrosis development. 14 , 50 These studies suggest that role of apoptosis is disease specific.
Macrophages are highly plastic and able to quickly adapt to their environment by adopting diverse activation states to adjust their intracellular metabolic pathways in response to extracellular stimulants. 51 In animal models of fibrosis, proinflammatory macrophages are present for up to 5‐7 days. There is subsequent change in the activation state to a profibrotic phenotype. Disease progression mediated by the prolonged survival of lung macrophages is associated with the polarization to a profibrotic phenotype. Studies reveal that a predominance of profibrotic macrophages can induce fibrotic remodeling after lung injury. 8 , 9 , 50 , 52 Profibrotic polarization accelerates the development of a fibrotic phenotype in vivo, 8 , 9 and lung macrophages are the primary source of TGF‐β1 in BAL fluid. 14 Profibrotic and anti‐fibrotic macrophages alter their metabolism to regulate function. While anti‐fibrotic macrophages utilize glycolytic metabolism, which can be rapidly activated to fuel response to injury, metabolic reprogramming that entails fatty acid oxidation and oxidative phosphorylation is a key feature of profibrotic macrophages. 7 , 8 Here we show that PPARGC1A, which regulates metabolic reprogramming to fatty acid oxidation, is increased in lung macrophages from IPF subjects. Moreover, the metabolic reprogramming in profibrotic macrophages is necessary to support long‐term cellular activities and promote apoptotic resistance. 13 , 30 , 53
The p38 MAPK is known to be a key mediator of stress signals and is often associated with apoptosis. Upstream activation of p38 by apoptosis signal‐regulating kinase 1 (ASK1) promotes apoptosis through mitochondria‐dependent caspase activation as well as in response to ROS. 54 Although p38 is well known for mediating apoptosis, it has also been connected with apoptosis resistance. 55 , 56 During endoplasmic reticulum stress or pro‐apoptotic signals, p38 is required for survival of macrophages by activating Akt. 57 In certain conditions, the Akt pathway negatively regulates MAPKs and ASK1 in vitro. 58 The p38 MAPK is activated downstream of inflammatory cytokines to phosphorylate PGC‐1α in muscle, 33 , 59 β3‐adrenergic stimulation in brown adipocytes, 60 and by exercise 61 and nitric oxide 62 ; however, the role of Akt1 in p38 MAPK‐mediated PGC‐1α activation has not been shown, especially in fibrosis. Here, we show that Akt1 plays a key role in monocyte‐derived macrophage apoptosis resistance by promoting the activation of the p38 MAPK to regulate the post‐translational modification of PGC‐1α by phosphorylation in a redox‐dependent manner. Although we cannot rule out that other proteins modulate PGC‐1α and may contribute to the increased expression seen after bleomycin injury, our data strongly implicates the crucial role of the post‐translational modification of PGC‐1α in fibrosis.
The master regulator of mitochondrial biogenesis, PGC‐1α, is a critical regulator of mtROS generation. PGC‐1α is a positive regulator of oxidative metabolism, stimulating mitochondrial electron transport, but it also inhibits ROS levels by increasing expression of antioxidant enzymes. 22 , 63 During a state of oxidative stress, cells increase production of PGC‐1α to increase the antioxidant defense. 22 Studies suggest that failure of this protective system may lead to some neurodegenerative diseases, such as Parkinson’s, Alzheimer’s, Huntington’s diseases, and amyotrophic lateral sclerosis. 22 , 64 Dysregulation of PGC‐1α has also been connected to type 2 diabetes, obesity, and heart failure. 65 , 66 , 67 , 68 In fibrosis models, overexpression of PGC‐1α in renal tubular cells protected mice from renal fibrosis, 69 and administration of the PPARγ agonist, rosiglitazone, protected mice against kidney fibrosis. 70 In the lung, bleomycin‐exposed Ppargc1a−/− mice were more susceptible to lung fibrosis. 71 Mitochondrial homeostasis was enhanced in alveolar epithelial cells via PGC‐1α in bleomycin‐exposed WT mice. These improvements were not observed in bleomycin‐treated Ppargc1a−/−mice; however, the role of mitochondrial homeostasis in pulmonary fibrosis has not been investigated using mice harboring a conditional deletion of PGC‐1α. Although MMP‐7, VCAM‐1, S100A12, ICAM‐1, and IL‐8 have been associated with disease outcome and progression in IPF subjects, we are the first to show that PGC‐1α expression correlated with reduced pulmonary function. 72 , 73
A limitation of our study is that the Cre driver we used to delete Akt1 and Akt2 in lung macrophages has also been suggested to target neutrophils, dendritic cells, and lung epithelial cells. 74 , 75 Although we did not see changes in expression of Akt1 or Akt2 in these cells, we cannot exclude the importance of these cells in the pathogenesis of fibrosis. The Csf1rMeriCreMer mice have been previously shown to target macrophage/monocytes, while lymphocytes and neutrophils are not affected. 35 Moreover, we did not evaluate the contribution of interstitial macrophages as evidence suggests that monocytes give rise to interstitial macrophages and then differentiate into monocyte‐derived macrophages. 10 To determine the mechanistic regulation of PGC‐1α, we utilized plasmid constructs in commercially available cell lines. Although we observed many similarities in IPF subjects and within our mouse models to the data presented using cell lines, we cannot rule out the possibility that PGC‐1α may be differentially regulated in primary cells.
Studies implicate Akt2 phosphorylates PGC‐1α leading to its inactivation, 31 and PGC‐1α has been shown to regulate Akt expression and its downstream signaling. 76 , 77 Our data do not support these findings; rather, we demonstrate the critical role of Akt1‐mediated mtROS in the regulation of PGC‐1α, thereby increasing mitochondrial biogenesis, and the conditional deletion of PGC‐1α in monocyte‐derived macrophages protected mice from bleomycin‐induced fibrosis. In aggregate, these observations suggest that the regulation of PGC‐1α in monocyte‐derived macrophages is a novel, druggable target that may protect against the development and/or progression of pulmonary fibrosis.
CONFLICT OF INTEREST
The authors declare no conflicts of interest exists.
AUTHOR CONTRIBUTIONS
J.L. Larson‐Casey and A. Brent Carter developed the concept and design of the study. J.L. Larson‐Casey, L. Gu, D. Davis, G.‐Q. Cai, and Q. Ding assisted with conducting experiments. J.L. Larson‐Casey, L. Gu, D. Davis, C. He, and A. A. Brent Carter acquired data. J.L. Larson‐Casey, Q. Ding, and A. A. Brent Carter provided reagents. J.L. Larson‐Casey and A. A. Brent Carter provided analysis and interpretation of experiments and results. J.L. Larson‐Casey and A. A. Brent Carter wrote the manuscript.
Supporting information
Fig S1‐S7
ACKNOWLEDGMENTS
We thank Dr. Roger Davis, University of Massachusetts, Worcester, MA for generously gifting the pcDNA‐MKK6(Glu) and pCMV‐Flag‐p38DN plasmids, Vidya Sagar Hanumanthu, Ana Leda F. Longhini, and the Comprehensive Flow Cytometry Core for assistance with flow cytometry, and Melissa Foley Chimento and the UAB High Resolution Imaging Facility for assistance with TEM. Research reported in this publication was supported by the National Institute of Health grants 2R01ES015981‐13, P01 HL114470‐7, and P42 ES027723, and 1 I01 CX001715‐01 to the Department of Veteran Affairs to ABC, and a Parker B. Francis fellowship and American Lung Association grant RG‐507440 to JLC. Support for the Comprehensive Flow Cytometry Core provided by NIH P30AR048311 and NIH P30AI27667.
Larson‐Casey JL, Gu L, Davis D, et al. Post‐translational regulation of PGC‐1α modulates fibrotic repair. The FASEB Journal. 2021;35:e21675. 10.1096/fj.202100339R
References
- 1. Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810‐816. [DOI] [PubMed] [Google Scholar]
- 2. Hutchinson JP, McKeever TM, Fogarty AW, Navaratnam V, Hubbard RB. Increasing global mortality from idiopathic pulmonary fibrosis in the twenty‐first century. Ann Am Thorac Soc. 2014;11:1176‐1185. [DOI] [PubMed] [Google Scholar]
- 3. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence‐based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46:795‐806. [DOI] [PubMed] [Google Scholar]
- 5. American Cancer Society Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015. [Google Scholar]
- 6. King TE Jr, Bradford WZ, Castro‐Bernardini S et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083‐2092. [DOI] [PubMed] [Google Scholar]
- 7. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071‐2082. [DOI] [PubMed] [Google Scholar]
- 8. He C, Ryan AJ, Murthy S, Carter AB. Accelerated development of pulmonary fibrosis via Cu, Zn‐superoxide dismutase‐induced alternative activation of macrophages. J Biol Chem. 2013;288:20745‐20757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murthy S, Larson‐Casey JL, Ryan AJ, He C, Kobzik L, Carter AB. Alternative activation of macrophages and pulmonary fibrosis are modulated by scavenger receptor, macrophage receptor with collagenous structure. FASEB J. 2015;29:3527‐3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Misharin AV, Morales‐Nebreda L, Reyfman PA, et al. Monocyte‐derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. 2017;214:2387‐2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McCubbrey AL, Barthel L, Mohning MP, et al. Deletion of c‐FLIP from CD11b(hi) macrophages prevents development of bleomycin‐induced lung fibrosis. Am J Respir Cell Mol Biol. 2018;58:66‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He C, Larson‐Casey JL, Davis D, et al. NOX4 modulates macrophage phenotype and mitochondrial biogenesis in asbestosis. JCI Insight. 2019;4(16). 10.1172/jci.insight.126551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Larson‐Casey JL, Vaid M, Gu L, et al. Increased flux through the mevalonate pathway mediates fibrotic repair without injury. J Clin Invest. 2019;129:4962‐4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Larson‐Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 kinase‐mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity. 2016;44:582‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty‐Wolf KE, Piantadosi CA. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest. 2007;117:3730‐3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ikeda Y, Shirakabe A, Maejima Y, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264‐278. [DOI] [PubMed] [Google Scholar]
- 18. Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans . Nature. 2015;521:525‐528. [DOI] [PubMed] [Google Scholar]
- 19. Mannam P, Shinn AS, Srivastava A, et al. MKK3 regulates mitochondrial biogenesis and mitophagy in sepsis‐induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;306:L604‐L619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase‐1 regulates cardiac mitochondrial biogenesis via Nrf2‐mediated transcriptional control of nuclear respiratory factor‐1. Circ Res. 2008;103:1232‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Larsson NG, Wang J, Wilhelmsson H, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231‐236. [DOI] [PubMed] [Google Scholar]
- 22. St‐Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC‐1 transcriptional coactivators. Cell. 2006;127:397‐408. [DOI] [PubMed] [Google Scholar]
- 23. He C, Murthy S, McCormick ML, Spitz DR, Ryan AJ, Carter AB. Mitochondrial Cu, Zn‐superoxide dismutase mediates pulmonary fibrosis by augmenting H2O2 generation. J Biol Chem. 2011;286:15597‐15607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3‐ and MKK6‐regulated gene expression is mediated by the p38 mitogen‐activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247‐1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Larson‐Casey JL, Murthy S, Ryan AJ, Carter AB. Modulation of the mevalonate pathway by akt regulates macrophage survival and development of pulmonary fibrosis. J Biol Chem. 2014;289:36204‐36219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator‐activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci USA. 2003;100:7111‐7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ichida M, Nemoto S, Finkel T. Identification of a specific molecular repressor of the peroxisome proliferator‐activated receptor gamma Coactivator‐1 alpha (PGC‐1alpha). J Biol Chem. 2002;277:50991‐50995. [DOI] [PubMed] [Google Scholar]
- 28. Gu L, Larson‐Casey JL, Carter AB. Macrophages utilize the mitochondrial calcium uniporter for profibrotic polarization. FASEB J. 2017;31:3072‐3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carter AB, Hunninghake GW. A constitutive active MEK ‐> ERK pathway negatively regulates NF‐kappa B‐dependent gene expression by modulating TATA‐binding protein phosphorylation. J Biol Chem. 2000;275:27858‐27864. [DOI] [PubMed] [Google Scholar]
- 30. Gu L, Larson Casey JL, Andrabi SA, et al. Mitochondrial calcium uniporter regulates PGC‐1alpha expression to mediate metabolic reprogramming in pulmonary fibrosis. Redox Biol. 2019;26:101307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC‐1alpha transcription coactivator. Nature. 2007;447:1012‐1016. [DOI] [PubMed] [Google Scholar]
- 32. Wu Z, Puigserver P, Andersson U, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC‐1. Cell. 1999;98:115‐124. [DOI] [PubMed] [Google Scholar]
- 33. Knutti D, Kressler D, Kralli A. Regulation of the transcriptional coactivator PGC‐1 via MAPK‐sensitive interaction with a repressor. Proc Natl Acad Sci USA. 2001;98:9713‐9718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bueno M, Lai Y‐C, Romero Y, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015;125(2):521‐538. 10.1172/jci74942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qian BZ, Li J, Zhang H, et al. CCL2 recruits inflammatory monocytes to facilitate breast‐tumour metastasis. Nature. 2011;475:222‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hughes R, Qian BZ, Rowan C, et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015;75:3479‐3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sheikh AQ, Saddouk FZ, Ntokou A, Mazurek R, Greif DM. Cell autonomous and non‐cell autonomous regulation of SMC progenitors in pulmonary hypertension. Cell Rep. 2018;23:1152‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scarpulla RC. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene. 2002;286:81‐89. [DOI] [PubMed] [Google Scholar]
- 39. Zhao C, Tan YC, Wong WC, et al. The CD14(+/low)CD16(+) monocyte subset is more susceptible to spontaneous and oxidant‐induced apoptosis than the CD14(+)CD16(−) subset. Cell Death Dis. 2010;1:e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alzaid F, Lagadec F, Albuquerque M, et al. IRF5 governs liver macrophage activation that promotes hepatic fibrosis in mice and humans. JCI Insight. 2016;1:e88689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Narula J, Haider N, Virmani R, et al. Apoptosis in myocytes in end‐stage heart failure. N Engl J Med. 1996;335:1182‐1189. [DOI] [PubMed] [Google Scholar]
- 42. Qiu W, Wu B, Wang X, et al. PUMA‐mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J Clin Invest. 2011;121:1722‐1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kaufmann T, Jost PJ, Pellegrini M, et al. Fatal hepatitis mediated by tumor necrosis factor TNFalpha requires caspase‐8 and involves the BH3‐only proteins Bid and Bim. Immunity. 2009;30:56‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Han L, Zhang EB, Yin DD, et al. Low expression of long noncoding RNA PANDAR predicts a poor prognosis of non‐small cell lung cancer and affects cell apoptosis by regulating Bcl‐2. Cell Death Dis. 2015;6:e1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shearn AI, Deswaerte V, Gautier EL, et al. Bcl‐x inactivation in macrophages accelerates progression of advanced atherosclerotic lesions in Apoe(−/−) mice. Arterioscler Thromb Vasc Biol. 2012;32:1142‐1149. [DOI] [PubMed] [Google Scholar]
- 46. Li MO, Sarkisian MR, Mehal WZ, Rakic P, Flavell RA. Phosphatidylserine receptor is required for clearance of apoptotic cells. Science. 2003;302:1560‐1563. [DOI] [PubMed] [Google Scholar]
- 47. Breunig C, Pahl J, Kublbeck M, et al. MicroRNA‐519a‐3p mediates apoptosis resistance in breast cancer cells and their escape from recognition by natural killer cells. Cell Death Dis. 2017;8:e2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Atreya R, Mudter J, Finotto S, et al. Blockade of interleukin 6 trans signaling suppresses T‐cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583‐588. [DOI] [PubMed] [Google Scholar]
- 49. Meinecke I, Cinski A, Baier A, et al. Modification of nuclear PML protein by SUMO‐1 regulates Fas‐induced apoptosis in rheumatoid arthritis synovial fibroblasts. Proc Natl Acad Sci USA. 2007;104:5073‐5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Redente EF, Keith RC, Janssen W, et al. Tumor necrosis factor‐alpha accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am J Respir Cell Mol Biol. 2014;50:825‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. 2016;17:34‐40. [DOI] [PubMed] [Google Scholar]
- 52. Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. 2011;6:275‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90‐94. [DOI] [PubMed] [Google Scholar]
- 55. Okamoto S, Krainc D, Sherman K, Lipton SA. Antiapoptotic role of the p38 mitogen‐activated protein kinase‐myocyte enhancer factor 2 transcription factor pathway during neuronal differentiation. Proc Natl Acad Sci USA. 2000;97:7561‐7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297:2048‐2051. [DOI] [PubMed] [Google Scholar]
- 57. Seimon TA, Wang Y, Han S, et al. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Invest. 2009;119:886‐898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Luyendyk JP, Schabbauer GA, Tencati M, Holscher T, Pawlinski R, Mackman N. Genetic analysis of the role of the PI3K‐Akt pathway in lipopolysaccharide‐induced cytokine and tissue factor gene expression in monocytes/macrophages. J Immunol. 2008;180:4218‐4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Puigserver P, Rhee J, Lin J, et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator‐1. Mol Cell. 2001;8:971‐982. [DOI] [PubMed] [Google Scholar]
- 60. Cao W, Medvedev AV, Daniel KW, Collins S. beta‐Adrenergic activation of p38 MAP kinase in adipocytes: cAMP induction of the uncoupling protein 1 (UCP1) gene requires p38 MAP kinase. J Biol Chem. 2001;276:27077‐27082. [DOI] [PubMed] [Google Scholar]
- 61. Akimoto T, Pohnert SC, Li P, et al. Exercise stimulates Pgc‐1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem. 2005;280:19587‐19593. [DOI] [PubMed] [Google Scholar]
- 62. Ptasinska A, Wang S, Zhang J, Wesley RA, Danner RL. Nitric oxide activation of peroxisome proliferator‐activated receptor gamma through a p38 MAPK signaling pathway. FASEB J. 2007;21:950‐961. [DOI] [PubMed] [Google Scholar]
- 63. Aquilano K, Vigilanza P, Baldelli S, Pagliei B, Rotilio G, Ciriolo MR. Peroxisome proliferator‐activated receptor gamma co‐activator 1alpha (PGC‐1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis. J Biol Chem. 2010;285:21590‐21599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Eschbach J, Schwalenstocker B, Soyal SM, et al. PGC‐1alpha is a male‐specific disease modifier of human and experimental amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22:3477‐3484. [DOI] [PubMed] [Google Scholar]
- 65. Arany Z, He H, Lin J, et al. Transcriptional coactivator PGC‐1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259‐271. [DOI] [PubMed] [Google Scholar]
- 66. Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466‐8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lin J, Wu PH, Tarr PT, et al. Defects in adaptive energy metabolism with CNS‐linked hyperactivity in PGC‐1alpha null mice. Cell. 2004;119:121‐135. [DOI] [PubMed] [Google Scholar]
- 68. Huss JM, Imahashi K, Dufour CR, et al. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25‐37. [DOI] [PubMed] [Google Scholar]
- 69. Han SH, Wu MY, Nam BY, et al. PGC‐1alpha protects from notch‐induced kidney fibrosis development. J Am Soc Nephrol. 2017;28:3312‐3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang L, Liu J, Zhou F, Wang W, Chen N. PGC‐1alpha ameliorates kidney fibrosis in mice with diabetic kidney disease through an antioxidative mechanism. Mol Med Rep. 2018;17:4490‐4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yu G, Tzouvelekis A, Wang R, et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med. 2018;24:39‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bauer Y, White ES, de Bernard S, et al. MMP‐7 is a predictive biomarker of disease progression in patients with idiopathic pulmonary fibrosis. ERJ Open Research. 2017;3(1):00074‐2016. 10.1183/23120541.00074-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Richards TJ, Kaminski N, Baribaud F, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;185:67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid‐Cre deleting strains using ROSA‐EYFP reporter mice. J Immunol Methods. 2014;408:89‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jakubzick C, Bogunovic M, Bonito AJ, Kuan EL, Merad M, Randolph GJ. Lymph‐migrating, tissue‐derived dendritic cells are minor constituents within steady‐state lymph nodes. J Exp Med. 2008;205:2839‐2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Geng T, Li P, Yin X, Yan Z. PGC‐1alpha promotes nitric oxide antioxidant defenses and inhibits FOXO signaling against cardiac cachexia in mice. Am J Pathol. 2011;178:1738‐1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Besse‐Patin A, Jeromson S, Levesque‐Damphousse P, Secco B, Laplante M, Estall JL. PGC1A regulates the IRS1:IRS2 ratio during fasting to influence hepatic metabolism downstream of insulin. Proc Natl Acad Sci USA. 2019;116:4285‐4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S7