

Abstract
AP-2α (encoded by TFAP2A) functions as a tumor suppressor and influences response to therapy in several cancer types. We aimed to characterize regulation of the transcriptome by AP-2α in colon cancer. CRISPR-Cas9 and shRNA were used to eliminate TFAP2A expression in HCT116 and a panel of colon cancer cell lines. AP-2α target genes were identified with RNA-seq and ChIP-seq. Effects on cell cycle were characterized in cells synchronized with aphidicolin and analyzed by FACS and Premo FUCCI. Effects on invasion and tumorigenesis were determined by invasion assay, growth of xenografts and phosphorylated histone H3 (PHH3). Knockout of TFAP2A induced significant alterations in the transcriptome including repression of TGM2, identified as a primary gene target of AP-2α. Loss of AP-2α delayed progression through S-phase into G2/M and decreased phosphorylation of AKT, effects that were mediated through regulation of TGM2. Buparlisib (BKM120) repressed in vitro invasiveness of HCT116 and a panel of colon cancer cell lines; however, loss of AP-2α induced resistance to Buparlisib. Similarly, Buparlisib repressed PHH3 and growth of tumor xenografts and increased overall survival of tumor-bearing mice, whereas, loss of AP-2α induced resistance to the effect of PI3K inhibition. Loss of AP-2α in colon cancer leads to prolonged S-phase through altered activation of AKT leading to resistance to the PI3K inhibitor, Buparlisib. The findings demonstrate an important role for AP-2α in regulating progression through the cell cycle and indicates that AP-2α is a marker for response to PI3K inhibitors.
INTRODUCTION
Colon cancer is one of the most common cancers with approximately 1.4 million new cases diagnosed each year and is responsible annually for 639,000 deaths (1). The incidence of colon cancer has decreased in certain countries, including the United States, largely due to routine screening and removal of precancerous lesions; however, the incidence is increasing in other countries and, despite the decreasing incidence in the United States, colon cancer remains the 3rd leading cause of cancer death (2). FOLFOX is the first line chemotherapy regimen for colon cancer and has been shown to be efficacious and successful at improving survival when used as adjuvant therapy (3,4); however, chemoresistance is still thought to be the cause of treatment failure in over 90% of patients with metastatic cancer (5). Hence, molecular characterization of tumors including next generation sequencing to identify gene mutations that alter response to chemotherapeutic drugs continues to be of great interest to optimize targeted, personalized therapies (6).
The use of gene expression profiling in colon cancer has been validated to predict recurrence and estimate prognosis, but is thus far unable to reliably predict response to chemotherapy (7). Currently, the National Comprehensive Cancer Network does not feel there is sufficient evidence regarding next generation sequencing to direct therapy and recommends testing only in select cases. For example, testing for mismatch repair gene status, HER2 amplification, KRAS and BRAF mutations in certain patients, such as those with metastatic disease, is encouraged to guide second line chemotherapeutic agents that offer clear benefit to patients with these mutations (8). These advancements have significantly improved survival in a subset of colorectal cancer patients and have avoided unnecessary therapy in patients likely to be resistant (9); however, the majority of patients currently have no targeted therapy options (10). Therefore, expanding our knowledge of genetic markers associated with sensitivity to specific chemotherapeutic agents is of utmost importance to improve outcomes and prevent chemoresistance.
Transcription Factor Activating Protein 2 Alpha (AP-2α), encoded by the TFAP2A gene, is one of five members of the family of AP-2 factors which are highly expressed early in differentiation of the ectoderm (11). The AP-2 family regulates the molecular phenotype of multiple cancer types, including breast, thyroid, melanoma and colon cancer (12–17). AP-2α functions as a tumor suppressor in many cancer types (18–20) and influences response to chemotherapy (21,22). In colon cancer, although the rate of somatic mutation is quite low, AP-2α has been shown to be involved in tumorigenesis (23,24), cell growth (14), and acts as a tumor suppressor (23,24), potentially through interactions with p53 (25). Overexpression of TFAP2A inhibited tumorigenesis in the colon cancer cell line SW480 (24) and in Apc(min) mice, in vivo gene delivery of AP-2α inhibited intestinal polyp formation (23). Given the importance of expanding our knowledge regarding gene expression profiling to direct chemotherapy and the growing body of evidence indicating AP-2α as an important regulator in colon cancer, we sought to examine the molecular changes in colon cancer when AP-2α is lost with a complete knockout (KO) and its effect on responsiveness to chemotherapeutic agents.
MATERIALS and METHODS
Cell Culture
The cell lines HCT116 (RRID:CVCL_0291), LoVo (RRID:CVCL_0399), SW48 (RRID:CVCL_1724), LS180 (RRID:CVCL_0397) and MDA-MB-231 (RRID:CVCL_0062) were purchased from the ATCC and were used at a low passage number (<10) with no further testing. HCT116 was maintained in McCoy’s 5A media, LoVo in F-12K media, SW48 in DMEM/F12, LS180 in EMEM and MDA-MB-231 in DMEM. Cell lines were tested for mycoplasma using LONZA MycoAlert Mycoplasma Detection Kit. All media were supplemented with 10% fetal bovine serum and 1% Plasmocin (InvivoGen, catalog no. ant-mpp).
CRISPR/Cas9-mediated TFAP2A knockout in HCT116 cell line.
To minimize off-target effect we used pCas9D10A-GFP plasmid for co-expression mutant CasD10A nickase (Addgene plasmid # 44720; http://n2t.net/addgene:44720; RRID:Addgene_44720) and two gRNAs cloned as gBlocks into pCR™4-TOPO™ Vector (Thermofisher) (Guide A: ATCAAACTGTAATTAAGAA and Guide B: TTCTACATGCTGCAACAAA) within exon 3 of TFAP2A. Lipofectamine 2000 was used for DNA transfection of HCT 116 CCL-247™ cells accordingly to the manufacturer protocol (molar ratio Cas9D10A/gRNAa/gRNAb was 1/5/5). After 24 hours GFP positive cells were sorted as a single cell per well in 96 well plates. Colonies were screened after 14 days for the presence of genomic alterations (insertion/deletion) using a pair of primers TFAP2A_EX3_F1_ctrl AGC TAG CCT GTT GGC ATT ACC and TFAP2A_EX3_R1_ctrl CCT CTT GAG TTG CAA AGC CC. Disruption of ORF was confirmed by sequencing and Western Blot with AP-2α antibody.
Gene knockdown
Cells were transfected using siRNA directed toward nontargeting #2 (Ambion by Life Technologies, catalog no. 4390846), TFAP2A (Stealth siRNA by Invitrogen ID:82654707) and TGM2 (Ambion by Life Technologies, catalog no. 4390824) with Lipofectamine RNAiMAX reagent (Thermo Fischer Scientific, catalog no. 13778150), as per manufacturer’s instructions. After 72 hours of incubation, cells were immediately analyzed or used in subsequent experiments.
Knockdown of TFAP2A was completed in HCT116, LoVo, SW48, LS180 and MDA-MB-231 with lentiviral delivered short hairpin (sh)RNA vector (Sigma Aldrich, catalog no. TRCN0000004926) at a multiplicity of infection (MOI) of 0.5. The same cell lines were transduced with nontargeting shRNA lentivirus (Sigma Aldrich, catalog no. SHC016) at the same MOI to serve as controls. Cells were cultured in Opti-MEM media (ThermoFisher Scientific, catalog no. 31985-062) for 24 hours with the respective lentivirus and then put under puromycin dihydrochloride (Sigma Aldrich, catalog no. P9620) selection at 1 μg/ml for 24 hours. Knockdown of TFAP2A was confirmed with Western blot.
Western blot
Protein was isolated in RIPA lysis buffer (Millipore, catalog no. 20–188), supplemented with protease inhibitor (Roche, catalog no. 11836170001) and PhosSTOP (Roche, catalog no. 4906845001). The following primary antibodies were used according to the manufacturer’s recommendations: AP-2α (Abcam, catalog no. ab108311), TGM2 (Abcam, catalog no. ab2386), CDK1+2+3 (Abcam, catalog no. ab32384), RAD51 (Santa Cruz, catalog no. sc-8349), CALB2 (Abnova, catalog no. MAB2741), GMNN (DSHB, catalog no. CPTC-GMNN-1-s), ACSS2 (Santa Cruz, catalog no. sc-398559), pan AKT (Cell Signaling Technology, catalog no. 4691), pAKT Thr308 (Cell Signaling Technology, catalog no. D25E6), and pAKT1/2/3 Ser473 (Santa Cruz, catalog no. sc-7985 R). GAPDH (Santa Cruz, catalog no. sc-47724) was used as a loading control. Secondary antibodies were used according to the manufacturer’s specification: anti-rabbit horseradish peroxidase (HRP) (Cell Signaling Technology, catalog no. 7074) and anti-mouse HRP (Cell Signaling Technology, catalog no. 7076). Protein was visualized with SuperSignal West Femto maximum sensitivity substrate (TFS, catalog no. 34095).
RNA Sequencing
Experimental setup and analyses were performed in accordance to ENCODE Guidelines and Best Practices for RNA Sequencing (RNA-seq). RNA was harvested using the RNeasy Mini Plus kit (Qiagen, catalog no. 74134) at a concentration of 100–200 ng/uL and biologic triplicates of each condition were sent to the University of Nebraska (Omaha, NE) for further processing. The RNA quality was confirmed by the receiving facility and was subsequently sequenced with specifications for gene differentiation (50 base pair, single-end reads). The RNA-seq analysis of the raw data was performed using the Galaxy web platform (RRID:SCR_006281) at usegalaxy.org with the built-in tools: HISAT2 (RRID:SCR_015530) and Cufflinks (RRID:SCR_014597). For gene expression comparisons, genes with significant expression changes as determined by Cufflinks data analysis were included, using an arbitrary log2fold change cutoff to allow for a reliable analysis of consistently altered gene expression.
RNA-seq data are available at the GEO database (National Center for Biotechnology Information, Bethesda, MD) under accession number GSE155169.
PIK3CA (H1047R) single nucleotide polymorphism analysis.
All obtained single cell knockout TFAP2A clones and maternal HCT 116 cell line were analyzed for the presence of potential heterozygosity c.3140A>G p.H1047R in Exon 20 of PIK3CA. Full sequence of Exon 20 was amplified using a pair of primers PIK3CA ex20_F1 GCC ACA CAC TAC ATC AGT GG and PIK3CA_ex20_R1 AGA GAT TGG CAT GCT GTC GAA. Obtained PCR product of 372 bp was cloned in pCR™4-TOPO™ Vector and at least 12 individual colonies were sequenced per each single cell clone or maternal cell line.
Chromatin immunoprecipitation sequencing
Chromatin immunoprecipitation sequencing (ChIP-seq) was accomplished with AP-2α 3B5 antibody (Santa Cruz, catalog no. sc-12726X) as described previously (26). AP-2γ (Santa Cruz, catalog no. sc-12762X) was used as the negative control. ChIP-Seq data are available in the GEO database under accession number GSE155169.
Analysis with FACS
For cell cycle analysis, cells in culture were synchronized with 2.5 ug/mL Aphidicolin (Sigma Aldrich, catalog no. A4487) in media supplemented with serum as previously described (26). After 36 hours of incubation, the Aphidicolin containing media was removed, cells were washed with PBS and 10% FBS containing media without Aphidicolin was replaced. Cells were harvested immediately after release from synchronization then at 4 and 8 hours after release and were fixed immediately with 70% ethanol. Fixed cells were stained within 24 hours of fixation with propidium iodide (Life Technologies, catalog no. P3566) and DNA quantification was performed on a FACS Becton Dickinson LSR II machine.
For the apoptosis assay, cells were seeded in 6-well plates (Corning, COSTAR, 3516) and allowed to attach overnight in full media. After attachment, cells were washed with DPBS (Gibco, Germany) three times and cell were incubated in serum free medium for 24 hours. Apoptosis was measured 24 and 48 hours after the cells were released from G0 cell arrest by adding complete medium. The FITC-Annexin V Apoptosis Detection Kit (BD Bioscience, Heidelberg, Germany) was used according to the manufacturer’s protocol. In brief, floating and harvested cells were mixed, washed twice with cold phosphate buffered saline (DPBS) and resuspended in binding buffer at a final density of 106 cells/ml. FITC-annexin (5 μl) and PI (5 μl) were added to 100 μL of the cell suspension containing 105 cells. The cell suspensions were mixed by gently vortexing and incubated for 15 min at room temperature in the dark. Subsequently, 400 μl of binding buffer were added and cells were analyzed by flow cytometry using FACS analysis.
Cell Cycle Protein Phospho Array
Cells were synchronized as above and harvested for protein 8 hours after release from synchronization. Protein quantification was completed using the Cell Cycle Phospho Antibody Array (Full Moon Biosystems, catalog no. PCC076) using the Antibody Array Assay Kit (Full Moon Biosystems, catalog no. KAS02) according to manufacturer’s instructions. Cy3-streptavidin (Invitrogen, catalog no. 434315) was used to allow for fluorescent imaging quantification. Levels of expression of the protein were normalized to the background.
Cell Cycle Progression with Immunofluorescence
Using the Premo FUCCI Cell Cycle Sensor BacMam 2.0 (ThermoFisher Scientific, catalog no. P36237) according to manufacturer’s instructions, unsynchronized cells in media supplemented with 10% FBS were incubated for 16–24 hours prior to imaging. An Olympus IX81 Inverted Microscope was used to obtain time lapse images for time lapse and levels of fluorescence. For each condition within the experiment, six randomly chosen locations with similar levels of cellular confluency were chosen for imaging.
Cell Viability Assay
Cells were plated in 96-well plates in technical triplicates and were incubated with MTT (Thermo Fisher Scientific, catalog no. M6494) according to manufacturer’s recommendations. Measurements to reflect viability were obtained on an Infinity 200 Pro (Tecan) plate reader at an absorbance of 570 nm. Measurements were normalized to the initial read 24 hours after plating to reflect changes in viability.
Clonogenicity Assay
A clonogenicity assay was performed as previously described by Franken, et al. (27). Each condition was completed as a biologic triplicate. Wells were imaged for analysis using a 55mm lens Canon camera.
Immunohistochemistry & TMA Analysis
Colon adenocarcinoma cases and their corresponding ‘best tumor block’ were identified in the diagnostic files of the University of Iowa Hospitals and Clinics. Tissue microarrays (TMAs) were constructed from 34 cases with sufficient paraffin embedded tumor. 4-μm thick sections of these TMA blocks were cut and used for immunohistochemistry (IHC). IHC was performed to detect expression of transcription factor AP-2-alpha (rabbit monoclonal antibody, clone EPR2688(2), RRID:AB_867683, AbCam, Cambridge, MA, 1:100 dilution) and TMG2 (mouse monoclonal antibody, clone CUB 7402, RRID:AB_2287299, AbCam, 1:200 dilution). Expression for each marker was evaluated for extent (0–100%) and intensity (0–3+) in each TMA core, and an overall H-score (mean extent × intensity) was calculated for by each tumor.
Invasion Assay
Invasion assay was completed as previously described (28). Cells were treated within an invasion chamber with either DMSO or 250 nM NVP-BKM120 (Selleck Chemicals, catalog no. S2247) immediately after plating. Cells were incubated for 24 hours prior to fixation and quantification of invasion.
Tumor xenografts
Following University of Iowa IACUC approval, twelve male and twelve female nu/J mice (RRID:IMSR_JAX:002019, Jackson Laboratory, Bar Harbor, ME, USA) were flank injected with 1×106 cells of either parental HCT116 or sub-clone KO-3 suspended in media/Matrigel (BD Biosciences) in a 1:1 ratio. As previously described (29), tumors were allowed to establish for seven days prior to starting treatments. Mice were randomized to a treatment group prior to flank injection so that each condition and respective treatment group had an equal number of male and female mice.
They received either 40 mg/kg NVP-BKM120 (Selleck Chemicals, catalog no. S2247) or control (water) for ten consecutive days by oral gavage. Tumor growth was monitored by calipers and tumor volume was calculated in mm3 using the width, length and height of tumors (16). Animals were euthanized when tumors reached 1000 mm3. Tumors were harvested, fixed in formalin and embedded in paraffin. Immunohistochemistry staining for pHH3 (Cell Marque, catalog no. 369A-14) was performed and quantified on all harvested tumors.
Statistical analysis
GraphPad Prism 8.0 (RRID:SCR_002798) was used to complete statistical analysis. Statistical comparisons between length of cell cycle, clonogenicity, protein expression, invasion and tumor volume were represented as mean ± standard error of the mean (SEM) and compared by Dunn’s multiple comparison test. Overall survival curves were compared by logrank test.
RESULTS
Characterization of cells with KO of TFAP2A.
Using CRISPR/Cas9 genome editing, clones of HCT116 colon carcinoma cells were generated with complete loss of AP-2α following disruption of exon 3 of the TFAP2A gene. We characterized three clones with KO of TFAP2A, after confirming complete absence of the AP-2α protein by western blot (Figure 1A). To determine changes in gene expression following loss of AP-2α, RNA was isolated from the parental HCT116 cell line, KO-1, KO-2 and KO-3, and was analyzed by RNA-seq, comparing gene expression in each TFAP2A KO clone to the parental HCT116 cell line. Differential expression analysis revealed 654 differentially expressed genes in KO-1, 394 differentially expressed genes in KO-2, and 1437 differentially expressed genes in KO-3 when compared to the parental HCT116 cell line. The significantly changed genes from all KO clones were compared and 131 commonly regulated genes were identified (Figure 1A). Differential expression of selected genes was confirmed by western blot (Figure 1A).
Figure 1. Identification of AP-2α target genes.
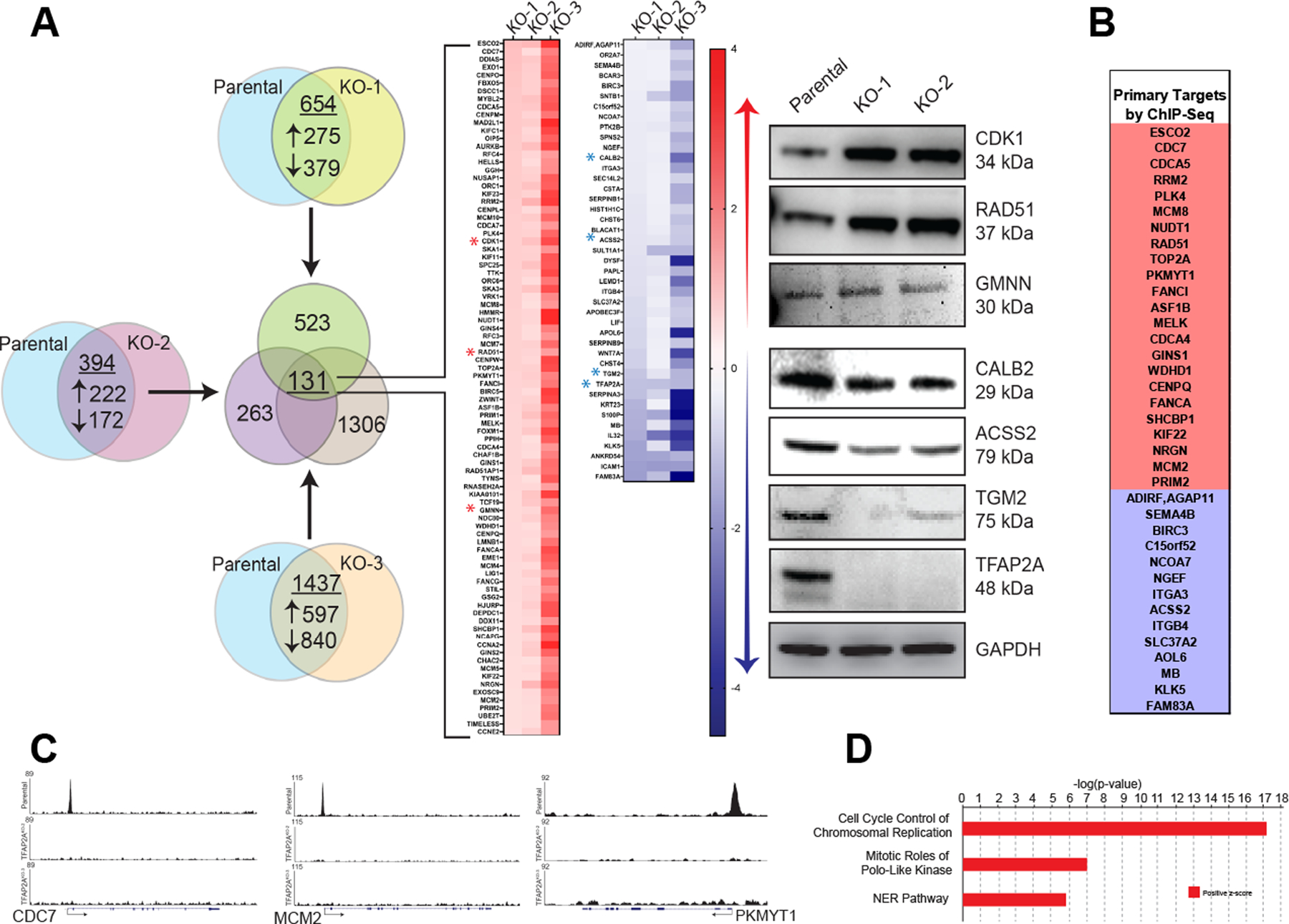
A. RNA sequencing of the parental colon cancer cell line HCT116 was compared to sequencing of three individual AP-2α knockout clones, yielding 131 genes whose expression was either significantly increased (red) or decreased (blue) in all three knockout clones. Gene expression changes were confirmed for select genes at the protein level using western blot (right panel). B. ChIP-seq analysis of the 131 commonly altered genes from by RNA-seq identified 37 as primary targets of AP-2α with select representation of peaks (C). D. Ingenuity Pathway Analysis identified three primary altered pathways based on gene expression after loss of AP-2α.
Loss of AP-2α alters gene expression through direct chromatin occupancy of regulatory regions of target genes and secondarily through downstream effects. To identify primary AP-2α target genes, ChIP-seq analysis was performed on HCT116, KO-2 and KO-3. Of the 131 genes consistently differentially expressed following complete KO of TFAP2A, 37 genes were identified using peak calling software that had AP-2α occupancy in the HCT116 parental cell line, but no AP-2α occupancy in TFAP2A KO clones (Figure 1B–C).
Loss of AP-2α alters cell cycle.
Ingenuity Pathway Analysis (IPA) software identified “Cell Cycle Control of Chromosomal Replication” as the most significantly altered pathway with loss of AP-2α (Figure 1D). To confirm that loss of AP-2α alters cell cycle progression, parental HCT116 cells and TFAP2A KO cells both transfected with siRNA to TFAP2A or non-targeting (NT) were synchronized in G1 phase with aphidicolin and allowed to progress through the cell cycle following drug withdrawal (Figure 2A). Appropriate reduction of AP-2α expression with siTFAP2A was confirmed by western blot (Supplemental Figure 1). Parental HCT116 cells expressing AP-2α progressed more rapidly through S phase and into G2/M phase compared to conditions with either knockdown or complete loss of AP-2α (Figure 2A). In cells with transient knockdown of TFAP2A four hours after release from synchronization, only 7% of cells had progressed to in G/2M compared to 22% of cells in HCT116 transfected with NT siRNA. Similarly, in KO-3 clone with KO of TFAP2A transfected with NT siRNA only 14% of cells had progressed to G2/M phase. As an additional control, treatment of KO-3 with siRNA targeting TFAP2A had no significant effect on cell cycle progression (Figure 2A). As cells were synchronized at the end of G1 phase, these results were consistent with a prolongation in S-phase after loss of AP-2α.
Figure 2. Cell cycle progression altered after loss of AP-2α.
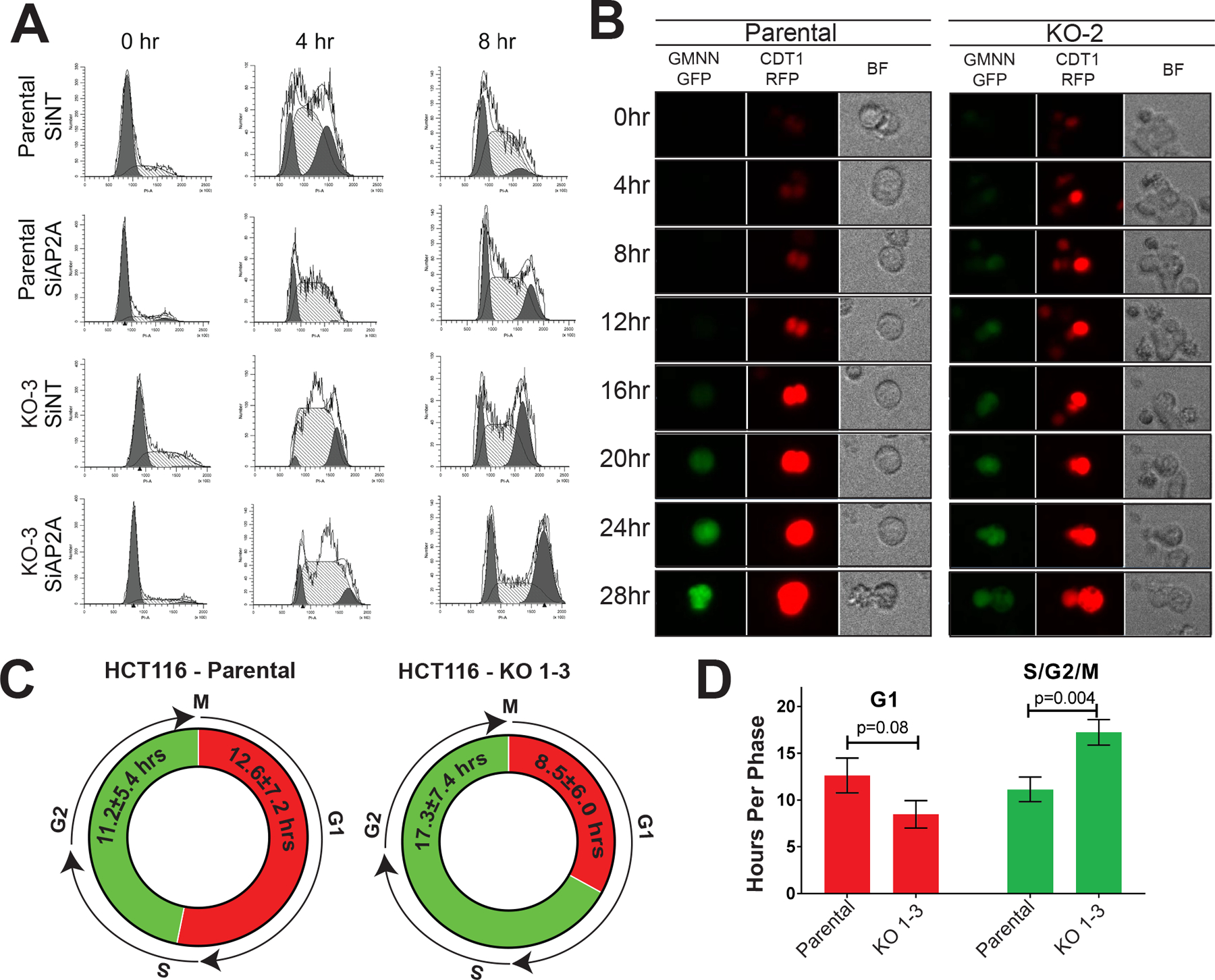
A. Progression through cell cycle after synchronization at G1-phase was analyzed in HCT116 parental cells with transient knockdown of TFAP2A were compared to cells treated with siNT and a CRISPR/Cas9 AP-2α KO clone similarly treated with siRNA to non-targeting (siNT) and TFAP2A (siAP2A). B. FUCCI Premo analysis of HCT116 parental cells vs. TFAP2A knockout clone (KO-2) using GMNN-GFP and CDT1-RFP demonstrate early entry into S-phase after loss of AP-2α, resulting in a shorter G1 phase (C) and increased time spent in S, G2 and M phase (D).
The prolongation of S-phase with loss of AP-2α was further confirmed on unsynchronized cells through imaging fluorescence. Parental HCT116 and all three KO clones were transduced with the Premo FUCCI Cell Cycle Sensor, which utilizes CDT1-RFP and GMNN-GFP to indicate the start of G1 phase and S phase, respectively. Parental HCT116 cells spent a mean of 12.6±7.2 hours in G1 phase and 11.2±5.4 hours progressing through S, G2 and M phase. All KO clones spent a mean of 8.5±6.0 hours in G1 phase and 17.3±7.4 hours in S and G2 phase until reaching M phase and dividing confirming that, as predicted, the KO clones spent a significantly longer period of time progressing through S and G2 phase compared to parental HCT116 cells expressing AP-2α (p=0.004) (Figure 2B–D).
The overall length of the cell cycle after loss of AP-2α did not significantly differ from the parental HCT116 cells despite the prolonged duration of S and G2 phase, which is likely due to a compensatory shortened G1 phase (Supplemental Video 1&2). To confirm this conclusion, we examined the growth and clonogenicity TFAP2A KO clones compared to parental HCT116. We found no differences in baseline cell growth or clonogenicity after KO of TFAP2A (Supplemental Figure 2). This was consistent with our findings identifying no difference in the overall length of the cell cycle after loss of AP-2α. Additionally, we confirmed that no difference existed in the level of early or late apoptosis after loss of AP-2α as KO clones progressed through cell cycle, as compared to parental HCT116 (Supplemental Figure S3).
AP-2α regulates AKT phosphorylation.
We sought to identify the mechanism by which loss of AP-2α prolongs S phase. Parental HCT116 and KO-3 clone were compared for alterations in phosphorylation of cell cycle regulatory proteins. Protein was harvested from parental HCT116 and KO-3 cells 8 hours after release from synchronization of G1 phase and a phospho-protein microarray examining common cell cycle regulators was performed. The level of protein and phosphorylated forms of these proteins were analyzed (Figure 3A). We determined that AKT expression was unaltered by KO of TFAP2A; however, the level of phosphorylation of AKT at Ser473 and Thr308, the two critical sites of phosphorylation activating AKT, were significantly reduced with KO of TFAP2A (Figure 3B–C). Likewise, phosphorylation of Ser124 of AKT reduced, but did not reach statistical significance (Figure 3C). Additionally, one of the downstream targets of AKT, CDC25A, showed reduced phosphorylation at Ser124 with KO of TFAP2A, consistent with decreased activity downstream of the AKT cascade after loss of AP-2α. To further confirm these findings, western blot analysis of unsynchronized parental HCT116 and TFAP2A KO clones revealed similar overall expression of AKT, but decreased phosphorylation of AKT at Ser473 and Thr308 with KO of TFAP2A (Figure 3D). A similar decreased level of phosphorylation of AKT was observed with transient knockdown of TFAP2A in the colon cancer cell line, LoVo, in unsynchronized cells (Figure 3E). AKT is phosphorylated at Thr308 by PDK1 through the activation of the PI3K cascade (30) and at Ser473 by mTOR (31). The decreased level of phosphorylation after loss of AP-2α, both at baseline and as cells actively progress through the cell cycle, suggests a disruption of the PI3K/mTOR/AKT pathway.
Figure 3. Alteration in phosphorylated cell cycle regulators.
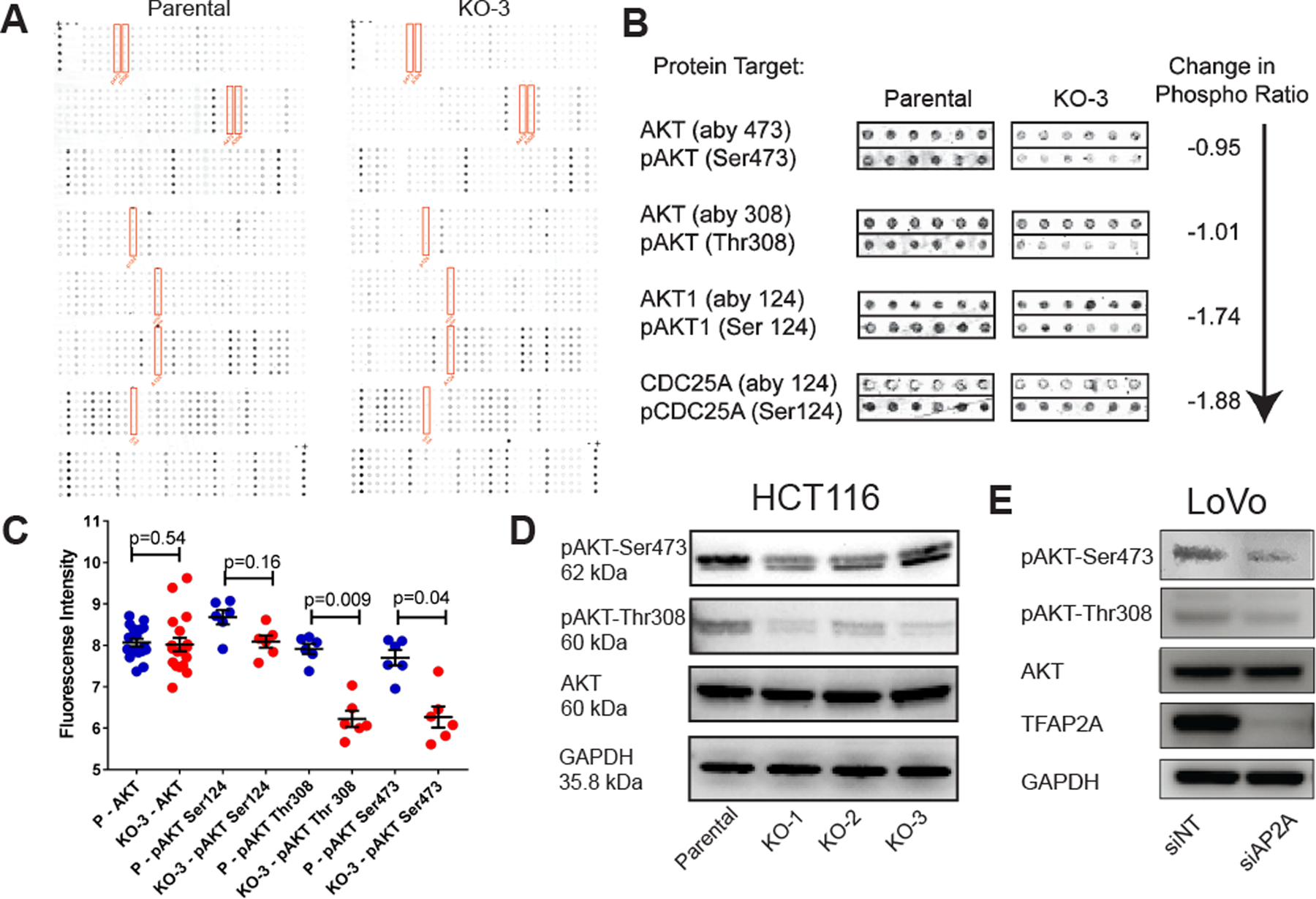
A. Cell cycle regulator phospho-protein microarray utilizing protein harvested 8 hours after release from G1 phase synchronization of HCT116 parental cells and clone KO-3 demonstrate significant decrease in the level of phosphorylated AKT without an overall change in the protein level of AKT (B & C).
D. Decreased phosphorylation of AKT after loss of AP-2α was confirmed in unsynchronized cells by western blot. E. Knockdown of TFAP2A in LoVo colon cancer cells demonstrate decreased pAKT at Ser473 and Thr308 by western blot.
The activation of the PI3K/AKT cascade is often driven by the presence of mutations in PIK3CA, encoding the catalytic subunit of PI3K. The colon cancer cell line HCT116 has a PIK3CA gene mutation c.3140A>G causing the missense mutation H1047R leading to its constitutive activation (32). The mutation status of all 3 AP-2α knockout clones was examined and was unchanged compared to the parental cell line (data not shown). We subsequently sought to identify further mechanisms for the alteration of PI3K/AKT cascade activity.
TGM2 is transcriptionally activated by AP-2α.
We identified the protein Tissue Transglutaminase (TGM2) as a potential mechanism for how AP-2α affects the activity of the PI3K/AKT cascade. TGM2, which is known to form a complex with PI3K, leading to its activation (33–35), was one of the 131 common genes whose level of expression was altered after loss of AP-2α. The level of TGM2 RNA and protein was decreased in KO-1 and KO-2 compared parental HCT116 (Figure 1) and ChIP-seq identified direct promoter occupancy by AP-2α in regulatory regions of the TGM2 gene (Figure 4A), confirmed by the presence of multiple AP-2α consensus sequences at the sites of observed AP-2α occupancy (Supplemental Figure S4).
Figure 4. TGM2 as AP-2α target gene and regulator of cell cycle.
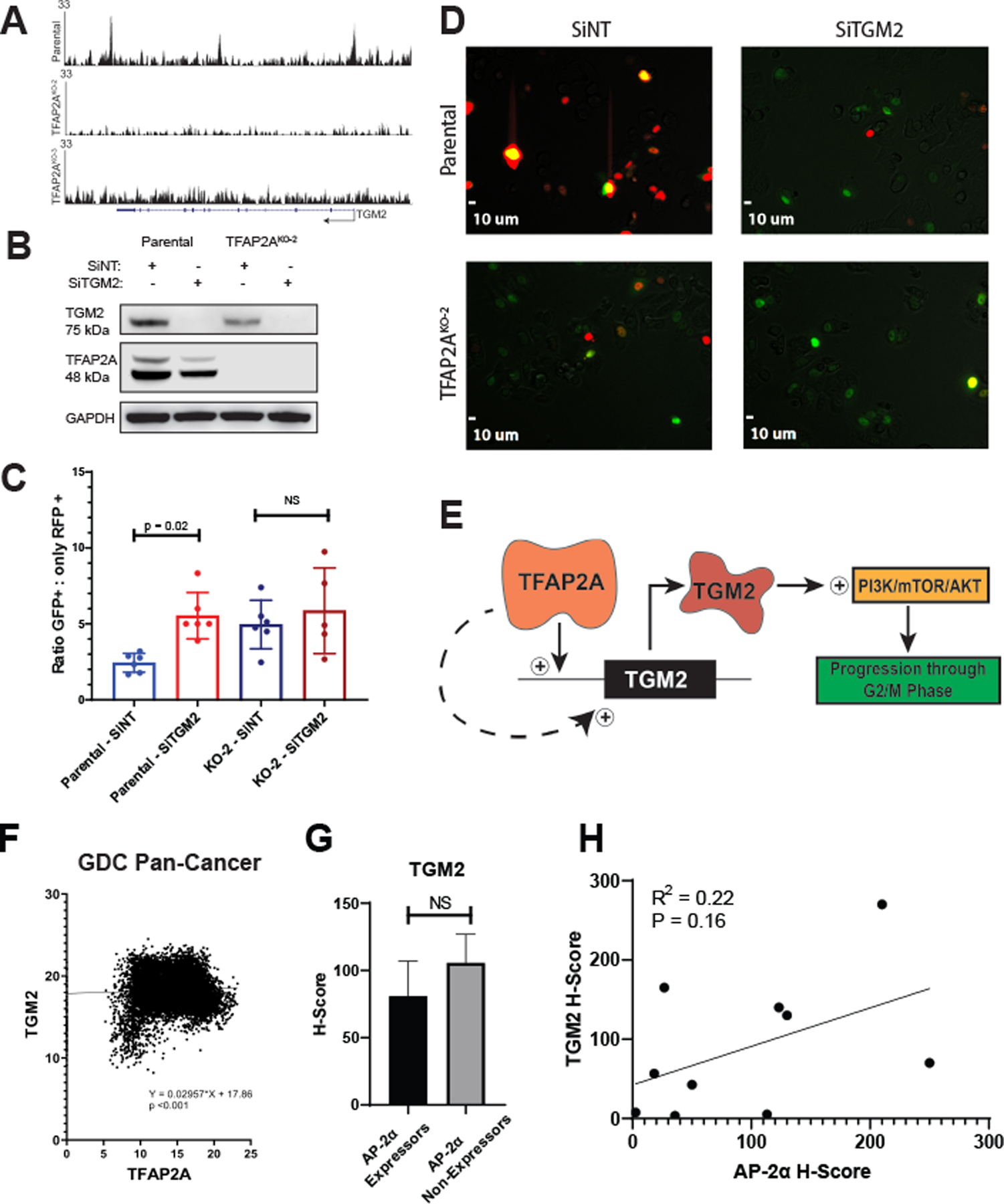
A. ChIP-Seq analysis identified TGM2 as a potential primary target of AP-2α with known AP-2α consensus binding sequences identified at the site of peaks within TGM2 (Supplementary Figure S4). B. Transient knockdown of TGM2 in parental HCT116 with siRNA resulted in a significant increase in the proportion of cells in S/G2/M phase (GFP+) vs. G1 phase (only RFP+) compared to cells treated with siNT, but no change was observed in KO-2 cells (C&D). E. Proposed model suggests TGM2 is a primary target of AP-2α, through which the PI3K/mTOR/AKT pathway is altered, leading to prolongation of S/G2/M phase. F. Data from the GDC Pan-Cancer database demonstrated a correlation between TFAP2A and TGM2 expression. Analysis of a TMA showed no significant difference in TGM2 protein expression comparing AP-2α expressors and non-expressors (G); however, there was a trend in the correlation of TGM2 and AP-2α protein in tumors expressing AP-2α (H).
We characterized the effect of TGM2 on cell cycle (Figure 4B–D). TGM2 was transiently knocked down with siRNA in parental HCT116 and KO clone KO-2. Unsynchronized cells at similar levels of confluency were transduced with the Premo FUCCI Cell Cycle Sensor, which utilizes CDT1-RFP and GMNN-GFP to fluorescently indicate the current phase of cell cycle.
Cells in G1 phase were identified by the sole expression of CDT1-RFP and cells in S/G2/M phase expressed either GMNN-GFP alone or GMNN-GFP + CDT1-RFP. The proportion of cells in S/G2/M phase compared to G1 phase was significantly higher after transient knockdown of TGM2 in parental HCT116 cells compared to cells transfected with siNT (Figure 4 C–D). As expected, there was no effect in the KO clone KO-2 with knockdown of TGM2. This supports the hypothesis that activation of TGM2 may be one of the mechanisms through which AP-2α regulates the activity of the PI3K/AKT cascade and subsequently cell cycle progression (Figure 4E). To determine if a correlation exists between TFAP2A and TGM2 expression in clinical cancer samples, expression of these genes was examined in published databases. A positive correlation was observed in the GDC Pan-Cancer database between TGM2 and TFAP2A expression (Figure 4F). This correlation was supported using a colon cancer tissue microarray (TMA), which identified 32% of tumors as expressing AP-2α, with a positive correlative trend between AP-2α and TGM2 expression in AP-2α expressing tumors (Figure 4G–H, Supplemental Figure S5).
AP-2α confers sensitivity to BKM120.
Given the alteration in PI3K/AKT activity observed after loss of AP-2α expression, we predicted that KO of TFAP2A will alter the response to a PI3K inhibitor. To test this hypothesis, we utilized the highly selective PI3K inhibitor, BKM120, also known as Buparlisib. The effect of BKM120 on the invasiveness of cells was tested in vitro using an invasion assay. After 24 hours of in vitro treatment, the parental HCT116 cells exposed to 250 nM BKM120 exhibited significantly less invasion compared to vehicle treated cells (Figure 5A&B). All three TFAP2A KO clones did not demonstrate any significant change in invasion with 250 nM BKM120 treatment compared to vehicle, suggesting that loss of AP-2α in colon cancer may confer resistance to this highly selective PI3K inhibitor.
Figure 5. Resistance to selective PI3K inhibitor BKM120 after loss of AP-2α.
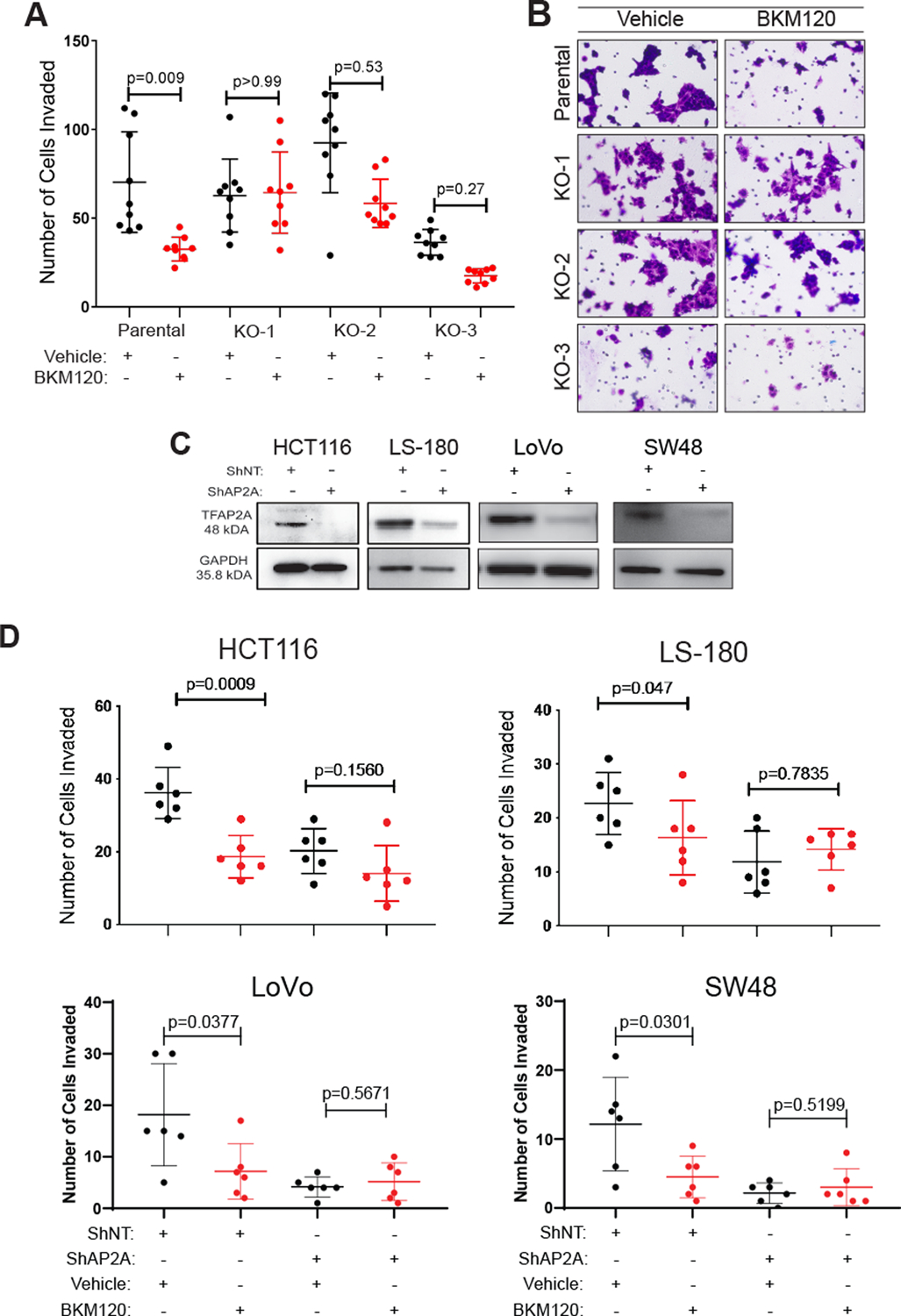
A & B. Invasion assay with BKM120 treatment revealed a decrease in invasion within 24 hours compared to vehicle treated cells in the HCT116 parental cell line, but no alteration in invasion occurred with BKM120 treatment in three AP-2α KO clones. C. Western blots showing stable knockdown of TFAP2A using shRNA in colon cancer cell lines HCT116, LS180, LoVo and SW40. D. Knockdown of TFAP2A induced resistance to the selective PI3K inhibitor BKM120, which led to a decrease in invasion compared to vehicle in shNT-treated cells.
We confirmed the effect of loss of AP-2α expression on response to BKM120 in two additional conditions. First, we performed an invasion assay which confirmed that stable knockdown of TFAP2A in HCT116 with shRNA conferred resistance to BKM120 (Figure 5C&D). Secondly, we analyzed the effect of stable knockdown of TFAP2A using shRNA transduction in a panel of colon cancer cell lines.. On invasion assay, BKM120 treated cells transduced with shNT demonstrated decreased invasion compared to those treated with vehicle, whereas loss of AP-2α in the panel of colon cancer cell lines conferred resistance to BKM120 (Figure 5C&D).
To further test the efficacy of BKM120 after loss of AP-2α, xenografts were established by inoculating immunocompromised Nu/J mice with either parental HCT116 cells or KO-3 in the subcutaneous tissue of their flanks. Tumors were allowed to establish over 7 days and mice were randomly assigned to be gavaged daily for 10 days with either vehicle or 40 mg/kg of BKM120. Tumors in mice with parental HCT116 xenografts treated with BKM120 demonstrated significantly reduced tumor growth (Figure 6A) and mice had longer disease specific survival (Figure 6B) compared to vehicle treated mice. Interestingly, mice inoculated with KO-3 had no difference in either tumor growth or tumor-specific survival when treated with BKM120 as compared to vehicle treated mice (Figure 6A&C). Tumors from all mice were analyzed for mitotic activity via immunohistochemistry for phosphorylated histone H3 (PHH3), which is known to decrease in vivo with PI3K inhibition (30). The number of PHH3 positive cells was significantly lower in BKM120 treated parental HCT116 tumors compared to vehicle treated tumors; however, there were no differences in the number of cells expressing PHH3 in KO-3 tumors that were treated with BKM120 as compared to vehicle treated tumors (Figure 6D–E). It is worth commenting that KO-3 xenografts did grow at a significantly slower rate than parental HCT116 and had a lower number of PHH3 positive cells (Figure 6A&D). This potentially may be reflective of the baseline change due to decreased PI3K/AKT activity after loss of AP-2α and that treatment with BKM120 may decrease the level of PI3K/AKT activity to that of the KO clone. These in vivo data further support that expression of AP-2α is a marker of BKM120 sensitivity.
Figure 6. Loss of AP-2α induces in vivo resistance to BKM120.
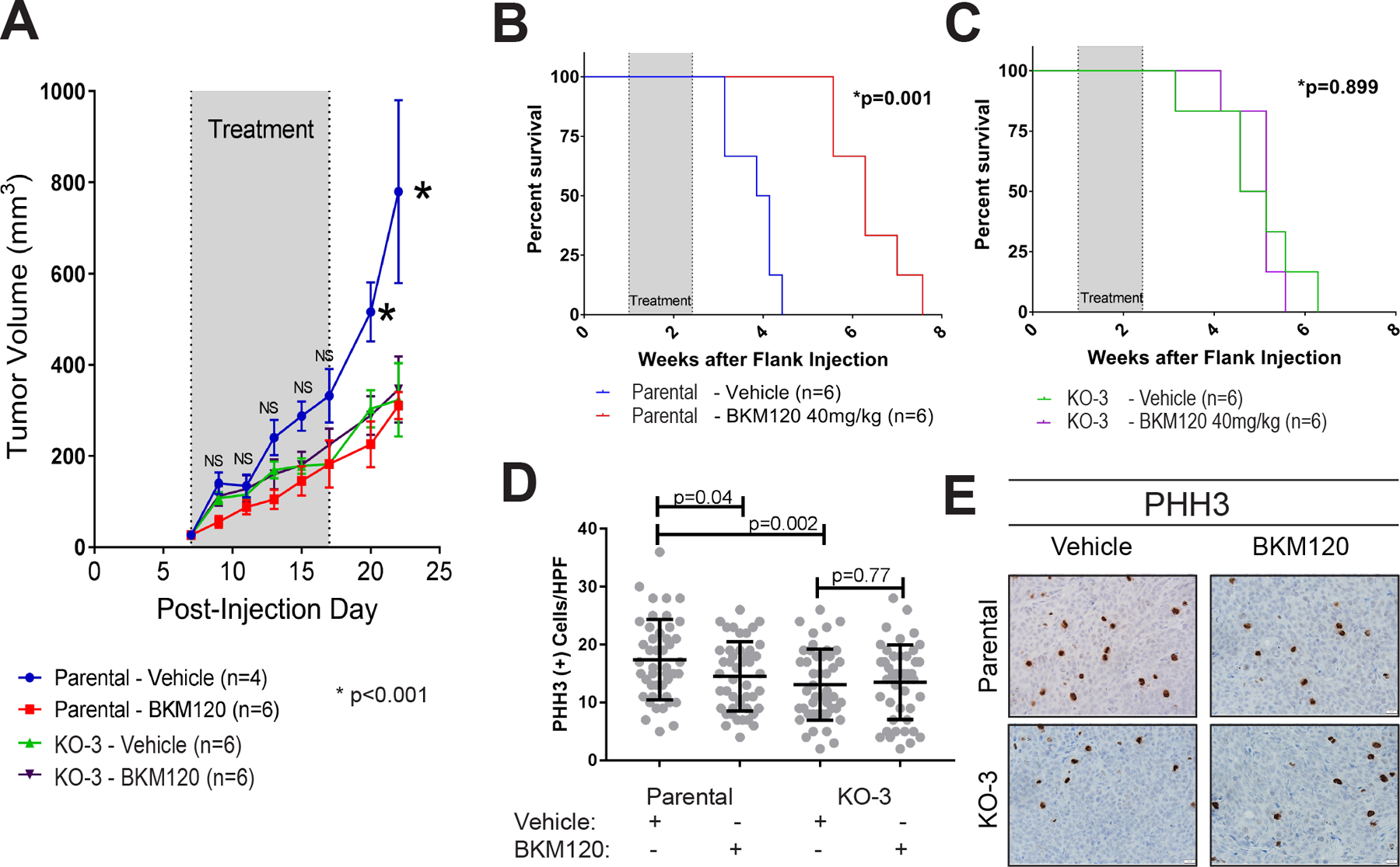
Mouse xenografts formed after flank injection of either HCT116 parental or KO-3. Xenografts were allowed 7 days to establish, then treatment with either vehicle (water) or BKM120 40 mg/kg oral gavage was initiated. A. BKM120 slowed tumor growth in parental HCT116 xenografts compared to vehicle, but had no effect in the KO-3 clone xenografts. B. Overall survival was significantly prolonged in BKM120 treated parental HCT116 xenografts, but no difference in survival was observed in KO-3 xenografts (C). D. Phosphorylated Histone H3 expression (PHH3) was significantly decreased in HCT116 parental xenografts treated with BKM120 compared to vehicle, but no change in PHH3 expression was observed in KO-3 xenografts. E. Examples of PHH3 staining in parental and KO-3 xenografts from mice treated with vehicle or BKM120.
Consistent with our findings, there is further evidence that TFAP2A expression correlates with sensitivity to BKM120 treatment in several cancer cell types. Analysis of RNA expression data in BKM120-sensitive vs. BKM120-resistant cells from published GeoDatabases (GSE69405, GSE98824, GSE49416), we found a statistically significant relationship between TFAP2A expression and response to BKM120 in lung adenocarcinoma (Supplemental Figure S6A). Furthermore, expression of TGM2 was also significantly predictive of response to BKM120. Similarly, we noted a trend in the relationship between TFAP2A and TGM2 expression and sensitivity to BKM120 in triple negative breast cancer, though this relationship did not reach statistical significance (Supplemental Figure S6A). We performed a transient knockdown of TFAP2A in a triple negative breast cancer cell line which demonstrated a loss of BKM120 sensitivity with knockdown of TFAP2A expression (Supplemental Figure S6B). Similar trends in the relationship between TFAP2A and TGM2 expression and BKM120 sensitivity were also seen in glioblastoma (Supplemental Figure S6A).
DISCUSSION
Previous studies have characterized changes in cell cycle progression after overexpression of AP-2α (25,36,37), and our findings both support these findings and add to our understanding of how AP-2α effects cell cycle progression. We identify a novel pathway by which AP-2α regulates the cell cycle and we have shown that the effect of AP-2α upon cell cycle regulation through the PI3K/AKT cascade alters the response to the highly specific PI3K inhibitor, BKM120.
Previous studies in the HCT116 colon cancer cell line demonstrated that AP-2α induced cell cycle arrest through p53-dependent activation of CDKN1A/p21 (25,38), and may also function in a p53-independent mechanism (38). Similar mechanisms of CDKN1A/p21 gene regulation by AP-2α have been demonstrated in breast, cervical carcinoma, hepatoblastoma, lung carcinoma and other colon cancer cell lines (14,36,38,39). Overexpression of AP-2α is also capable of inducing growth arrest and apoptosis characterized by hyperphosphorylation of Rb and cleavage of PARP (38). Although it is unlikely to be the sole pathway through which AP-2α alters cell cycle progression, the present study identified TGM2 as a primary target of AP-2α and a potential mechanism through which AP-2α alters the PI3K/AKT cascade. The previous studies and our current data support the finding that AP-2α plays a clear role in the progression of cancer cells through S-phase.
The role of AP-2α in the regulation of the PI3K/AKT cascade is relatively unexplored, although there are previous data that support this relationship. A study by Fertig et al. utilized the LINCS (library of integrated network-based cellular signatures) database and identified that AP-2α gene expression signatures were increased with the inhibition of MEK, PI3K and mTOR pathways indicating a positive feedback mechanism (40). Additionally, in a previous study knockdown of TFAP2C, encoding the AP-2γ family member, resulted in a decrease in AKT activation in breast cancer indicating that other AP-2 factors may have similar activity (41). Retinoic acid has been shown to repress the PI3K/AKT pathway in several cell types (42–44), and can induce expression of TFAP2A (45,46); however, the role of AP-2α in response to retinoic acid was not examined in these studies. Additionally, a study in mouse muscle myoblasts reported that AP-2α can repress activation of AKT through miR-25-3p, suggesting competing effects in different cell types that may be influenced by miR expression (47).
Recent data on the use of BKM120 in clinical trials in patients with colorectal cancer has highlighted the need for the identification of biomarkers to predict response (48). Identifying biomarkers for sensitivity to BKM120 will expand and improve patient directed therapy. This will allow for the early identification of patients who will receive the most benefit from BKM120 and avoid unnecessary toxicity in patients unlikely to demonstrate a favorable response. This in turn will help to establish the true efficacy of BKM120 in treating colon cancer. Previous studies have identified that, although BKM120 was efficacious in multiple colon cancer cell lines, it exhibited a greater level of apoptosis in those cell lines with a PI3KCA mutation (49). PIK3CA is one of the most commonly mutated genes in cancer (5% of cancers) and individualized treatment based on mutational profiles and in vitro drug testing are becoming more common methods to guide therapy (50). Our study identified that loss of AP-2α conferred resistance to PI3K inhibition in the presence of a PIK3CA mutation and expression of AP-2α may serve as a biomarker for response in other cancer types.
In conclusion, we identified that loss of AP-2α results in prolongation of S-phase, which is mediated through the PI3K cascade and activation of AKT. Additionally, TGM2 is an AP-2α target gene that influences activation of the PI3K/AKT pathway. The role of AP-2α in regulating the PI3K/AKT cascade in colon cancer is of particular importance as we identified that AP-2α is a marker of sensitivity to the highly selective PI3K inhibitor, BKM120, which is currently being used in multiple clinical trials. Therefore, AP-2α may be utilized as a biomarker for identifying patients likely to benefit from BKM120 therapy.
Supplementary Material
Implications:
AP-2alpha regulated cell cycle through the PI3K cascade and activation of AKT mediated through TGM2. AP-2alpha induced sensitivity to Buparlisib/BKM120, indicating that AP-2alpha is a biomarker predictive of response to PI3K inhibitors.
ACKNOWLEDGEMENTS
The data presented herein were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine/Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran’s Administration Medical Center. We thank Dr. Alejandro A Pezzulo at the University of Iowa for his assistance in analysis allowing for quantification of fluorescent imaging and Drs. Andrew Bellizzi and Anand Rajan for assistance with immunohistochemistry staining. Additionally, the authors would like to acknowledge use of the University of Iowa Central Microscopy Research Facility, a core resource supported by the University of Iowa Vice President for Research, and the Carver College of Medicine. This work was supported by the NIH grants R01CA183702 (PI: R.J.W.) and T32CA148062 (P.I.: R.J.W.) A.C.B, E.C., D.T.T., K.E.K., and V.T.W. were supported by the NIH grant T32CA148062.
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136(5):E359–86 doi 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Siegel RL, Ward EM, Jemal A. Global Cancer Incidence and Mortality Rates and Trends--An Update. Cancer Epidemiol Biomarkers Prev 2016;25(1):16–27 doi 10.1158/1055-9965.EPI-15-0578. [DOI] [PubMed] [Google Scholar]
- 3.Andre T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350(23):2343–51 doi 10.1056/NEJMoa032709. [DOI] [PubMed] [Google Scholar]
- 4.Andre T, Boni C, Navarro M, Tabernero J, Hickish T, Topham C, et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol 2009;27(19):3109–16 doi 10.1200/JCO.2008.20.6771. [DOI] [PubMed] [Google Scholar]
- 5.Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol 2005;205(2):275–92 doi 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- 6.Dietel M, Johrens K, Laffert MV, Hummel M, Blaker H, Pfitzner BM, et al. A 2015 update on predictive molecular pathology and its role in targeted cancer therapy: a review focussing on clinical relevance. Cancer Gene Ther 2015;22(9):417–30 doi 10.1038/cgt.2015.39. [DOI] [PubMed] [Google Scholar]
- 7.O’Connell MJ, Lavery I, Yothers G, Paik S, Clark-Langone KM, Lopatin M, et al. Relationship between tumor gene expression and recurrence in four independent studies of patients with stage II/III colon cancer treated with surgery alone or surgery plus adjuvant fluorouracil plus leucovorin. J Clin Oncol 2010;28(25):3937–44 doi 10.1200/JCO.2010.28.9538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benson AB 3rd, Venook AP, Cederquist L, Chan E, Chen YJ, Cooper HS, et al. Colon Cancer, Version 1.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15(3):370–98 doi 10.6004/jnccn.2017.0036. [DOI] [PubMed] [Google Scholar]
- 9.Cremolini C, Schirripa M, Antoniotti C, Moretto R, Salvatore L, Masi G, et al. First-line chemotherapy for mCRC-a review and evidence-based algorithm. Nat Rev Clin Oncol 2015;12(10):607–19 doi 10.1038/nrclinonc.2015.129. [DOI] [PubMed] [Google Scholar]
- 10.Sveen A, Kopetz S, Lothe RA. Biomarker-guided therapy for colorectal cancer: strength in complexity. Nat Rev Clin Oncol 2020;17(1):11–32 doi 10.1038/s41571-019-0241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitchell PJ, Timmons PM, Hebert JM, Rigby PW, Tjian R. Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev 1991;5(1):105–19 doi 10.1101/gad.5.1.105. [DOI] [PubMed] [Google Scholar]
- 12.Bosher JM, Williams T, Hurst HC. The developmentally regulated transcription factor AP-2 is involved in c-erbB-2 overexpression in human mammary carcinoma. Proc Natl Acad Sci U S A 1995;92(3):744–7 doi 10.1073/pnas.92.3.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McPherson LA, Baichwal VR, Weigel RJ. Identification of ERF-1 as a member of the AP2 transcription factor family. Proc Natl Acad Sci U S A 1997;94(9):4342–7 doi 10.1073/pnas.94.9.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeng YX, Somasundaram K, el-Deiry WS. AP2 inhibits cancer cell growth and activates p21WAF1/CIP1 expression. Nat Genet 1997;15(1):78–82 doi 10.1038/ng0197-78. [DOI] [PubMed] [Google Scholar]
- 15.Bogachek MV, Chen Y, Kulak MV, Woodfield GW, Cyr AR, Park JM, et al. Sumoylation pathway is required to maintain the basal breast cancer subtype. Cancer Cell 2014;25(6):748–61 doi 10.1016/j.ccr.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Andrade JP, Lorenzen AW, Wu VT, Bogachek MV, Park JM, Gu VW, et al. Targeting the SUMO pathway as a novel treatment for anaplastic thyroid cancer. Oncotarget 2017;8(70):114801–15 doi 10.18632/oncotarget.21954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mobley AK, Braeuer RR, Kamiya T, Shoshan E, Bar-Eli M. Driving transcriptional regulators in melanoma metastasis. Cancer Metastasis Rev 2012;31(3–4):621–32 doi 10.1007/s10555-012-9358-8. [DOI] [PubMed] [Google Scholar]
- 18.Gee JM, Robertson JF, Ellis IO, Nicholson RI, Hurst HC. Immunohistochemical analysis reveals a tumour suppressor-like role for the transcription factor AP-2 in invasive breast cancer. J Pathol 1999;189(4):514–20 doi . [DOI] [PubMed] [Google Scholar]
- 19.Ruiz M, Troncoso P, Bruns C, Bar-Eli M. Activator protein 2alpha transcription factor expression is associated with luminal differentiation and is lost in prostate cancer. Clin Cancer Res 2001;7(12):4086–95. [PubMed] [Google Scholar]
- 20.Su W, Xia J, Chen X, Xu M, Nie L, Chen N, et al. Ectopic expression of AP-2alpha transcription factor suppresses glioma progression. Int J Clin Exp Pathol 2014;7(12):8666–74. [PMC free article] [PubMed] [Google Scholar]
- 21.Nordentoft I, Dyrskjot L, Bodker JS, Wild PJ, Hartmann A, Bertz S, et al. Increased expression of transcription factor TFAP2alpha correlates with chemosensitivity in advanced bladder cancer. BMC Cancer 2011;11:135 doi 10.1186/1471-2407-11-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang H, Zeng L, Wang J, Zhang X, Ruan Q, Wang J, et al. Reversal of 5-fluorouracil resistance by EGCG is mediate by inactivation of TFAP2A/VEGF signaling pathway and down-regulation of MDR-1 and P-gp expression in gastric cancer. Oncotarget 2017;8(47):82842–53 doi 10.18632/oncotarget.20666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Q, Lohr CV, Dashwood RH. Activator protein 2alpha suppresses intestinal tumorigenesis in the Apc(min) mouse. Cancer Lett 2009;283(1):36–42 doi 10.1016/j.canlet.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz B, Melnikova VO, Tellez C, Mourad-Zeidan A, Blehm K, Zhao YJ, et al. Loss of AP-2alpha results in deregulation of E-cadherin and MMP-9 and an increase in tumorigenicity of colon cancer cells in vivo. Oncogene 2007;26(28):4049–58 doi 10.1038/sj.onc.1210193. [DOI] [PubMed] [Google Scholar]
- 25.McPherson LA, Loktev AV, Weigel RJ. Tumor suppressor activity of AP2alpha mediated through a direct interaction with p53. J Biol Chem 2002;277(47):45028–33 doi 10.1074/jbc.M208924200. [DOI] [PubMed] [Google Scholar]
- 26.Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A 2009;106(29):11960–5 doi 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006;1(5):2315–9 doi 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 28.Wu VT, Kiriazov B, Koch KE, Gu VW, Beck AC, Borcherding N, et al. A TFAP2C Gene Signature Is Predictive of Outcome in HER2-Positive Breast Cancer. Mol Cancer Res 2020;18(1):46–56 doi 10.1158/1541-7786.MCR-19-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang S, Li X, Guan W, Qian M, Yao Z, Yin X, et al. NVP-BKM120 inhibits colon cancer growth via FoxO3a-dependent PUMA induction. Oncotarget 2017;8(47):83052–62 doi 10.18632/oncotarget.20943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol 1997;7(10):776–89 doi 10.1016/s0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- 31.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005;307(5712):1098–101 doi 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 32.Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknaes M, Hektoen M, et al. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013;2:e71 doi 10.1038/oncsis.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boroughs LK, Antonyak MA, Cerione RA. A novel mechanism by which tissue transglutaminase activates signaling events that promote cell survival. J Biol Chem 2014;289(14):10115–25 doi 10.1074/jbc.M113.464693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verma A, Wang H, Manavathi B, Fok JY, Mann AP, Kumar R, et al. Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res 2006;66(21):10525–33 doi 10.1158/0008-5472.CAN-06-2387. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Ande SR, Mishra S. Phosphorylation of transglutaminase 2 (TG2) at serine-216 has a role in TG2 mediated activation of nuclear factor-kappa B and in the downregulation of PTEN. BMC Cancer 2012;12:277 doi 10.1186/1471-2407-12-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scibetta AG, Wong PP, Chan KV, Canosa M, Hurst HC. Dual association by TFAP2A during activation of the p21cip/CDKN1A promoter. Cell Cycle 2010;9(22):4525–32 doi 10.4161/cc.9.22.13746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lian W, Zhang L, Yang L, Chen W. AP-2alpha reverses vincristine-induced multidrug resistance of SGC7901 gastric cancer cells by inhibiting the Notch pathway. Apoptosis 2017;22(7):933–41 doi 10.1007/s10495-017-1379-x. [DOI] [PubMed] [Google Scholar]
- 38.Wajapeyee N, Somasundaram K. Cell cycle arrest and apoptosis induction by activator protein 2alpha (AP-2alpha) and the role of p53 and p21WAF1/CIP1 in AP-2alpha-mediated growth inhibition. J Biol Chem 2003;278(52):52093–101 doi 10.1074/jbc.M305624200. [DOI] [PubMed] [Google Scholar]
- 39.Orso F, Penna E, Cimino D, Astanina E, Maione F, Valdembri D, et al. AP-2alpha and AP-2gamma regulate tumor progression via specific genetic programs. FASEB J 2008;22(8):2702–14 doi 10.1096/fj.08-106492. [DOI] [PubMed] [Google Scholar]
- 40.Fertig EJ, Ozawa H, Thakar M, Howard JD, Kagohara LT, Krigsfeld G, et al. CoGAPS matrix factorization algorithm identifies transcriptional changes in AP-2alpha target genes in feedback from therapeutic inhibition of the EGFR network. Oncotarget 2016;7(45):73845–64 doi 10.18632/oncotarget.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spanheimer PM, Cyr AR, Gillum MP, Woodfield GW, Askeland RW, Weigel RJ. Distinct pathways regulated by RET and estrogen receptor in luminal breast cancer demonstrate the biological basis for combination therapy. Ann Surg 2014;259(4):793–9 doi 10.1097/SLA.0b013e3182a6f552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park EY, Wilder ET, Chipuk JE, Lane MA. Retinol decreases phosphatidylinositol 3-kinase activity in colon cancer cells. Mol Carcinog 2008;47(4):264–74 doi 10.1002/mc.20381. [DOI] [PubMed] [Google Scholar]
- 43.Feng Y, Hua X, Niu R, Du Y, Shi C, Zhou R, et al. ROS play an important role in ATPR inducing differentiation and inhibiting proliferation of leukemia cells by regulating the PTEN/PI3K/AKT signaling pathway. Biol Res 2019;52(1):26 doi 10.1186/s40659-019-0232-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang L, Wang X, Zheng Y, Liu Y. Alltransretinoic acid modulates TGFbetainduced apoptosis, proliferation, migration and extracellular matrix synthesis of conjunctival fibroblasts by inhibiting PI3K/AKT signaling. Mol Med Rep 2019;20(3):2929–35 doi 10.3892/mmr.2019.10507. [DOI] [PubMed] [Google Scholar]
- 45.Williams T, Admon A, Luscher B, Tjian R. Cloning and expression of AP-2, a cell-type-specific transcription factor that activates inducible enhancer elements. Genes Dev 1988;2(12A):1557–69 doi 10.1101/gad.2.12a.1557. [DOI] [PubMed] [Google Scholar]
- 46.Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res 2002;43(11):1773–808 doi 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 47.Zhang F, Chen K, Tao H, Kang T, Xiong Q, Zeng Q, et al. miR-25–3p, Positively Regulated by Transcription Factor AP-2alpha, Regulates the Metabolism of C2C12 Cells by Targeting Akt1. Int J Mol Sci 2018;19(3) doi 10.3390/ijms19030773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodwin R, Jonker D, Chen E, Kennecke H, Cabanero M, Tsao MS, et al. A phase Ib study of a PI3Kinase inhibitor BKM120 in combination with panitumumab in patients with KRAS wild-type advanced colorectal cancer. Invest New Drugs 2019. doi 10.1007/s10637-019-00814-3. [DOI] [PubMed] [Google Scholar]
- 49.Mueller A, Bachmann E, Linnig M, Khillimberger K, Schimanski CC, Galle PR, et al. Selective PI3K inhibition by BKM120 and BEZ235 alone or in combination with chemotherapy in wild-type and mutated human gastrointestinal cancer cell lines. Cancer Chemother Pharmacol 2012;69(6):1601–15 doi 10.1007/s00280-012-1869-z. [DOI] [PubMed] [Google Scholar]
- 50.Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, Sboner A, et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov 2017;7(5):462–77 doi 10.1158/2159-8290.CD-16-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.