
Figure 1 .
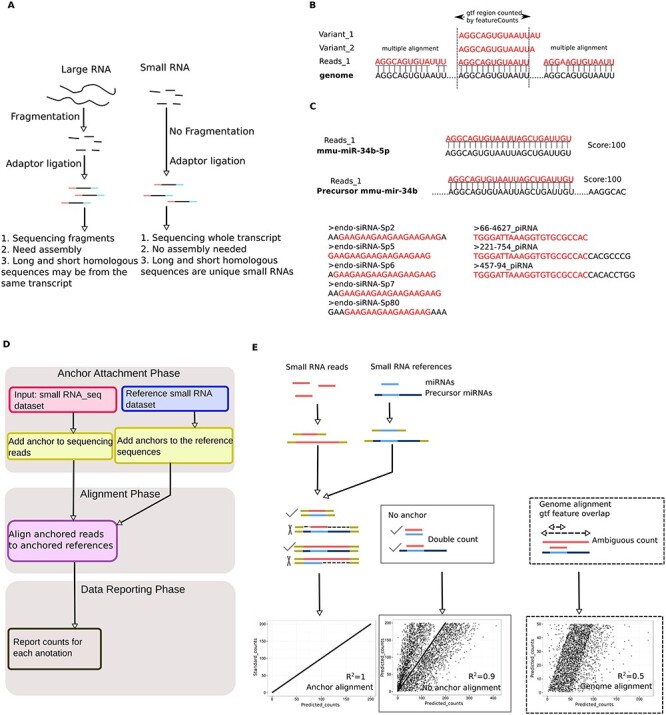
Development of the anchor alignment algorithm for sncRNA annotation. (A) Schematic illustration of the differences in large and small RNA library construction methods. Note that adaptors are directly added to the small RNAs for sncRNA-Seq, whereas fragmentation is needed before adaptor ligation for large RNA sequencing. (B) Issues associated with direct sncRNA alignment to the genome: multiple alignment of sncRNAs to the genome due to their small sizes (20–40 nt), and inability to recognize sncRNA variants (e.g., homologous piRNAs, endo-siRNAs, mitosRNAs, etc.). (C) Issues associated with the direct sncRNA–sncRNA alignment algorithm: repetitive counting of mature miRNA reads (because they can be mapped to both mature and precursor miRNA references), and certain sncRNA reads (e.g., endo-siRNAs and piRNAs, due to the presence of multiple, staggered sncRNA homologs in the reference databases, which differ by only several nucleotides). (D) Workflow of the AASRA pipeline. (E) Schematic illustration of anchor alignment algorithm. Anchors are added to both ends of the sequencing reads and the reference sncRNAs. Gap opening penalty can prevent mature miRNA sequence reads from mapping to the precursor miRNA reference sequences. Perfect alignment and correct annotation of both mature and precursor miRNAs were achieved for the simulation data using the anchor alignment algorithm (R2 = 1), whereas direct alignment of the simulation data to either the sncRNA references (R2 = 0.9), or the genome (R2 = 0.5) led to partial alignment.