

Abstract
During viral replication, herpesviruses utilize a unique strategy, termed nuclear egress, to translocate capsids from the nucleus into the cytoplasm. This initial budding step transfers a newly formed capsid from within the nucleus, too large to fit through nuclear pores, through the inner nuclear membrane to the perinuclear space. The perinuclear enveloped virion must then fuse with the outer nuclear membrane to be released into the cytoplasm for further maturation, undergoing budding once again at the trans-Golgi network or early endosomes, and ultimately exit the cell non-lytically to spread infection. This first budding process is mediated by two conserved viral proteins, UL31 and UL34, that form a heterodimer called the nuclear egress complex (NEC). This chapter focuses on what we know about how the NEC mediates capsid transport to the perinuclear space, including steps prior to and after this budding event. Additionally, we discuss the involvement of other viral proteins in this process and how NEC-mediated budding may be regulated during infection.
Introduction: overview of nuclear egress
Herpesviruses replicate their double-stranded-DNA genomes and package them into capsids within the host cell nucleus. These progeny capsids must then exit the nucleus to form mature virions within the cytoplasm (Figure 1). The nucleus is surrounded by the nuclear envelope - a double membrane bilayer consisting of the inner nuclear membrane (INM) and the outer nuclear membrane (ONM) - and most transport in and out of the nucleus occurs through nuclear pores, which have an inner channel diameter of ~39-nm (Pante and Kann, 2002). But at ~125 nm in diameter, herpesviral capsids are too large to fit through the nuclear pores and must use a more complex route, termed nuclear egress, to exit the nucleus. Multiple lines of evidence support the model whereby capsids first become enveloped at the INM, pinching off into the perinuclear space (PNS) (primary envelopment) [reviewed in (Bigalke and Heldwein, 2016; Crump, 2018; Roller and Baines, 2017a)]. These perinuclear enveloped virions (PEVs) then fuse with the ONM, releasing the capsids into the cytoplasm (de-envelopment). The capsids then undergo another, final envelopment, gaining tegument proteins (layer between capsid and the envelope) and a final lipid envelope. The mature virion then hijacks the secretory pathway and leaves the cell by exocytosis.
Figure 1. Overview of herpesvirus egress.
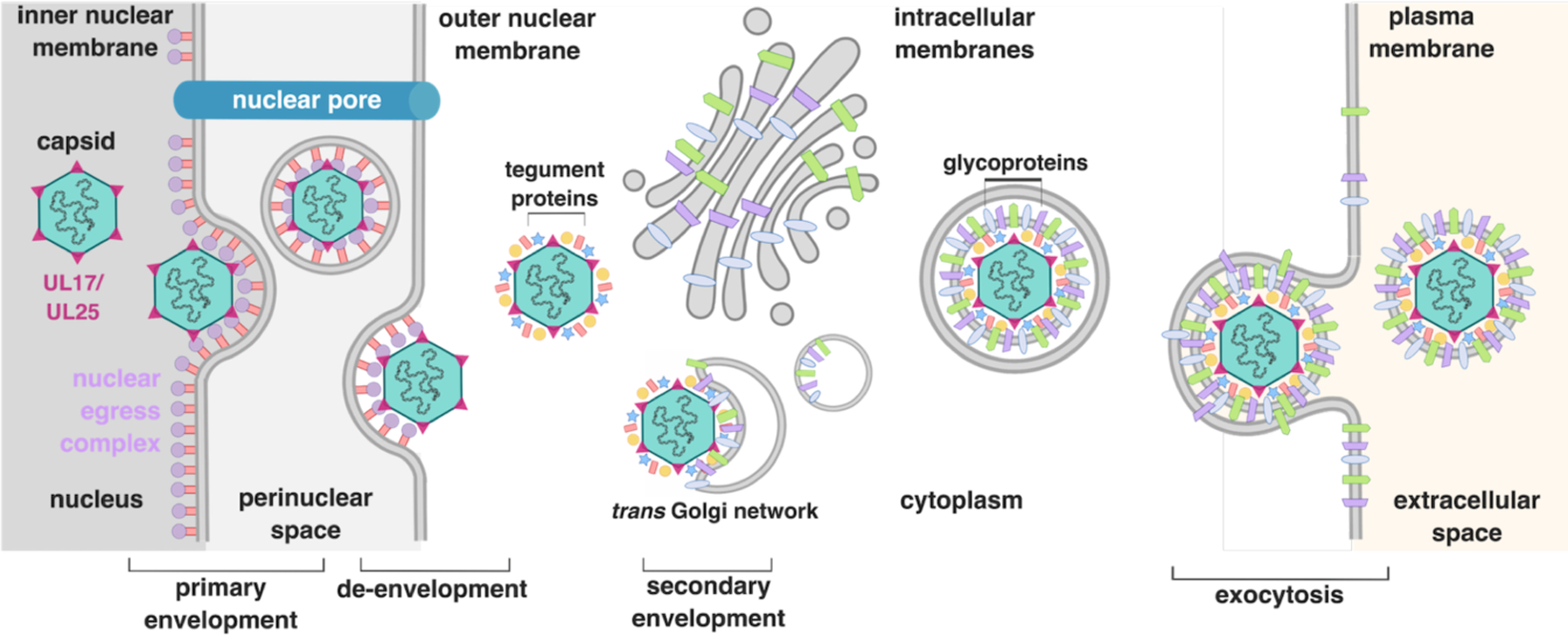
Capsids undergo primary envelopment (nuclear budding) at the inner nuclear membrane where they pinch off into the perinuclear space in a process mediated by the nuclear egress complex. The resulting perinuclear enveloped vesicles fuse with the outer nuclear membrane and release the capsid into the cytoplasm (de-envelopment). The resulting virion gains necessary tegument proteins before undergoing secondary envelopment during which it acquires a glycoprotein envelope derived from the trans Golgi network or early endosomes. The mature virion then hijacks the host secretory pathway and is released into the extracellular space via exocytosis to spread infection.
This envelopment/de-envelopment model of nuclear egress is the most widely accepted model for capsid escape from the nucleus and is backed up by substantial data. First, enveloped virions in the PNS have been observed in cells infected with herpesvirus for several decades (Fuchs et al., 2002; Granzow et al., 1997; Reynolds et al., 2002; Stackpole, 1969). The phenotypes of viral gene deletions likewise provide compelling evidence in favor of the envelopment/de-envelopment model. Deletion of the US3 kinase gene from either HSV-1 or from the closely related pseudorabies virus (PRV) results in the accumulation of PEVs (Klupp et al., 2001; Reynolds et al., 2002; Ryckman and Roller, 2004; Sehl et al., 2020), which points to budding into the PNS being an intermediate stage in nuclear egress. Second, PEVs differ from mature virions in their morphology and protein composition. For example, UL31 and UL34, two viral proteins critical for nuclear egress, are absent from mature HSV-1 and PRV virions (Fuchs et al., 2002; Reynolds et al., 2002). Conversely, most tegument proteins found in mature virions are not found in the nucleus or in PEVs (Gershon et al., 1994; Granzow et al., 2001; Klupp et al., 2000; Skepper et al., 2001). Furthermore, mature virions are studded with glycoprotein spikes and have a thick tegument while PEVs have a smooth envelope and thin tegument (Gershon et al., 1994; Granzow et al., 2001), consistent with the two types of particles being formed during different budding events. Third, deletions of particular tegument proteins, such as HSV-1 UL36 and UL37, or the glycoproteins gD and gE from either HSV-1 or PRV result in unenveloped capsids accumulating in the cytoplasm (Brack et al., 2000; Desai et al., 2001; Desai, 2000; Farnsworth et al., 2003; Leege et al., 2009; Roberts et al., 2009) suggesting that a secondary envelopment step in the cytoplasm is necessary for egress. Lastly, the envelopes of mature viral particles from both HSV-1 and varicella-zoster virus (VZV), another alphaherpesvirus, are derived from cytoplasmic membranes, rather than nuclear membranes, as shown by immunogold electron microscopy and transmission electron microscopy radioautography experiments, respectively (Gershon et al., 1994; Skepper et al., 2001). Collectively, these observations make a strong case for a model of nuclear egress in which nuclear capsids gain their initial, temporary envelope at the INM, escape into the cytoplasm as unenveloped capsids, and then acquire the final envelope at the trans Golgi network (Henaff et al., 2012) or early endosomes (Hollinshead et al., 2012) (see Chapter 9).
The nuclear egress complex
The nuclear egress complex is essential for nuclear egress
Nuclear egress is mediated by two conserved viral proteins, UL31 and UL34, that form the nuclear egress complex (NEC) (Fuchs et al., 2002; Reynolds et al., 2001; Roller et al., 2000) [reviewed in (Bigalke and Heldwein, 2016; Hellberg et al., 2016; Roller and Baines, 2017a)]. This complex is conserved across all members of the Herpesviridae family (see Chapter 3), and both the UL31 and UL34 proteins are essential for this process (Fuchs et al., 2002) (Bubeck et al., 2004; Farina et al., 2005; Reynolds et al., 2001). While UL31 and UL34 homologs are known under different names in other herpesviruses, throughout this chapter, alphaherpesvirus nomenclature will be used in keeping with the subject of this book. UL31 is a soluble nuclear phosphoprotein that is 306-aa long in HSV-1 (Chang and Roizman, 1993; Reynolds et al., 2001). UL34, which is 275-aa long in HSV-1, has a single-spanning C-terminal transmembrane (TM) region that anchors the complex to the INM such that the NEC faces the nucleoplasm (Reynolds et al., 2001; Shiba et al., 2000).
UL31 homologs have nuclear localization signals that target the protein from its site of synthesis in the cytosol to the nucleus (Funk et al., 2015; Li et al., 2015; Passvogel et al., 2015). In the absence of HSV-1 UL34, UL31 is diffusely distributed throughout the nucleoplasm (Reynolds et al., 2001; Zhu et al., 1999). In the absence of UL31, UL34 localizes to the ER membranes, probably, because most UL34 homologs do not have a nuclear localization signal. UL31 and UL34 localize to the INM only when both proteins are present, either during infection or in uninfected cells expressing UL31 and UL34, which suggests that proper localization of the two proteins at the INM requires formation of the NEC (Fuchs et al., 2002; Funk et al., 2015; Reynolds et al., 2001) [reviewed in (Hellberg et al., 2016; Roller and Baines, 2017a)].
In the absence of either UL31 or UL34, viral replication is reduced by three to four orders of magnitude and capsids accumulate within the nucleus (Fuchs et al., 2002) (Bubeck et al., 2004; Chang and Roizman, 1993; Klupp et al., 2000; Reynolds et al., 2001; Roller et al., 2000), which indicates that both proteins are required for efficient nuclear egress. Moreover, PRV NEC expressed in uninfected cells results in the formation of capsidless perinuclear vesicles, demonstrating that UL31 and UL34 are the only viral proteins necessary for nuclear budding (Klupp et al., 2007). Formation of capsidless perinuclear vesicles was also observed for NEC homologs from gammaherpesviruses Epstein-Barr virus (EBV) (Farina et al., 2005; Gonnella et al., 2005) and Kaposi’s sarcoma-associated herpesvirus (KSHV) (Desai et al., 2012; Luitweiler et al., 2013) expressed in insect cells using recombinant baculoviruses. Together, these findings implicated the NEC in the trafficking of capsids out of the nucleus through a nuclear budding mechanism.
High-resolution structures of the NEC
Within the last few years, five crystal structures of the NEC from subfamilies of alphaherpesviruses and betaherpesviruses have been determined: one from HSV-1 (PDB ID: 4ZXS) (Bigalke and Heldwein, 2015), two from PRV (PDB IDs: 4Z3U and 5E8C) (Bigalke and Heldwein, 2015; Zeev-Ben-Mordehai et al., 2015), and two from the human cytomegalovirus (HCMV) (PDB IDs: 5DOB and 5D5N) (Lye et al., 2015; Walzer et al., 2015). The NEC has an elongated cylindrical shape (Figure 2). UL34, located at the base of the complex, forms a globular pedestal for UL31. UL31, located at the top of the complex, is composed of a globular core that rests on UL34 and an N-terminal hook-like extension that projects downwards and wraps around UL34. Both UL31 and UL34 have unique folds according to Dali structural similarity algorithms (Holm and Rosenstrom, 2010). In all of the structures determined thus far, UL31 and UL34 form an extensive interface that can be subdivided into two regions: one that involves the globular core of UL31 (interface 1) and the other formed by the hook-like extension of UL31 (interface 2) (Figure 2). Prior to structural studies, genetic studies performed using in-frame deletion mutants pinpointed regions of UL34 from HSV-1 (Liang and Baines, 2005) and UL31 homolog from HCMV (Sam et al., 2009; Schnee et al., 2012) necessary for complex formation. While these regions, indeed, participate in UL31/UL34 interactions, they map to different interfaces and do not contact one another within the structure (Bigalke and Heldwein, 2015). Additionally, point mutations within HSV-1, PRV, and HCMV UL34 suggested specific residues necessary for complex formation (Bjerke et al., 2003; Bubeck et al., 2004; Milbradt et al., 2012; Passvogel et al., 2014; Passvogel et al., 2015; Passvogel et al., 2013; Roller et al., 2010).
Figure 2. Structures of the HSV-1 (PDB: 4ZXS), PRV (PDB: 4Z3U), and HCMV (PDB: 5D5N) nuclear egress complexes.
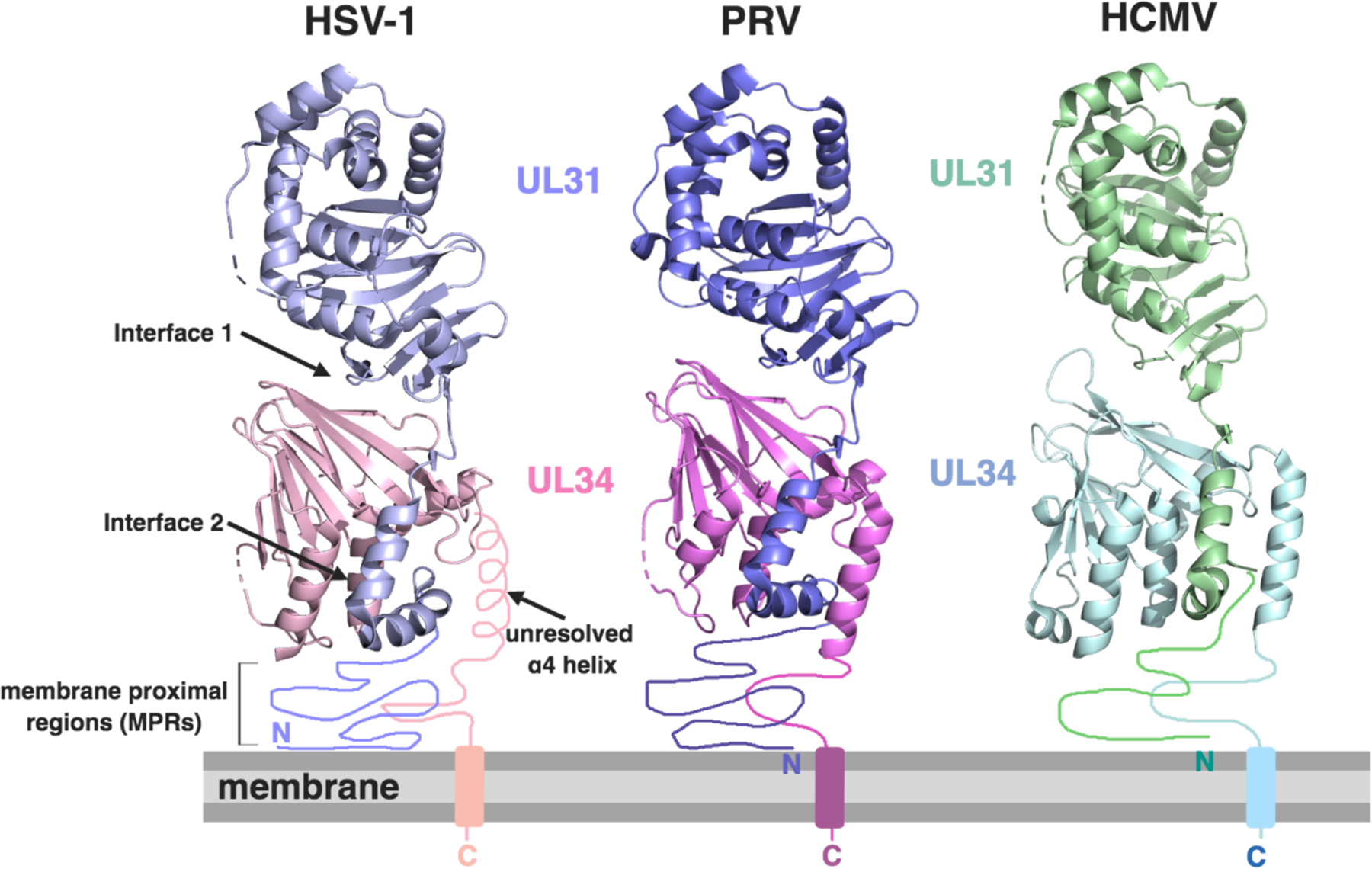
UL31 (purple and green) and UL34 (pink and light blue) form an elongated cylindrical molecule with the hook-like extension of UL31 wrapping around UL34 at the base of the molecules. The unresolved membrane proximal regions (MPRs) and transmembrane regions are drawn in schematically, with the MPRs located closest to the membrane. The approximate location of the unresolved α4 helix is schematically drawn for HSV-1. Interfaces are indicated by arrows. The space above the membrane is the nucleoplasm.
All five NEC structures lack the N terminus of UL31 and the C terminus of UL34, which are predicted to be unstructured and were left out of the crystallized constructs to promote crystallization (Bigalke and Heldwein, 2015; Lye et al., 2015; Walzer et al., 2015; Zeev-Ben-Mordehai et al., 2015). These segments contain sequences necessary for HSV-1 NEC interaction with membranes (Bigalke et al., 2014), along with the TM anchor of UL34, and are expected to be located near the membrane. These so-called membrane-proximal regions (MPRs) map to the base of the NEC complex (Figure 2), which places UL34 at the membrane-proximal and UL31 at the membrane-distal end of the NEC cylinder.
The NEC homologs have remarkably similar structures despite low sequence identity. The biggest difference among the five structures is the conserved C-terminal alpha helix α4 that was unresolved in the HSV-1 UL34 structure (Figure 2) (Bigalke and Heldwein, 2015), possibly due to differences in the construct boundaries, because the HSV-1 UL34 used for crystallization is five amino acid residues shorter than the PRV UL34. Truncated helix α4 in HSV-1 UL34 may be less stable and, as a result, higher flexibility of this region would cause it to be unresolved in the structure. No structural information is yet available on the NEC homologs from gammaherpesviruses such as EBV and KSHV. However, from secondary structure predictions, they are expected to share similar folds with their counterparts from alpha and betaherpesviruses.
A recent molecular dynamics simulation using the available NEC crystal structures investigated the relative flexibilities of NEC heterodimers from HSV-1 and HCMV (Diewald et al., 2018). For these studies, the PRV α4 helix was docked into the HSV-1 structure corresponding to the missing HSV-1 α4 helix region, prior to simulations. Molecular dynamics suggests that isolated NEC heterodimers have different flexibilities, with HCMV NEC being more flexible than HSV-1 NEC. Regions of increased flexibility map to the UL31/UL34 interface 1 (Figure 2), with UL31 capable of twisting around UL34. The differences in flexibility likely stem from low conservation of the interface regions, and it is tempting to speculate that the increased rotational freedom at this UL31/UL34 interface of the HCMV heterodimer could play a role in the nuclear budding process of betaherpesviruses.
Stages in primary envelopment
During primary envelopment, the first step in nuclear egress, capsids bud at the INM to form PEVs in the PNS. This process, at a minimum, requires the following events to take place: the nuclear lamina is dissolved, capsid docks at the INM, membrane is deformed around the capsid to form a nascent bud, and the bud is pinched off the INM releasing the PEV into the perinuclear space. Nuclear budding is a complex process that involves several viral and cellular participants, some of which have not yet been identified. It is also subject to regulation to prevent budding in the absence of the capsid and to ensure that the mature, DNA-containing C-capsids, rather than the immature procapsids, the empty A-capsids, or the scaffold-containing B-capsids, described in more detail below, preferentially undergo budding. These undercharacterized regulatory mechanisms ensure quality control and conservation of resources.
Nuclear lamina alteration
Nuclear lamina is a dense filamentous protein network underneath the INM that provides shape and mechanical stability to the nuclear envelope. Nuclear lamina is primarily composed of two types of intermediate filament proteins called lamins A and B that oligomerize into filaments [reviewed in (Dechat et al., 2008; Gruenbaum and Foisner, 2015)]. The A-type lamins, A and C, which are the products of alternative splicing of the LMNA gene, are expressed mainly in differentiated cells whereas the B-type lamins, B1 and B2, are expressed in all cells (Gruenbaum and Foisner, 2015). The B-type lamins tether the lamina to the INM by binding integral membrane proteins such as the lamin B receptor, lamina-associated proteins (LAPs) and emerin [reviewed in (Goldberg et al., 2008; Schirmer and Foisner, 2007; Wilson and Foisner, 2010)]. The A-type lamins, A and C, add additional stiffness to the nuclear envelope [reviewed in (Dechat et al., 2008; Gruenbaum and Foisner, 2015)].
The dense, overlapping filamentous networks of B-type and A-type lamins create a barrier to capsid docking at the INM and would probably impede membrane deformation. Efficient capsid budding thus requires that the nuclear lamina be disassembled. Complete lamina disassembly in uninfected cells occurs during mitosis [reviewed in (Hetzer, 2010)] and is achieved by phosphorylation of lamins and LAPs by cellular mitotic kinases CDK1 (cyclin dependent kinase 1), PKA (protein kinase A), PKC and MAPK (mitogen-activated protein kinase) (Fields and Thompson, 1995; Goss et al., 1994; Guttinger et al., 2009; Heald and McKeon, 1990; Likhacheva and Bogachev, 2001; Margalit et al., 2005; Peter et al., 1992). However, during herpesvirus infection, no massive disassembly of the lamina is observed, likely, because it is important for the virus to preserve the integrity of the nucleus since this is where viral gene transcription and genome replication take place. Instead, the lamina is disassembled locally (Granzow et al., 1997) through phosphorylation of lamins and LAPs by the viral US3 kinase and likely by cellular kinases that are different from the mitotic ones (Hertel, 2011; Lv et al., 2019a, b; Roller and Baines, 2017b) [reviewed in (Roller and Baines, 2017a)].
Lamins A/C are phosphorylated by PKC which is recruited to the INM, along with the host protein facilitator of nuclear egress p32, by the HSV-1 gamma(1)34.5 protein (Wang et al., 2014; Wu et al., 2016). PKC is also involved in hyperphosphorylation of emerin, along with US3, which also contributes to lamina disassembly (Leach et al., 2007; Morris et al., 2007). UL13 is another viral kinase that phosphorylates lamina in HSV-2 (Cano-Monreal et al., 2009) and HCMV (Marschall et al., 2005; Sharma et al., 2014) but in HSV-1, instead, appears to regulate the distribution of UL31 and UL34 by phosphorylating US3 (Kato et al., 2006). Further mechanistic studies are necessary to delineate the exact contributions of both viral and host proteins facilitating lamina disruption during nuclear egress.
Capsid docking to the INM
Herpesviral capsids are icosahedral, with T = 16, and consist of the major capsid protein, VP5 (UL19) in alphaherpesviruses, arranged into 11 pentons and 150 hexons, plus additional proteins UL18, UL38, and UL35 (Dai and Zhou, 2018) (see Chapter 3). In each capsid, one of the 12 vertices contains a single portal composed of the portal protein UL6 (McElwee et al., 2018) through which viral DNA is encapsidated during assembly (Newcomb et al., 2001) and, presumably, ejected early in infection (Newcomb et al., 2007). The capsid vertex specific complex (CVSC)/capsid-associated tegument complex (CATC) consists of the capsid proteins UL17 and UL25 and ensures proper DNA retention in the mature capsid (Dai and Zhou, 2018; Salmon et al., 1998; Stow, 2001) likely by stabilizing the capsid to help it withstand the internal pressure of several atmospheres from the encapsidated DNA genome (Bauer et al., 2013).
During assembly, a spherical scaffold-containing procapsid forms initially and matures into genome-containing icosahedral C-capsids [reviewed in (Baines, 2011)]. Two additional icosahedral capsid types observed in infected cells are the empty A-capsids and the scaffold-containing B-capsids, which are thought to be the defective by-products of capsid assembly [reviewed in (Baines, 2011)].
How the capsids reach the INM is yet unclear, but they likely dock to the lamina-free, exposed areas of the INM by binding to the UL31 component of the NEC (Funk et al., 2015; Leelawong et al., 2011). UL31 likely interacts with the CVSC/CATC at the capsid vertices because CVSC/CATC components UL25 and UL17 bind UL31 (Yang and Baines, 2011). Furthermore, UL25 mediates interactions between capsids purified from HSV-1 infected cells and NEC expressed in vitro (Takeshima et al., 2019). A putative capsid-binding site of UL31 has been mapped to a conserved surface patch at the membrane-distal end composed of residues (HSV-1 D275-D282, PRV D238-D245) (Bigalke and Heldwein, 2015). In HSV-1, residues R281 and D282 of UL31 have been implicated in NEC/capsid binding (Takeshima et al., 2019) whereas in PRV, residue K242 (HSV-1 K279) is important for the primary capsid envelopment (Ronfeldt et al., 2020; Ronfeldt et al., 2017) but may not interact with the capsid directly (Ronfeldt et al., 2020). Although capsids isolated from UL25-null HSV-1 or PRV can interact with UL31 (Leelawong et al., 2011; Yang et al., 2014), this interaction is insufficient for nuclear egress because in the absence of UL25, HSV-1 and PRV capsids can only dock at the INM but cannot exit the nucleus (Klupp et al., 2006; Kuhn et al., 2008). Taken together, these findings suggest that while UL25/UL31 interaction is not required for capsid docking at the INM, it is necessary for egress.
Although several capsid types are present in the nucleus, primarily C-capsids undergo primary envelopment at the INM whereas A- and B-capsids typically do not (Klupp et al., 2011; Roizman and Furlong, 1974; Stackpole, 1969). These observations imply the existence of a capsid selection mechanism that ensures only mature, DNA-filled capsids exit the nucleus, which could, in principle, increase the yield of infectious virions by reducing unproductive budding of the A- and B-capsids. The nature of this selection mechanism has not yet been identified but likely involve interactions between the CVSC/CATC and the NEC. Although CVSC/CATC is present on all three capsid types, A- and B- capsids have fewer CVSC/CATC copies on the capsid surface (Newcomb et al., 2006). It is also possible that the CVSC/CATC on C-capsids adopts a conformation distinct from those on A- or B-capsids (Newcomb et al., 2017). Multiple NEC binding sites on a mature C-capsid could produce avidity effects, providing the driving force for creating a PEV.
Membrane deformation and budding by the NEC
The NEC is a self-contained budding machine
While the studies discussed above suggested the NEC is necessary for capsid budding at the INM and sufficient for vesiculation of the INM, they left unclear the exact function of the NEC during nuclear egress. Was the NEC mediating the budding process at the INM directly or recruiting yet unknown host proteins? To answer this question, recombinant soluble HSV-1 NEC (lacking the C-terminal TM) was purified and incubated in vitro with synthetic lipid membranes in the form of giant unilamellar vesicles (Bigalke et al., 2014). The NEC vesiculated the membranes, forming intraluminal vesicles within the GUVs, in the absence of any other factors or ATP. This observation was subsequently recapitulated with recombinant PRV NEC anchored to the membranes through the TM of UL34 (Lorenz et al., 2015). These findings demonstrated that the NEC is a self-contained membrane budding machine. The fact that soluble HSV-1 NEC mediates budding shows that, at least, in vitro, the TM is dispensable for budding. Moreover, this region can be replaced with a heterologous TM without any ill effects on egress in vivo (Schuster et al., 2012). This suggested that while the TM anchors the NEC to the INM in vivo, other regions of the NEC are responsible for membrane interactions required for budding.
HSV-1 NEC-mediated budding in vitro requires the presence of acidic lipids (Bigalke et al., 2014), suggesting the potential involvement of electrostatic interactions. In the case of PRV NEC, in-vitro budding required the presence of sphingomyelin or cholesterol (Lorenz et al., 2015). Thus, lipid composition appears to play an important role in budding.
NEC-mediated membrane budding proceeds rapidly in vitro (Bigalke et al., 2014). In addition to generating a scaffold on the inner surface of the nascent bud, the NEC also mediates scission of its neck. The ability of the NEC to mediate the entire budding process is a unique property of this protein complex. Other enveloped viruses encode proteins that mediate budding, yet most must also recruit cellular ESCRT [endosomal sorting complexes required for transport (ESCRT)] proteins [reviewed in (Alonso et al., 2016; Hurley, 2015; Votteler and Sundquist, 2013)]. For example, the HIV matrix protein Gag forms a scaffold on the membranes of buds and recruits ESCRT proteins (Carlson and Hurley, 2012; Flower et al., 2020) [reviewed in (Hurley and Cada, 2018)], notably ESCRT-III proteins that mediate scission by assembling a spiral-like polymer on the inward face of the membrane neck and constricting it (Effantin et al., 2013; Nguyen et al., 2020). Likewise, in Hepatitis C and yellow fever viruses from the Flaviviridae family, membrane deformation is driven by the formation of an external viral glycoprotein coat, but scission requires ESCRT machinery (Barouch-Bentov et al., 2016; Carpp et al., 2011; Corless et al., 2010). Not all enveloped viruses, however, recruit ESCRT proteins for membrane budding. Influenza A virus, for example, solely uses virally encoded proteins M1 and M2 that mediate scaffolding and neck scission, respectively (Rossman et al., 2010) [reviewed in (Rossman and Lamb, 2011, 2013)]. How NEC accomplishes neck scission is unknown, but according to one proposed scenario (Bigalke et al., 2014), formation of the hexagonal NEC lattice could narrow the neck to the point where it could undergo spontaneous scission. Alternatively, the NEC could alter local lipid composition to membrane curvature conducive to scission, as has been proposed for the Influenza M2 protein (Rossman et al., 2010). Additional experiments are needed to elucidate the mechanism behind NEC scission.
HSV-1 nuclear egress is thought to be independent of the ESCRT-III machinery in HEK293 cells because it is insensitive to a dominant negative mutation of Vps4, an ATPase required for ESCRT-III-mediated scission (Crump et al., 2007), considered to be “a gold standard” for ESCRT-III involvement (Alonso et al., 2016). However, a more recent report challenged that assertion by showing that nuclear egress was reduced in the presence of the Vps4 dominant-negative mutant in HeLa cells, rather than in HEK293 cells (Arii et al., 2018). Moreover, depletion of CHMP4B, an ESCRT-III protein, resulted in accumulation of PEVs, apparently, due to impaired scission (Arii et al., 2018). Thus, while HSV-1 NEC is capable of mediating scission in vitro, it may recruit ESCRT-III machinery in vivo to increase the efficiency of scission. A similar concept is at play in Ebola virus, where the viral matrix protein VP40 mediates membrane budding in vitro (Soni and Stahelin, 2014) yet recruits ESCRT machinery in vivo (Licata et al., 2003; Silvestri et al., 2007) [reviewed in (Gordon et al., 2019)]. Due to the discordant findings regarding the role of ESCRTs in HSV-1 nuclear egress, whether the ESCRT-III machinery is universally important for nuclear egress remains unclear. Future studies with different viral strains and host cell types are needed to resolve this question.
NEC assembly into a hexagonal lattice is important for budding
Cryogenic electron microscopy and tomography (cryo-EM/T) demonstrated that during budding in vitro, the soluble HSV-1 NEC heterodimer forms hexagonal “honeycomb” coats on the inner surface of lipid vesicles (Figure 3B) (Bigalke et al., 2014). Given that the NEC cannot cross the membrane bilayer, these budded vesicles appear to be the products of budding and scission, because this would be the only way for the protein to get inside the vesicles. NEC also forms hexagonal coats on the inner surface of capsidless perinuclear vesicles formed in uninfected cells expressing PRV NEC (Figure 3C) (Hagen et al., 2015). Hexagonal symmetry evident in the coats formed as the result of budding both in vitro and in vivo suggested that the hexagonal arrangement is biologically relevant.
Figure 3. Comparison of NEC hexagonal lattice formed by HSV-1 (crystal lattice), PRV (model of cryo-ET coat) and HCMV (crystal lattice).
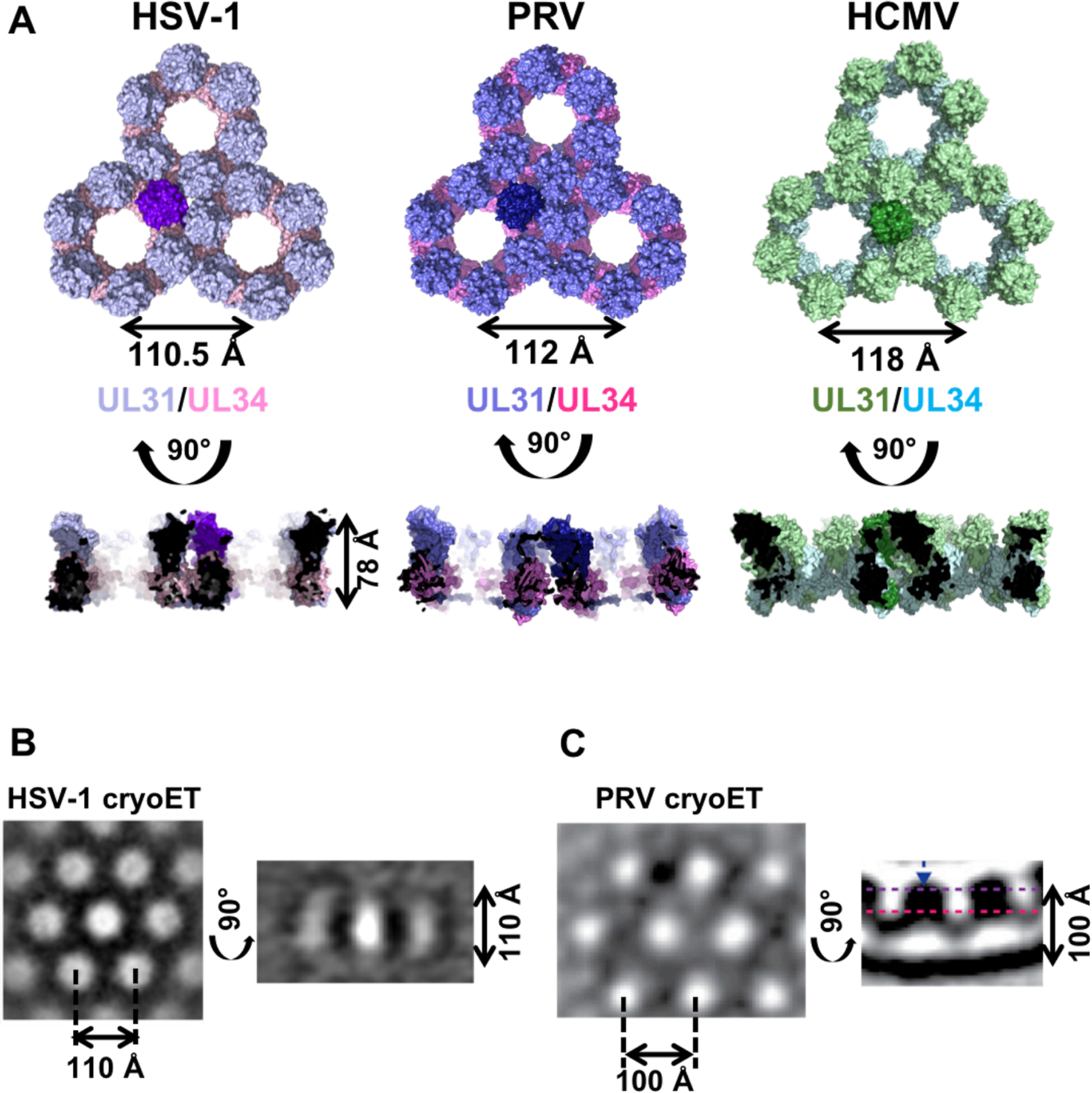
A) Three hexameric rings shown in top and side views with UL31 (purple/green) sitting on top of UL34 (pink/blue). One NEC heterodimer is highlighted in each lattice. The HSV-1 NEC crystal lattice is 78 Å thick. The PRV lattice in the side view is curved since it was determined from a membrane coat and not a planar crystal lattice. Both the HSV-1 NEC (B) and the PRV NEC (C) cryo-ET coats are shown for comparison. The dimensions of the PRV coat are slightly smaller due to curvature. The missing ~30 Å from the crystal lattice is present in the cryo-ET coat. The HSV-1 NEC cryo-ET coat is reprinted from (Bigalke and Heldwein, 2015). The PRV NEC cryo-ET coat is reprinted from (Hagen et al., 2015).
A high-resolution view of interactions involved in NEC oligomerization was afforded by the crystal structure of HSV-1 NEC (Bigalke and Heldwein, 2015). In the crystals, NEC assembled into a hexagonal lattice built from ~110-Å hexameric rings (Figure 3A). (Bigalke and Heldwein, 2015). The symmetry and the dimensions of the hexagonal crystal lattice are very similar to those of the hexagonal NEC coats formed in vitro (Bigalke et al., 2014) and in vivo (Figures 3B and C) (Hagen et al., 2015). Therefore, NEC interactions within the crystal lattice model interactions within the hexagonal coats enabling a systematic exploration of the role of oligomerization in the NEC budding mechanism. Additionally, HCMV NEC also packed into a hexagonal lattice in one of the crystal forms (Walzer et al., 2015), which suggests that the NEC from alpha- and betaherpesviruses use similar strategies for coat formation and budding. Although no structural information is yet available for NEC from any gammaherpesviruses, these principles likely apply to them as well.
The hexagonal lattice is formed by two types of oligomeric interactions: those within the hexameric rings (intra-hexameric) and those between adjacent hexamers (inter-hexameric) (Bigalke and Heldwein, 2015). Mutations disrupting oligomeric interfaces reduce budding both in vitro (Bigalke and Heldwein, 2015) and in vivo (Arii et al., 2019; Bjerke et al., 2003; Roller et al., 2010). For example, the dominant negative (DN) mutation in the UL34 (D35A/E37A) that blocks capsid budding in vivo (Roller et al., 2010), maps to the hexameric interface. Accordingly, this mutant is defective in both budding and coat formation in vitro (Bigalke et al., 2014). Therefore, the NEC oligomerization into a hexagonal coat is not only necessary for efficient budding, and ultimately nuclear egress, but is also the driving force behind the process.
Formation of spherical NEC coats
The NEC crystal lattice is flat while the coat formed on vesicles is curved. While the dimensions of the flat lattice are similar to those observed in the coat, the coat must allow a degree of flexibility that would permit the formation of a curved and ultimately, spherical array. Cryo-ET in combination with cryosectioning (CEMOVIS) and focused ion beam cryomilling (cryo-FIB) were used to analyze the structure of the NEC coats in capsidless perinuclear vesicles formed in uninfected cells expressing PRV NEC (Hagen et al., 2015). The PRV NEC-coated perinuclear vesicles were relatively uniform in size (~100–117 nm in diameter). Interestingly, budded intraluminal vesicles formed by soluble HSV-1 NEC in vitro range widely in size (Bigalke et al., 2014), suggesting that the C terminus of UL34 containing the TM (absent from the HSV- 1 NEC construct used in in vitro studies) may somehow determine the curvature of the NEC scaffold and, ultimately, the size of the budded vesicle. The PRV NEC-coated perinuclear vesicles formed in uninfected cells (Hagen et al., 2015) were smaller than the capsid [~125 nm in diameter (Liu et al., 2017a)] or the PEVs isolated from cells infected with HSV-1 US3-null mutant (Newcomb et al., 2017). This suggests that while the NEC oligomerization favors formation of a scaffold of relatively uniform curvature, the capsid defines the size of the NEC scaffold formed in infected cells.
Aside from flexibility, a purely hexagonal array is flat. Curvature can be achieved by the regular inclusion of another shape - typically a pentagon - within the hexagonal array, which leads to the formation of a polyhedral structure. Consider, for example, a soccer ball, in which a spherical shape is achieved by the inclusion of 12 pentagons within an otherwise hexagonal arrangement, a structure referred to as an icosahedron. Many viruses, including herpesviruses, employ this strategy to form their capsids, and it is reasonable to expect that the NEC coat may form an icosahedral structure. Although icosahedral symmetry was not observed in NEC coats formed either in vitro or in NEC-expressing cells (Bigalke et al., 2014; Hagen et al., 2015), these coats were formed in the absence of capsids, leaving open the possibility that formation of an icosahedral NEC coat requires the presence of an icosahedral capsid. Ideally, this would require in situ imaging of PEVs in infected cells at a sufficiently high resolution, which has not yet been achievable. Instead, NEC coats formed in the presence of capsids were visualized in PEVs isolated from cells infected with an HSV-1 US3-null virus (Newcomb et al., 2017). Deletion of the US3, a viral kinase, results in an accumulation of PEVs within the perinuclear space (Klupp et al., 2001; Reynolds et al., 2002; Ryckman and Roller, 2004). No icosahedral symmetry was evident in reconstructions of NEC coats in PEVs. However, these PEVs were formed in the absence of US3, which could have affected the coat structure. Therefore, further studies with wild-type HSV-1 or PRV are required to conclusively rule out the icosahedral symmetry in the NEC coats.
In the absence of icosahedral arrangement, a curved coat can alternatively be formed through the inclusion of irregular defects into the hexagonal NEC lattice. Such non-icosahedral arrangement has been observed for the immature HIV-1 capsid formed by the Gag matrix protein (Briggs et al., 2009; Schur et al., 2015) and for the early poxvirus envelope (Heuser, 2005) formed by D13 protein (Hyun et al., 2011). Although such irregular defects have not yet been reported for the HSV-1 NEC coats, molecular dynamics simulations of the hexagonal NEC lattice show it to be very flexible (Diewald et al., 2018). Such flexibility may allow for effortless incorporation of irregular defects within the coat. Ultimately, determining the presence of irregular defects within the NEC coat awaits high-resolution cryo-ET reconstructions.
Regulation of NEC budding
HSV-1 and PRV NEC-mediated budding is robust in vitro (Bigalke et al., 2014; Lorenz et al., 2015) and in uninfected cells expressing the PRV NEC (Klupp et al., 2007). However, during infection, capsidless perinuclear vesicles are rarely observed, which suggests that NEC-mediated budding at the INM is negatively regulated to prevent unproductive budding in the absence of C-capsids [reviewed in (Hellberg et al., 2016; Roller and Baines, 2017a)]. One way to regulate budding could be by maintaining the NEC at the INM in an inactive form prior to contact with the capsid. Phosphorylation of UL31 could be one potential mechanism of downregulating the NEC budding activity. The N terminus of HSV-1 UL31 contains six serines phosphorylated by the US3 kinase (Mou et al., 2009). Replacement of these UL31 serine residues with glutamates, which mimic phosphorylated serines, prevented capsid budding (Mou et al., 2009). By contrast, replacing these serines with alanines, which mimics a constitutively dephosphorylated state, resulted in the accumulation of the PEVs in the perinuclear space (Mou et al., 2009). Deletion or inactivation of the US3 kinase results in a similar phenotype (Bjerke et al., 2003; Klupp et al., 2007; Klupp et al., 2001; Reynolds et al., 2002; Ryckman and Roller, 2004), which suggests that PEVs accumulate whenever US3 cannot phosphorylate UL31. While this could mean that phosphorylation of UL31 is important for the de-envelopment process (Mou et al., 2009), the alternative possibility is that in the absence of negative regulation of budding leads to overproduction of the PEVs.
If phosphorylation of UL31, indeed, inhibits the NEC activity prior to C-capsid docking, dephosphorylation would then be required to relieve inhibition. Herpesviral genomes do not encode any phosphatases (Bernard Roizman, 2013) but could, potentially, recruit host phosphatases for this purpose. Indeed, PRV US3 activates host phosphatases to rearrange cytoplasmic actin (Jacob et al., 2013) (see Chapter 3). Altogether, these findings implicate US3 in nuclear egress regulation.
A number of tegument proteins, described later in this Chapter, have been implicated in nuclear egress and could, in principle, regulate the NEC activity either positively or negatively. Additionally, the nonstructural ICP22 (Maruzuru et al., 2014) protein interacts with the NEC but is not critical for egress. Another nonstructural HSV-1 protein, UL24, has also been implicated in contributing to nuclear egress (Lymberopoulos et al., 2011), yet not much is known about this phenotype.
De-envelopment
During de-envelopment, the PEVs lose their envelopes and NEC coats at the ONM, and the naked capsids are released into the cytoplasm. This poorly understood process must include fusion of the envelope with the ONM as well as the disassembly of the NEC scaffold around the capsid, which could occur concomitantly or sequentially. To ensure efficient nuclear egress, PEVs must preferentially fuse with the ONM rather than the INM, but how this selection is achieved or whether backfusion events occur is yet unknown. In uninfected cells expressing UL31 and UL34 from HSV-1 or PRV, de-envelopment of the capsidless perinuclear vesicles either does not take place or is very inefficient because PEVs accumulate in herniations (Klupp et al., 2007). This suggests that de-envelopment requires viral proteins besides UL31 and UL34.
Defects in de-envelopment typically manifest as the accumulation of PEVs in the PNS, with a concomitant reduction in viral titer. Several proteins, as described below, have been implicated in de-envelopment. Yet, proteins that mediate membrane fusion during de-envelopment have not yet been conclusively identified. The conserved viral glycoproteins gB and gH, both of which are essential for membrane fusion during herpesviral entry, have been reported as important for efficient de-envelopment in HSV-1 because deletion of both gB and gH impedes nuclear egress in HSV-1 (Farnsworth et al., 2007). Intact fusion loops of gB are important for the de-envelopment (Wright et al., 2009). However, single deletions of gB or gH have minimal effects on de-envelopment (Farnsworth et al., 2007) implying functional redundancy, which is inconsistent with both glycoproteins being essential for fusion during HSV-1 entry (see Chapter 2). This raises the possibility that instead of mediating fusion, gB and gH instead contribute to de-envelopment through a different mechanism. In any case, gB and gH are simultaneously dispensable for de-envelopment in the closely related PRV (Granzow et al., 2001; Klupp et al., 2008). Overexpression of another viral glycoprotein gK from HSV-1, which localizes to the ER during infection, results in the accumulation of PEVs (Hutchinson and Johnson, 1995), but its precise role in the de-envelopment remains unclear.
An outer tegument protein, UL51, has also been implicated in de-envelopment in HSV-1 (Nozawa et al., 2005). However, UL51 localizes to the Golgi and probably contributes to de-envelopment indirectly. Treatment of PRV-infected cells with the inhibitor of Golgi-to-ER transport, brefeldin A, causes the accumulation of PEVs (Whealy et al., 1991), indicating that UL51 could potentially facilitate de-envelopment indirectly by helping traffic yet unidentified membrane fusogen(s) to ER and the ONM. Nonetheless, UL51 is dispensable for de-envelopment in HCMV, PRV and Bovine Herpesvirus 1 (Klupp et al., 2005c; Raza et al., 2016; Schauflinger et al., 2011; Womack and Shenk, 2010). The identification of viral or cellular fusogens mediating de-envelopment and the elucidation of their mechanisms is an important area for future investigation.
During de-envelopment, the hexagonal NEC coats within the PEVs need to be disassembled to expose the surface of the capsid for subsequent interactions with the tegument proteins in the cytoplasm. The fate of the NEC following nuclear egress remains unclear. What triggers disassembly is also unknown, but phosphorylation of UL31 by US3 may play a role in the disassembly of the NEC coat. HSV-1 US3 is present in the PEVs (Reynolds et al., 2002), and it is tempting to speculate that phosphorylation of UL31 within the PEVs could lead to structural rearrangements that would disrupt the NEC interactions within the hexagonal lattice and promote the disassembly of the NEC coat. However, the N terminal region of UL31, targeted by US3, is thought to be located near the membrane due to its apparent requirement for NEC/membrane interactions, and it is unclear how US3 would access this region sandwiched between the NEC coat and the membrane. Therefore, disassembly of the NEC coat could, instead, be facilitated by perturbations to the envelope during fusion with the ONM.
Phosphorylation of UL31 by US3 could alternatively serve as a selection mechanism for mature C-capsids. According to this scenario, in the absence of phosphorylation, A- and B-capsids could undergo primary envelopment, along with C-capsids, yet be incapable of undergoing de-envelopment and, therefore, accumulate in the PNS as PEVs.
gB, another target of US3, contains only one US3 phosphorylation site located within the gB cytoplasmic domain (Wisner et al., 2009). Mutation of this phosphorylation site results in the accumulation of PEVs when gH is absent (Wisner et al., 2009). The phosphorylation of the cytoplasmic domain could potentially regulate the function of gB by controlling its oligomerization state or by triggering a fusogenic conformational change, with the caveat that gB may not be mediating fusion during de-envelopment (described above).
Collectively, these findings point to the involvement of US3 kinase activity in the de-envelopment process. Nevertheless, the modest defect in viral growth, only 10-fold, observed in viruses lacking US3 kinase (Mou et al., 2009) argues that while US3 kinase activity is important for efficient de-envelopment, it is not essential, at least under the conditions of cell culture.
Tegument and nuclear egress
The tegument is a multiprotein layer sandwiched between the capsid and the envelope, which is a unique feature of herpesvirus particles. Although tegument proteins are thought to be recruited to cytoplasmic capsids, some tegument proteins localize to the nucleus and a few of these, UL36, UL47, UL16, UL11 and UL21, are implicated in nuclear egress.
At ~300-kDa, UL36 is the largest protein conserved across the Herpesviridae family. The best studied role of UL36 is its interaction with cellular motor proteins dynein and kinesin that is essential for directed trafficking of HSV-1 and PRV capsids post-entry and during egress (Shanda and Wilson, 2008; Zaichick et al., 2013) (see Chapters 3 and 9). In both HSV-1 and PRV, UL36 is dispensable for nuclear egress (Desai, 2000; Luxton et al., 2006; Roberts et al., 2009), but in PRV, the absence, capsids egress from the nucleus is less efficient in the absence of UL36 (Luxton et al., 2006). Infections with PRV UL36 dually labeled with fluorescent proteins at both N and C termini showed that nuclear UL36 is proteolytically cleaved (Leelawong et al., 2012), and only the C-terminal fragment of PRV UL36 localizes to the nucleus and co-purifies with nuclear C-capsids (Leelawong et al., 2012). The C-terminal fragment of PRV UL36 is also sufficient for increased levels of nuclear egress compared to a UL36-null virus (Leelawong et al., 2012). Association of the C-terminal fragment of UL36 with nuclear capsids has also been reported for HSV-1 (Radtke et al., 2010). Indeed, UL36 is a component of the aforementioned CVSC/CATC located at the capsid vertices. In addition to two copies of UL25 and one copy of UL17, each CVSC/CATC also contains two copies of the C-terminal helix of UL36 (Dai and Zhou, 2018; Liu et al., 2017b). Large numbers of B-capsids were observed in the cytoplasm of cells infected with UL36-null HSV-1 (Kharkwal et al., 2016), which suggests that UL36 contributes to the selection of mature C-capsids for nuclear egress.
Several outer tegument proteins have also been implicated in nuclear egress. UL11, UL16, and UL21 are important for efficient cytoplasmic budding of capsids that leads to the formation of infectious virions but during infection also localize to the nucleus. UL16 is a conserved, ~40-kDa tegument protein that colocalizes with HSV-1 capsids within the nucleus of infected cells but is not found on HSV-1 nor HSV-2 capsids after purification (Meckes and Wills, 2007; Nalwanga et al., 1996; Oshima et al., 1998), potentially due to a weak association between UL16 and the capsid. Infections with HSV-1 or HSV-2 lacking UL16 increases the proportion of A- and B-capsids relative to C-capsids within the nucleus, suggesting a defect in viral DNA packaging (Gao et al., 2018; Oshima et al., 1998). While both HSV-1 and HSV-2 lacking UL16 have reduced ability to generate C-capsids, they differ in their replicative capacity. In HSV-1 strains F and KOS, viral titers were reduced 10-fold when most of the UL16 gene was deleted (Gao et al., 2018) whereas in several HSV-2 strains, viral titers were reduced 100-fold (Gao et al., 2017; Gao et al., 2018), and in PRV, UL16 is dispensable for replication (Klupp et al., 2005b). These findings suggest a species-specific role for UL16 during infection such that the differences in replication defects could reflect varying roles of UL16 in the viral replication life cycle. In the absence of UL16, HSV-1 C-capsids relocate into the cytoplasm whereas HSV-2 C-capsids are retained in the nucleus (Gao et al., 2017; Gao et al., 2018). Perhaps, UL16 deletion has a lesser effect on the HSV-1 titers because more C-capsids can enter the maturation pathway.
UL11 is the smallest among conserved tegument proteins. During infection, HSV-1 UL11 localizes to both nuclear and cytoplasmic membranes but does not interact with intranuclear capsids, as assessed by immunogold electron microscopy (Baines et al., 1995). Nonetheless, a C-terminal deletion of ~2/3 of HSV-1 UL11 results in lower viral titers and a ~10% increase in capsid retention at the INM (Baines and Roizman, 1992). The deletion does not affect UL11/16 interactions indicating that this phenotype is due to a loss of some other function of UL11 (Loomis et al., 2003b). To date, this phenotype appears specific to HSV-1 because UL11-null PRV has no obvious defects in nuclear egress (Klupp et al., 2005b) (Klupp et al., 2005a; Kopp et al., 2003).
UL21 is a ~61-kDa capsid-associated tegument protein that interacts with UL11 and UL16 (de Wind et al., 1992; Harper et al., 2010; Klupp et al., 2005b; Loomis et al., 2003a). In HSV-2 strain 186, deletion of UL21 results in capsid retention within the nucleus and a defect in cell-to-cell spread (Finnen and Banfield, 2018; Le Sage et al., 2013). Some studies examining HSV-1 UL21 found it to be dispensable for replication (Baines et al., 1994; Mbong et al., 2012) while another reported a 100-fold decrease in replication, albeit due to the presence of A- and B-capsids in the cytoplasm rather than a defect in nuclear egress (Sarfo et al., 2017). While all PRV strains exhibit a 10-fold decrease in replication, PRV strain NIA-3 lacking UL21 results in A- and B-capsids representing 50% of cytoplasmic capsids whereas the Kaplan strain exhibits no defects in viral maturation (de Wind et al., 1992; Klopfleisch et al., 2006; Klupp et al., 2005b; Klupp et al., 1995; Wagenaar et al., 2001). Strain-specific differences could be the result of differences among the strains, the presence of additional unknown mutations in the virus, or due to differences in the cell lines used.
While mainly studied in HSV-1, UL47 may play an important role during nuclear egress. UL47-null HSV-1 results in a 50-fold reduction in PEVs and accumulation of capsids in the nucleus (Liu et al., 2014). The block in nuclear egress may be the result of disrupting a capsid-NEC interaction, as UL47 associates with both UL31/UL34 as well as UL17, which is found on capsids (Liu et al., 2014; Scholtes et al., 2010). UL47 homologs are not required for nuclear egress in PRV (Kopp et al., 2002) or bovine herpes virus 1 (Lobanov et al., 2010).
The gamma(1)34.5 protein interacts with the NEC (Jing et al., 2004; Wu et al., 2016), and its deletion results in drastically reduced viral replication due to a defect in nuclear lamina dissolution (described above) that ultimately prevents capsid egress (Wu et al., 2016).
Nuclear membrane alterations
When properly assembled, the NEC functions as a complete membrane budding nanomachine, forming perfectly sized vesicles encapsulating herpesvirus capsids in infected cells. However, when the proper assembly of the NEC is disrupted by mutations or when the capsid is absent, the perturbation of the membrane budding activity of the NEC results in oddly shaped membrane structures.
For example, tightly packed multilayered membranous structures at the nuclear envelope were observed in insect cells heterologously expressing EBV BFRF1, a homolog of HSV-1 UL34 (Gonnella et al., 2005). These structures were also observed in insect cells co-expressing BFRF1 and BFLF2, a homolog of HSV-1 UL31. However, some of the structures are less tightly packed and “were in concentric whorls” (Gonnella et al., 2005). The increased spacing between the INM and ONM was also seen in Vero cells infected with a baculovirus encoding only HSV-1 UL34 (Ye et al., 2000). The mechanism of UL34-mediated membrane alterations could be due to alternations of the nuclear lamina, which provides structural support to the nuclear envelope (Reynolds et al., 2004). While expression of UL34 alone resulted in nuclear membrane alterations, aberrant membrane vesiculation has been observed during infections where interaction between UL31 and UL34 were perturbed by mutagenesis (Roller et al., 2011). Disruption of the UL31/UL34 interaction also resulted in poor co-localization to the nuclear rim (Roller et al., 2011). These findings suggest that while UL34 alone is capable of altering the structure of the nuclear envelope, only the NEC can mediate productive budding. Tightly packed multilayered membranes have also been observed upon infection with PRV lacking UL11, UL16, and UL21 (Klupp et al., 2005b), although the mechanism behind this phenotype is yet unknown.
Nuclear budding in uninfected cells
Although the process of nuclear egress was once thought to be unique to herpesviruses, we now know that a topologically similar process is also utilized during the nuclear export of large cellular synaptic ribonucleoproteins (RNPs) in Drosophila (Speese et al., 2012) [reviewed in (Fradkin and Budnik, 2016)]. These RNPs cannot exit the nucleus through the nuclear pores and, instead, bud at the INM to form RNP cargo containing vesicles in the perinuclear space that subsequently fuse at the ONM like herpesviruses. Torsin ATPases are essential for nuclear budding in uninfected cells, and in their absence, vesicles formed in the perinuclear space remain attached to the INM (Goodchild et al., 2005; Jokhi et al., 2013; Kim et al., 2010). Abundant PEVs containing granules that resemble RNPs, some in the process of fusion with the ONM have also been observed in sea urchin gastrula, suggesting that nuclear egress may be involved in nucleocytoplasmic transfer during sea urchin development (LaMassa et al., 2018). Thus, the nuclear egress may be more common in host cells than previously thought.
The existence of the nuclear egress pathway in uninfected cells may imply that herpesviruses have hijacked it. However, the absence of both isoforms of torsin has only a modest effect on the efficiency of nuclear egress in HSV-1 (Turner et al., 2015). While this does not rule out the involvement of other host proteins in herpesvirus nuclear egress, it suggests that herpesviruses utilize a distinct nuclear egress pathway. Further studies are clearly necessary to delineate the contributions of host factors to nuclear egress of herpesviruses vs. large RNPs and mechanistic similarities and differences between these two processes.
Alternative routes of nuclear egress: nuclear envelope breakdown and nuclear pore enlargement
The NEC is essential for nuclear egress, and deletions of either UL31 or UL34 reduce titers at least 1000-fold (Fuchs et al., 2002; Reynolds et al., 2001; Roller et al., 2000). Nevertheless, some infectious virus can be recovered even in the absence of UL31 or UL34. This suggests that while the envelopment/de-envelopment route is preferred, other, far less efficient nuclear escape routes also exist. Two such routes have been proposed: the nuclear envelope breakdown (NEBD) whereby the nuclear envelope is ruptured (Grimm et al., 2012; Klupp et al., 2011; Maric et al., 2014; Stackpole, 1969) and the dilation of nuclear pores (Leuzinger et al., 2005; Wild et al., 2009), either of which would allow the capsid to access the cytoplasm directly.
The NEBD phenomenon was discovered when upon passaging in culture, PRV lacking UL31 or UL34 acquired the ability to induce NEBD (Grimm et al., 2012; Klupp et al., 2011) by manipulating mitosis-related processes (Grimm et al., 2012). NEBD, along with reduced viral titers, was also observed during HSV-1 infections in cells lacking the cellular ATPase torsinA (Maric et al., 2014).
Studies using a combination of scanning and transmission electron microscopy of HSV-1 infected Vero and Hela cells showed instances of enlarged nuclear pores (~100 nm) (Wild et al., 2009), yet biochemical studies by other groups using fluorescent microscopy (Nagel et al., 2008) and live-cell imaging (Hofemeister and O’Hare, 2008) were unable to corroborate these findings. This suggests that while there may be instances of nuclear pore enlargement under certain experimental conditions, this route of capsid egress from the nucleus appears atypical.
It is unlikely that either of these alternative nuclear exit routes contribute significantly to nuclear egress under normal circumstances because they would eliminate an important checkpoint in the capsid quality control. NEC-mediated budding as an important quality control mechanism ensuring that primarily DNA-filled C-capsids undergo maturation and are released from the cells.
Many viruses that replicate in the nucleus, e.g., adenoviruses, parvoviruses, and polyomaviruses, escape the nucleus by rupturing the nuclear envelope (Cohen et al., 2011; Raghava et al., 2013; Tollefson et al., 1996) [reviewed in (Kobiler et al., 2012; Mettenleiter, 2015)]. By contrast, herpesviruses clearly favor the envelopment/de-envelopment pathway and exit by an alternative route only when this preferred route of nuclear egress is disrupted.
Discussion and future trends
The unusual nuclear escape route used by all herpesviruses, which involves capsids budding at the INM (primary envelopment) followed by fusion of the resulting perinuclear enveloped virions with the ONM (de-envelopment), is supported by substantial evidence. Yet, our mechanistic understanding of this process remains in infancy. The significant advances made in the field over the last decade have clarified some aspects of the nuclear egress mechanism. For example, we now know that the key player in nuclear egress, the NEC, is a membrane budding machine that scaffolds the membrane by forming the striking hexagonal coats on its surface and interacts with the capsid. At the same time, the number of proteins involved in nuclear egress has increased, and with it, the complexity of this process. In contrast, we know very little about the mechanism by which perinuclear enveloped virions fuse with the ONM or the proteins involved in this process. Therefore, we envision clarification of the mechanisms that control capsid budding and NEC activity as one major direction of inquiry for the next decade while another is the elucidation of the mechanism of the subsequent membrane fusion step. Technological advances in in situ imaging of infected cells by cryo-EM and cryo-ET will be essential for visualizing the process of nuclear egress and the interactions that shape it at high resolution.
References
- Alonso YAM, Migliano SM, and Teis D (2016). ESCRT-III and Vps4: a dynamic multipurpose tool for membrane budding and scission. FEBS J 283, 3288–3302. [DOI] [PubMed] [Google Scholar]
- Arii J, Takeshima K, Maruzuru Y, Koyanagi N, Kato A, and Kawaguchi Y (2019). Roles of the Interhexamer Contact Site for Hexagonal Lattice Formation of the Herpes Simplex Virus 1 Nuclear Egress Complex in Viral Primary Envelopment and Replication. J Virol 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arii J, Watanabe M, Maeda F, Tokai-Nishizumi N, Chihara T, Miura M, Maruzuru Y, Koyanagi N, Kato A, and Kawaguchi Y (2018). ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat Commun 9, 3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines JD (2011). Herpes simplex virus capsid assembly and DNA packaging: a present and future antiviral drug target. Trends Microbiol 19, 606–613. [DOI] [PubMed] [Google Scholar]
- Baines JD, Jacob RJ, Simmerman L, and Roizman B (1995). The herpes simplex virus 1 UL11 proteins are associated with cytoplasmic and nuclear membranes and with nuclear bodies of infected cells. J Virol 69, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines JD, Koyama AH, Huang T, and Roizman B (1994). The UL21 gene products of herpes simplex virus 1 are dispensable for growth in cultured cells. Journal of virology 68, 2929–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines JD, and Roizman B (1992). The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J Virol 66, 5168–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barouch-Bentov R, Neveu G, Xiao F, Beer M, Bekerman E, Schor S, Campbell J, Boonyaratanakornkit J, Lindenbach B, Lu A, et al. (2016). Hepatitis C Virus Proteins Interact with the Endosomal Sorting Complex Required for Transport (ESCRT) Machinery via Ubiquitination To Facilitate Viral Envelopment. MBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DW, Huffman JB, Homa FL, and Evilevitch A (2013). Herpes virus genome, the pressure is on. J Am Chem Soc 135, 11216–11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard Roizman DMK, and Whitley Richard J. (2013). Herpes Simplex Viruses. In Fields Virology, David PH Knipe M ed. (Wolters Kluwer Lippincott Williams and Wilkins; ). [Google Scholar]
- Bigalke JM, and Heldwein EE (2015). Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J 34, 2921–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigalke JM, and Heldwein EE (2016). Nuclear Exodus: Herpesviruses Lead the Way. Annu Rev Virol 3, 387–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigalke JM, Heuser T, Nicastro D, and Heldwein EE (2014). Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5, 4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerke SL, Cowan JM, Kerr JK, Reynolds AE, Baines JD, and Roller RJ (2003). Effects of charged cluster mutations on the function of herpes simplex virus type 1 UL34 protein. J Virol 77, 7601–7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AR, Klupp BG, Granzow H, Tirabassi R, Enquist LW, and Mettenleiter TC (2000). Role of the cytoplasmic tail of pseudorabies virus glycoprotein E in virion formation. J Virol 74, 4004–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, and Krausslich HG (2009). Structure and assembly of immature HIV. Proc Natl Acad Sci U S A 106, 11090–11095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck A, Wagner M, Ruzsics Z, Lotzerich M, Iglesias M, Singh IR, and Koszinowski UH (2004). Comprehensive mutational analysis of a herpesvirus gene in the viral genome context reveals a region essential for virus replication. J Virol 78, 8026–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano-Monreal GL, Wylie KM, Cao F, Tavis JE, and Morrison LA (2009). Herpes simplex virus 2 UL13 protein kinase disrupts nuclear lamins. Virology 392, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson LA, and Hurley JH (2012). In vitro reconstitution of the ordered assembly of the endosomal sorting complex required for transport at membrane-bound HIV-1 Gag clusters. Proceedings of the National Academy of Sciences of the United States of America 109, 16928–16933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpp LN, Galler R, and Bonaldo MC (2011). Interaction between the yellow fever virus nonstructural protein NS3 and the host protein Alix contributes to the release of infectious particles. Microbes Infect 13, 85–95. [DOI] [PubMed] [Google Scholar]
- Chang YE, and Roizman B (1993). The product of the UL31 gene of herpes simplex virus 1 is a nuclear phosphoprotein which partitions with the nuclear matrix. J Virol 67, 6348–6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Marr AK, Garcin P, and Pante N (2011). Nuclear envelope disruption involving host caspases plays a role in the parvovirus replication cycle. J Virol 85, 4863–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corless L, Crump CM, Griffin SD, and Harris M (2010). Vps4 and the ESCRT-III complex are required for the release of infectious hepatitis C virus particles. J Gen Virol 91, 362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump C (2018). Virus Assembly and Egress of HSV. Adv Exp Med Biol 1045, 23–44. [DOI] [PubMed] [Google Scholar]
- Crump CM, Yates C, and Minson T (2007). Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. Journal of virology 81, 7380–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, and Zhou ZH (2018). Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wind N, Wagenaar F, Pol J, Kimman T, and Berns A (1992). The pseudorabies virus homology of the herpes simplex virus UL21 gene product is a capsid protein which is involved in capsid maturation. J Virol 66, 7096–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, and Goldman RD (2008). Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 22, 832–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P, Sexton GL, McCaffery JM, and Person S (2001). A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J Virol 75, 10259–10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai PJ (2000). A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J Virol 74, 11608–11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai PJ, Pryce EN, Henson BW, Luitweiler EM, and Cothran J (2012). Reconstitution of the Kaposi’s sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J virol 86, 594–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diewald B, Socher E, Soldner CA, and Sticht H (2018). Conformational Dynamics of Herpesviral NEC Proteins in Different Oligomerization States. International journal of molecular sciences 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effantin G, Dordor A, Sandrin V, Martinelli N, Sundquist WI, Schoehn G, and Weissenhorn W (2013). ESCRT-III CHMP2A and CHMP3 form variable helical polymers in vitro and act synergistically during HIV-1 budding. Cellular microbiology 15, 213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina A, Feederle R, Raffa S, Gonnella R, Santarelli R, Frati L, Angeloni A, Torrisi MR, Faggioni A, and Delecluse HJ (2005). BFRF1 of Epstein-Barr virus is essential for efficient primary viral envelopment and egress. J Virol 79, 3703–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth A, Goldsmith K, and Johnson DC (2003). Herpes simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J Virol 77, 8481–8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, and Johnson DC (2007). Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci U S A 104, 10187–10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields AP, and Thompson LJ (1995). The regulation of mitotic nuclear envelope breakdown: a role for multiple lamin kinases. Prog Cell Cycle Res 1, 271–286. [DOI] [PubMed] [Google Scholar]
- Finnen RL, and Banfield BW (2018). CRISPR/Cas9 Mutagenesis of UL21 in Multiple Strains of Herpes Simplex Virus Reveals Differential Requirements for pUL21 in Viral Replication. Viruses 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower TG, Takahashi Y, Hudait A, Rose K, Tjahjono N, Pak AJ, Yokom AL, Liang X, Wang HG, Bouamr F, et al. (2020). A helical assembly of human ESCRT-I scaffolds reverse-topology membrane scission. Nat Struct Mol Biol 27, 570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradkin LG, and Budnik V (2016). This bud’s for you: mechanisms of cellular nucleocytoplasmic trafficking via nuclear envelope budding. Curr Opin Cell Biol 41, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs W, Klupp BG, Granzow H, Osterrieder N, and Mettenleiter TC (2002). The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J Virol 76, 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk C, Ott M, Raschbichler V, Nagel CH, Binz A, Sodeik B, Bauerfeind R, and Bailer SM (2015). The Herpes Simplex Virus Protein pUL31 Escorts Nucleocapsids to Sites of Nuclear Egress, a Process Coordinated by Its N-Terminal Domain. PLoS Pathog 11, e1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Hay TJM, and Banfield BW (2017). The product of the herpes simplex virus 2 UL16 gene Is critical for the egress of capsids from the nuclei of infected cells. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Yan X, and Banfield BW (2018). Comparative Analysis of UL16 Mutants Derived from Multiple Strains of Herpes Simplex Virus 2 (HSV-2) and HSV-1 Reveals Species-Specific Requirements for the UL16 Protein. J Virol 92, e00629–00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershon AA, Sherman DL, Zhu Z, Gabel CA, Ambron RT, and Gershon MD (1994). Intracellular transport of newly synthesized varicella-zoster virus: final envelopment in the trans-Golgi network. J Virol 68, 6372–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MW, Fiserova J, Huttenlauch I, and Stick R (2008). A new model for nuclear lamina organization. Biochem Soc Trans 36, 1339–1343. [DOI] [PubMed] [Google Scholar]
- Gonnella R, Farina A, Santarelli R, Raffa S, Feederle R, Bei R, Granato M, Modesti A, Frati L, Delecluse HJ, et al. (2005). Characterization and intracellular localization of the Epstein-Barr virus protein BFLF2: interactions with BFRF1 and with the nuclear lamina. J Virol 79, 3713–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild RE, Kim CE, and Dauer WT (2005). Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron 48, 923–932. [DOI] [PubMed] [Google Scholar]
- Gordon TB, Hayward JA, Marsh GA, Baker ML, and Tachedjian G (2019). Host and Viral Proteins Modulating Ebola and Marburg Virus Egress. Viruses 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss VL, Hocevar BA, Thompson LJ, Stratton CA, Burns DJ, and Fields AP (1994). Identification of nuclear beta II protein kinase C as a mitotic lamin kinase. J Biol Chem 269, 19074–19080. [PubMed] [Google Scholar]
- Granzow H, Klupp BG, Fuchs W, Veits J, Osterrieder N, and Mettenleiter TC (2001). Egress of alphaherpesviruses: comparative ultrastructural study. J Virol 75, 3675–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzow H, Weiland F, Jons A, Klupp BG, Karger A, and Mettenleiter TC (1997). Ultrastructural analysis of the replication cycle of pseudorabies virus in cell culture: a reassessment. J Virol 71, 2072–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm KS, Klupp BG, Granzow H, Muller FM, Fuchs W, and Mettenleiter TC (2012). Analysis of viral and cellular factors influencing herpesvirus-induced nuclear envelope breakdown. J Virol 86, 6512–6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbaum Y, and Foisner R (2015). Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem 84, 131–164. [DOI] [PubMed] [Google Scholar]
- Guttinger S, Laurell E, and Kutay U (2009). Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat Rev Mol Cell Biol 10, 178–191. [DOI] [PubMed] [Google Scholar]
- Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, Klupp BG, Siebert CA, Vasishtan D, Bauerlein FJ, et al. (2015). Structural Basis of Vesicle Formation at the Inner Nuclear Membrane. Cell 163, 1692–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper AL, Meckes DG Jr., Marsh JA, Ward MD, Yeh PC, Baird NL, Wilson CB, Semmes OJ, and Wills JW (2010). Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J Virol 84, 2963–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heald R, and McKeon F (1990). Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 61, 579–589. [DOI] [PubMed] [Google Scholar]
- Hellberg T, Passvogel L, Schulz KS, Klupp BG, and Mettenleiter TC (2016). Nuclear Egress of Herpesviruses: The Prototypic Vesicular Nucleocytoplasmic Transport. Adv Virus Res 94, 81–140. [DOI] [PubMed] [Google Scholar]
- Henaff D, Radtke K, and Lippe R (2012). Herpesviruses exploit several host compartments for envelopment. Traffic 13, 1443–1449. [DOI] [PubMed] [Google Scholar]
- Hertel L (2011). Herpesviruses and intermediate filaments: close encounters with the third type. Viruses 3, 1015–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzer MW (2010). The nuclear envelope. Cold Spring Harb Perspect Biol 2, a000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J (2005). Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J Cell Biol 169, 269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofemeister H, and O’Hare P (2008). Nuclear pore composition and gating in herpes simplex virus-infected cells. J Virol 82, 8392–8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollinshead M, Johns HL, Sayers CL, Gonzalez-Lopez C, Smith GL, and Elliott G (2012). Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J 31, 4204–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, and Rosenstrom P (2010). Dali server: conservation mapping in 3D. Nucleic acids research 38, W545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH (2015). ESCRTs are everywhere. EMBO J 34, 2398–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, and Cada AK (2018). Inside job: how the ESCRTs release HIV-1 from infected cells. Biochem Soc Trans 46, 1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson L, and Johnson DC (1995). Herpes simplex virus glycoprotein K promotes egress of virus particles. J Virol 69, 5401–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun JK, Accurso C, Hijnen M, Schult P, Pettikiriarachchi A, Mitra AK, and Coulibaly F (2011). Membrane remodeling by the double-barrel scaffolding protein of poxvirus. PLoS Pathog 7, e1002239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob T, Van den Broeke C, van Troys M, Waterschoot D, Ampe C, and Favoreel HW (2013). Alphaherpesviral US3 kinase induces cofilin dephosphorylation to reorganize the actin cytoskeleton. J Virol 87, 4121–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing X, Cerveny M, Yang K, and He B (2004). Replication of herpes simplex virus 1 depends on the gamma 134.5 functions that facilitate virus response to interferon and egress in the different stages of productive infection. J Virol 78, 7653–7666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokhi V, Ashley J, Nunnari J, Noma A, Ito N, Wakabayashi-Ito N, Moore MJ, and Budnik V (2013). Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep 3, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Yamamoto M, Ohno T, Tanaka M, Sata T, Nishiyama Y, and Kawaguchi Y (2006). Herpes simplex virus 1-encoded protein kinase UL13 phosphorylates viral Us3 protein kinase and regulates nuclear localization of viral envelopment factors UL34 and UL31. J Virol 80, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharkwal H, Smith CG, and Wilson DW (2016). Herpes Simplex Virus Capsid Localization to ESCRT-VPS4 Complexes in the Presence and Absence of the Large Tegument Protein UL36p. J Virol 90, 7257–7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CE, Perez A, Perkins G, Ellisman MH, and Dauer WT (2010). A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc Natl Acad Sci U S A 107, 9861–9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfleisch R, Klupp BG, Fuchs W, Kopp M, Teifke JP, and Mettenleiter TC (2006). Influence of pseudorabies virus proteins on neuroinvasion and neurovirulence in mice. J Virol 80, 5571–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp B, Altenschmidt J, Granzow H, Fuchs W, and Mettenleiter TC (2008). Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J Virol 82, 6299–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Bottcher S, Granzow H, Kopp M, and Mettenleiter TC (2005a). Complex formation between the UL16 and UL21 tegument proteins of pseudorabies virus. J Virol 79, 1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Bottcher S, Granzow H, Kopp M, and Mettenleiter TC (2005b). Complex formation between the UL16 and UL21 tegument proteins of pseudorabies virus. J Virol 79, 1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, and Mettenleiter TC (2007). Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci U S A 104, 7241–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Keil GM, and Mettenleiter TC (2006). The capsid-associated UL25 protein of the alphaherpesvirus pseudorabies virus is nonessential for cleavage and encapsidation of genomic DNA but is required for nuclear egress of capsids. J Virol 80, 6235–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Klopfleisch R, Fuchs W, Kopp M, Lenk M, and Mettenleiter TC (2005c). Functional analysis of the pseudorabies virus UL51 protein. J Virol 79, 3831–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, and Mettenleiter TC (2000). Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J Virol 74, 10063–10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, and Mettenleiter TC (2001). Effect of the pseudorabies virus US3 protein on nuclear membrane localization of the UL34 protein and virus egress from the nucleus. J Gen Virol 82, 2363–2371. [DOI] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, and Mettenleiter TC (2011). Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. Journal of virology 85, 8285–8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Lomniczi B, Visser N, Fuchs W, and Mettenleiter TC (1995). Mutations affecting the UL21 gene contribute to avirulence of pseudorabies virus vaccine strain Bartha. Virology 212, 466–473. [DOI] [PubMed] [Google Scholar]
- Kobiler O, Drayman N, Butin-Israeli V, and Oppenheim A (2012). Virus strategies for passing the nuclear envelope barrier. Nucleus 3, 526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp M, Granzow H, Fuchs W, Klupp BG, Mundt E, Karger A, and Mettenleiter TC (2003). The pseudorabies virus UL11 protein is a virion component involved in secondary envelopment in the cytoplasm. J Virol 77, 5339–5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp M, Klupp BG, Granzow H, Fuchs W, and Mettenleiter TC (2002). Identification and characterization of the pseudorabies virus tegument proteins UL46 and UL47: role for UL47 in virion morphogenesis in the cytoplasm. J Virol 76, 8820–8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn J, Leege T, Klupp BG, Granzow H, Fuchs W, and Mettenleiter TC (2008). Partial functional complementation of a pseudorabies virus UL25 deletion mutant by herpes simplex virus type 1 pUL25 indicates overlapping functions of alphaherpesvirus pUL25 proteins. J Virol 82, 5725–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMassa N, Arenas-Mena C, and Phillips GR (2018). Electron microscopic characterization of nuclear egress in the sea urchin gastrula. J Morphol 279, 609–615. [DOI] [PubMed] [Google Scholar]
- Le Sage V, Jung M, Alter JD, Wills EG, Johnston SM, Kawaguchi Y, Baines JD, and Banfield BW (2013). The herpes simplex virus 2 UL21 protein is essential for virus propagation. J Virol 87, 5904–5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach N, Bjerke SL, Christensen DK, Bouchard JM, Mou F, Park R, Baines J, Haraguchi T, and Roller RJ (2007). Emerin is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both UL34 and US3. J Virol 81, 10792–10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leege T, Granzow H, Fuchs W, Klupp BG, and Mettenleiter TC (2009). Phenotypic similarities and differences between UL37-deleted pseudorabies virus and herpes simplex virus type 1. J Gen Virol 90, 1560–1568. [DOI] [PubMed] [Google Scholar]
- Leelawong M, Guo D, and Smith GA (2011). A physical link between the pseudorabies virus capsid and the nuclear egress complex. Journal of virology 85, 11675–11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelawong M, Lee JI, and Smith GA (2012). Nuclear Egress of Pseudorabies Virus Capsids Is Enhanced by a Subspecies of the Large Tegument Protein That Is Lost upon Cytoplasmic Maturation. Journal of virology 86, 6303–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzinger H, Ziegler U, Schraner EM, Fraefel C, Glauser DL, Heid I, Ackermann M, Mueller M, and Wild P (2005). Herpes simplex virus 1 envelopment follows two diverse pathways. J Virol 79, 13047–13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Nguyen CC, Ryckman BJ, Britt WJ, and Kamil JP (2015). A viral regulator of glycoprotein complexes contributes to human cytomegalovirus cell tropism. Proc Natl Acad Sci U S A 112, 4471–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, and Baines JD (2005). Identification of an essential domain in the herpes simplex virus 1 UL34 protein that is necessary and sufficient to interact with UL31 protein. J Virol 79, 3797–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata JM, Simpson-Holley M, Wright NT, Han Z, Paragas J, and Harty RN (2003). Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J Virol 77, 1812–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhacheva EV, and Bogachev SS (2001). Lamins and their functions in cell cycle. Membr Cell Biol 14, 565–577. [PubMed] [Google Scholar]
- Liu YT, Jiang J, Bohannon KP, Dai X, Gant Luxton GW, Hui WH, Bi GQ, Smith GA, and Zhou ZH (2017a). A pUL25 dimer interfaces the pseudorabies virus capsid and tegument. J Gen Virol 98, 2837–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YT, Jiang J, Bohannon KP, Dai X, Gant Luxton GW, Hui WH, Bi GQ, Smith GA, and Zhou ZH (2017b). A pUL25 dimer interfaces the pseudorabies virus capsid and tegument. J Gen Virol 98, 2837–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, and Kawaguchi Y (2014). Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88, 4657–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanov VA, Maher-Sturgess SL, Snider MG, Lawman Z, Babiuk LA, and van Drunen Littel-van den Hurk S (2010). A UL47 gene deletion mutant of bovine herpesvirus type 1 exhibits impaired growth in cell culture and lack of virulence in cattle. J Virol 84, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis JS, Courtney RJ, and Wills JW (2003a). Binding partners for the UL11 tegument protein of herpes simplex virus type 1. Journal of virology 77, 11417–11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis JS, Courtney RJ, and Wills JW (2003b). Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J Virol 77, 11417–11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz M, Vollmer B, Unsay JD, Klupp BG, Garcia-Saez AJ, Mettenleiter TC, and Antonin W (2015). A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J Biol Chem 290, 6962–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luitweiler EM, Henson BW, Pryce EN, Patel V, Coombs G, McCaffery JM, and Desai PJ (2013). Interactions of the Kaposi’s Sarcoma-Associated Herpesvirus Nuclear Egress Complex: ORF69 Is a Potent Factor for Remodeling Cellular Membranes. J virol 87, 3915–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, and Smith GA (2006). The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J Virol 80, 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Y, Zhou S, Gao S, and Deng H (2019a). Remodeling of host membranes during herpesvirus assembly and egress. Protein Cell 10, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Y, Zhou S, Gao S, and Deng H (2019b). Remodeling of host membranes during herpesvirus assembly and egress. Protein Cell 10, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye MF, Sharma M, El Omari K, Filman DJ, Schuermann JP, Hogle JM, and Coen DM (2015). Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J 34, 2937–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymberopoulos MH, Bourget A, Ben Abdeljelil N, and Pearson A (2011). Involvement of the UL24 protein in herpes simplex virus 1-induced dispersal of B23 and in nuclear egress. Virology 412, 341–348. [DOI] [PubMed] [Google Scholar]
- Margalit A, Liu J, Fridkin A, Wilson KL, and Gruenbaum Y (2005). A lamin-dependent pathway that regulates nuclear organization, cell cycle progression and germ cell development. Novartis Found Symp 264, 231–240; discussion 240–235. [PubMed] [Google Scholar]
- Maric M, Haugo AC, Dauer W, Johnson D, and Roller RJ (2014). Nuclear envelope breakdown induced by herpes simplex virus type 1 involves the activity of viral fusion proteins. Virology 460–461, 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschall M, Marzi A, aus dem Siepen P, Jochmann R, Kalmer M, Auerochs S, Lischka P, Leis M, and Stamminger T (2005). Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J Biol Chem 280, 33357–33367. [DOI] [PubMed] [Google Scholar]
- Maruzuru Y, Shindo K, Liu Z, Oyama M, Kozuka-Hata H, Arii J, Kato A, and Kawaguchi Y (2014). Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J Virol 88, 7445–7454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbong EF, Woodley L, Frost E, Baines JD, and Duffy C (2012). Deletion of UL21 causes a delay in the early stages of the herpes simplex virus 1 replication cycle. J Virol 86, 7003–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElwee M, Vijayakrishnan S, Rixon F, and Bhella D (2018). Structure of the herpes simplex virus portal-vertex. PLoS biology 16, e2006191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meckes DG Jr., and Wills JW (2007). Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J Virol 81, 13028–13036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettenleiter TC (2015). Breaching the Barrier-The Nuclear Envelope in Virus Infection. J Mol Biol. [DOI] [PubMed] [Google Scholar]
- Milbradt J, Auerochs S, Sevvana M, Muller YA, Sticht H, and Marschall M (2012). Specific residues of a conserved domain in the N terminus of the human cytomegalovirus pUL50 protein determine its intranuclear interaction with pUL53. J Biol Chem 287, 24004–24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JB, Hofemeister H, and O’Hare P (2007). Herpes simplex virus infection induces phosphorylation and delocalization of emerin, a key inner nuclear membrane protein. J Virol 81, 4429–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou F, Wills E, and Baines JD (2009). Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J Virol 83, 5181–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel CH, Dohner K, Fathollahy M, Strive T, Borst EM, Messerle M, and Sodeik B (2008). Nuclear egress and envelopment of herpes simplex virus capsids analyzed with dual-color fluorescence HSV1(17+). J Virol 82, 3109–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalwanga D, Rempel S, Roizman B, and Baines JD (1996). The UL 16 gene product of herpes simplex virus 1 is a virion protein that colocalizes with intranuclear capsid proteins. Virology 226, 236–242. [DOI] [PubMed] [Google Scholar]
- Newcomb WW, Booy FP, and Brown JC (2007). Uncoating the herpes simplex virus genome. Journal of molecular biology 370, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb WW, Fontana J, Winkler DC, Cheng N, Heymann JB, and Steven AC (2017). The Primary Enveloped Virion of Herpes Simplex Virus 1: Its Role in Nuclear Egress. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb WW, Homa FL, and Brown JC (2006). Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. Journal of virology 80, 6286–6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb WW, Juhas RM, Thomsen DR, Homa FL, Burch AD, Weller SK, and Brown JC (2001). The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. Journal of virology 75, 10923–10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HC, Talledge N, McCullough J, Sharma A, Moss FR 3rd, Iwasa JH, Vershinin MD, Sundquist WI, and Frost A (2020). Membrane constriction and thinning by sequential ESCRT-III polymerization. Nat Struct Mol Biol 27, 392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa N, Kawaguchi Y, Tanaka M, Kato A, Kato A, Kimura H, and Nishiyama Y (2005). Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J Virol 79, 6947–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima S, Daikoku T, Shibata S, Yamada H, Goshima F, and Nishiyama Y (1998). Characterization of the UL16 gene product of herpes simplex virus type 2. Arch Virol 143, 863–880. [DOI] [PubMed] [Google Scholar]
- Pante N, and Kann M (2002). Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol Biol Cell 13, 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passvogel L, Janke U, Klupp BG, Granzow H, and Mettenleiter TC (2014). Identification of conserved amino acids in pUL34 which are critical for function of the pseudorabies virus nuclear egress complex. J Virol 88, 6224–6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passvogel L, Klupp BG, Granzow H, Fuchs W, and Mettenleiter TC (2015). Functional characterization of nuclear trafficking signals in pseudorabies virus pUL31. J Virol 89, 2002–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passvogel L, Trube P, Schuster F, Klupp BG, and Mettenleiter TC (2013). Mapping of Sequences in Pseudorabies Virus pUL34 That Are Required for Formation and Function of the Nuclear Egress Complex. Journal of virology 87, 4475–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M, Sanghera JS, Pelech SL, and Nigg EA (1992). Mitogen-activated protein kinases phosphorylate nuclear lamins and display sequence specificity overlapping that of mitotic protein kinase p34cdc2. Eur J Biochem 205, 287–294. [DOI] [PubMed] [Google Scholar]
- Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, Karger A, and Sodeik B (2010). Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog 6, e1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghava S, Giorda KM, Romano FB, Heuck AP, and Hebert DN (2013). SV40 late protein VP4 forms toroidal pores to disrupt membranes for viral release. Biochemistry 52, 3939–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza S, Deng M, Shahin F, Yang K, Hu C, Chen Y, Chen H, and Guo A (2016). A bovine herpesvirus 1 pUL51 deletion mutant shows impaired viral growth in vitro and reduced virulence in rabbits. Oncotarget 7, 12235–12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Liang L, and Baines JD (2004). Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes U(L)31 and U(L)34. J Virol 78, 5564–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, and Roller RJ (2001). U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75, 8803–8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, and Baines JD (2002). Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76, 8939–8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AP, Abaitua F, O’Hare P, McNab D, Rixon FJ, and Pasdeloup D (2009). Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J Virol 83, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, and Furlong D (1974). The Replication of Herpesviruses. In Comprehensive Virology, Fraenkel-Conrat H, and Wagner RR, eds. (Boston, MA: Springer; ), pp. 229–403. [Google Scholar]
- Roller RJ, and Baines JD (2017a). Herpesvirus Nuclear Egress. Adv Anat Embryol Cell Biol 223, 143–169. [DOI] [PubMed] [Google Scholar]
- Roller RJ, and Baines JD (2017b). Herpesvirus Nuclear Egress. In Advances in Anatomy, Embryology, and Cell Biology, Osteirrieder K, ed. (Springer; ), pp. 143–169. [DOI] [PubMed] [Google Scholar]
- Roller RJ, Bjerke SL, Haugo AC, and Hanson S (2010). Analysis of a charge cluster mutation of herpes simplex virus type 1 UL34 and its extragenic suppressor suggests a novel interaction between pUL34 and pUL31 that is necessary for membrane curvature around capsids. J Virol 84, 3921–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roller RJ, Haugo AC, and Kopping NJ (2011). Intragenic and extragenic suppression of a mutation in herpes simplex virus 1 UL34 that affects both nuclear envelope targeting and membrane budding. J Virol 85, 11615–11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roller RJ, Zhou Y, Schnetzer R, Ferguson J, and DeSalvo D (2000). Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J Virol 74, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronfeldt S, Franzke K, Holper JE, Klupp BG, and Mettenleiter TC (2020). Mutational Functional Analysis of the Pseudorabies Virus Nuclear Egress Complex-Nucleocapsid Interaction. J Virol 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronfeldt S, Klupp BG, Franzke K, and Mettenleiter TC (2017). Lysine 242 within Helix 10 of the Pseudorabies Virus Nuclear Egress Complex pUL31 Component Is Critical for Primary Envelopment of Nucleocapsids. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Jing X, Leser GP, and Lamb RA (2010). Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142, 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, and Lamb RA (2011). Influenza virus assembly and budding. Virology 411, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, and Lamb RA (2013). Viral membrane scission. Annu Rev Cell Dev Biol 29, 551–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckman BJ, and Roller RJ (2004). Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J Virol 78, 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon B, Cunningham C, Davison AJ, Harris WJ, and Baines JD (1998). The herpes simplex virus type 1 U(L)17 gene encodes virion tegument proteins that are required for cleavage and packaging of viral DNA. J Virol 72, 3779–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam MD, Evans BT, Coen DM, and Hogle JM (2009). Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J Virol 83, 2996–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfo A, Starkey J, Mellinger E, Zhang D, Chadha P, Carmichael J, and Wills JW (2017). The UL21 Tegument Protein of Herpes Simplex Virus 1 Is Differentially Required for the Syncytial Phenotype. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauflinger M, Fischer D, Schreiber A, Chevillotte M, Walther P, Mertens T, and von Einem J (2011). The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J Virol 85, 3821–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer EC, and Foisner R (2007). Proteins that associate with lamins: many faces, many functions. Exp Cell Res 313, 2167–2179. [DOI] [PubMed] [Google Scholar]
- Schnee M, Wagner FM, Koszinowski UH, and Ruzsics Z (2012). A cell free protein fragment complementation assay for monitoring the core interaction of the human cytomegalovirus nuclear egress complex. Antiviral Res 95, 12–18. [DOI] [PubMed] [Google Scholar]
- Scholtes LD, Yang K, Li LX, and Baines JD (2010). The capsid protein encoded by U(L)17 of herpes simplex virus 1 interacts with tegument protein VP13/14. J Virol 84, 7642–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schur FK, Hagen WJ, Rumlova M, Ruml T, Muller B, Krausslich HG, and Briggs JA (2015). Structure of the immature HIV-1 capsid in intact virus particles at 8.8 A resolution. Nature 517, 505–508. [DOI] [PubMed] [Google Scholar]
- Schuster F, Klupp BG, Granzow H, and Mettenleiter TC (2012). Structural determinants for nuclear envelope localization and function of pseudorabies virus pUL34. J Virol 86, 2079–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehl J, Portner S, Klupp BG, Granzow H, Franzke K, Teifke JP, and Mettenleiter TC (2020). Roles of the Different Isoforms of the Pseudorabies Virus Protein Kinase pUS3 in Nuclear Egress. J Virol 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanda SK, and Wilson DW (2008). UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J Virol 82, 7388–7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Kamil JP, Coughlin M, Reim NI, and Coen DM (2014). Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J Virol 88, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba C, Daikoku T, Goshima F, Takakuwa H, Yamauchi Y, Koiwai O, and Nishiyama Y (2000). The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J Gen Virol 81, 2397–2405. [DOI] [PubMed] [Google Scholar]
- Silvestri LS, Ruthel G, Kallstrom G, Warfield KL, Swenson DL, Nelle T, Iversen PL, Bavari S, and Aman MJ (2007). Involvement of vacuolar protein sorting pathway in Ebola virus release independent of TSG101 interaction. The Journal of infectious diseases 196 Suppl 2, S264–270. [DOI] [PubMed] [Google Scholar]
- Skepper JN, Whiteley A, Browne H, and Minson A (2001). Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment --> deenvelopment --> reenvelopment pathway. J Virol 75, 5697–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni SP, and Stahelin RV (2014). The Ebola virus matrix protein VP40 selectively induces vesiculation from phosphatidylserine-enriched membranes. J Biol Chem 289, 33590–33597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, et al. (2012). Nuclear Envelope Budding Enables Large Ribonucleoprotein Particle Export during Synaptic Wnt Signaling. Cell 149, 832–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackpole CW (1969). Herpes-type virus of the frog renal adenocarcinoma. I. Virus development in tumor transplants maintained at low temperatures. J Virol 4, 75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stow ND (2001). Packaging of genomic and amplicon DNA by the herpes simplex virus type 1 UL25-null mutant KUL25NS. J Virol 75, 10755–10765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima K, Arii J, Maruzuru Y, Koyanagi N, Kato A, and Kawaguchi Y (2019). Identification of the Capsid Binding Site in the Herpes Simplex Virus 1 Nuclear Egress Complex and Its Role in Viral Primary Envelopment and Replication. J Virol 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollefson AE, Scaria A, Hermiston TW, Ryerse JS, Wold LJ, and Wold WS (1996). The adenovirus death protein (E3-11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J Virol 70, 2296–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner EM, Brown RS, Laudermilch E, Tsai PL, and Schlieker C (2015). The Torsin Activator LULL1 Is Required for Efficient Growth of Herpes Simplex Virus 1. J Virol 89, 8444–8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votteler J, and Sundquist WI (2013). Virus Budding and the ESCRT Pathway. Cell Host Microbe 14, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar F, Pol JM, de Wind N, and Kimman TG (2001). Deletion of the UL21 gene in Pseudorabies virus results in the formation of DNA-deprived capsids: an electron microscopy study. Vet Res 32, 47–54. [DOI] [PubMed] [Google Scholar]
- Walzer SA, Egerer-Sieber C, Sticht H, Sevvana M, Hohl K, Milbradt J, Muller YA, and Marschall M (2015). Crystal Structure of the Human Cytomegalovirus pUL50-pUL53 Core Nuclear Egress Complex Provides Insight into a Unique Assembly Scaffold for Virus-Host Protein Interactions. J Biol Chem 290, 27452–27458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang Y, Wu S, Pan S, Zhou C, Ma Y, Ru Y, Dong S, He B, Zhang C, et al. (2014). p32 is a novel target for viral protein ICP34.5 of herpes simplex virus type 1 and facilitates viral nuclear egress. J Biol Chem 289, 35795–35805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whealy ME, Card JP, Meade RP, Robbins AK, and Enquist LW (1991). Effect of brefeldin A on alphaherpesvirus membrane protein glycosylation and virus egress. J Virol 65, 1066–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild P, Senn C, Manera CL, Sutter E, Schraner EM, Tobler K, Ackermann M, Ziegler U, Lucas MS, and Kaech A (2009). Exploring the nuclear envelope of herpes simplex virus 1-infected cells by high-resolution microscopy. J Virol 83, 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KL, and Foisner R (2010). Lamin-binding Proteins. Cold Spring Harb Perspect Biol 2, a000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, and Johnson DC (2009). Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J Virol 83, 3115–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack A, and Shenk T (2010). Human cytomegalovirus tegument protein pUL71 is required for efficient virion egress. MBio 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CC, Wisner TW, Hannah BP, Eisenberg RJ, Cohen GH, and Johnson DC (2009). Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J Virol 83, 11847–11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Pan S, Zhang L, Baines J, Roller R, Ames J, Yang M, Wang J, Chen D, Liu Y, et al. (2016). Herpes Simplex Virus 1 Induces Phosphorylation and Reorganization of Lamin A/C through the gamma134.5 Protein That Facilitates Nuclear Egress. J Virol 90, 10414–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, and Baines JD (2011). Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc Natl Acad Sci U S A 108, 14276–14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Wills E, Lim HY, Zhou ZH, and Baines JD (2014). Association of herpes simplex virus pUL31 with capsid vertices and components of the capsid vertex-specific complex. J Virol 88, 3815–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye GJ, Vaughan KT, Vallee RB, and Roizman B (2000). The herpes simplex virus 1 U(L)34 protein interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. J Virol 74, 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, and Smith GA (2013). The Herpesvirus VP1/2 Protein Is an Effector of Dynein-Mediated Capsid Transport and Neuroinvasion. Cell host and microbe 13, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeev-Ben-Mordehai T, Weberruss M, Lorenz M, Cheleski J, Hellberg T, Whittle C, El Omari K, Vasishtan D, Dent KC, Harlos K, et al. (2015). Crystal Structure of the Herpesvirus Nuclear Egress Complex Provides Insights into Inner Nuclear Membrane Remodeling. Cell Rep 13, 2645–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu HY, Yamada H, Jiang YM, Yamada M, and Nishiyama Y (1999). Intracellular localization of the UL31 protein of herpes simplex virus type 2. Arch Virol 144, 1923–1935. [DOI] [PubMed] [Google Scholar]
Web Resources
1. HSV-1 NEC PDB ID: 4ZXS (Bigalke and Heldwein, 2015)
2. PRV NEC PDB ID: 4Z3U (Bigalke and Heldwein, 2015)
3. PRV NEC PDB ID: 5E8C (Zeev-Ben-Mordehai et al., 2015)
4. HCMV NEC PDB ID: 5DOB (Lye et al., 2015)
5. HCMV NEC PDB ID: 5D5N (Walzer et al., 2015)