

Abstract
Electrochemical reduction of carbon dioxide (CO2RR) is an attractive route to close the carbon cycle and potentially turn CO2 into valuable chemicals and fuels. However, the highly selective generation of multicarbon products remains a challenge, suffering from poor mechanistic understanding. Herein, we used operando Raman spectroscopy to track the potential-dependent reduction of Cu2O nanocubes and the surface coverage of reaction intermediates. In particular, we discovered that the potential-dependent intensity ratio of the Cu–CO stretching band to the CO rotation band follows a volcano trend similar to the CO2RR Faradaic efficiency for multicarbon products. By combining operando spectroscopic insights with Density Functional Theory, we proved that this ratio is determined by the CO coverage and that a direct correlation exists between the potential-dependent CO coverage, the preferred C–C coupling configuration, and the selectivity to C2+ products. Thus, operando Raman spectroscopy can serve as an effective method to quantify the coverage of surface intermediates during an electrocatalytic reaction.
Keywords: operando Raman spectroscopy, CO2 reduction, Cu2O nanocubes, C-C coupling
Introduction
The electrochemical reduction of carbon dioxide (CO2RR), powered by renewable electricity, is an attractive route to convert CO2 into valuable products, thereby closing the anthropogenic carbon cycle and transforming intermittent energy into chemical energy to provide fuels and feedstocks.1 Although numerous efforts have been made, the highly effective and selective generation of economically desirable products remains a great challenge, especially for multicarbon chemicals (C2+) such as ethylene and ethanol with higher energy density and wider applicability.2−6 Concurrently, processes involving C–C bond formation are of great interest and significance to fundamental research.
Cu-based materials are known to be the most active catalysts for CO2RR to yield C2+ products in significant amounts.2,7 Thus, many experimental and theoretical studies have focused on understanding the C–C coupling mechanism on Cu surfaces.8−12 From a modeling perspective, CO is considered as one of the key intermediates in CO2RR since it can dimerize to form OCCO species or be hydrogenated to form CHO species.9,13,51 Experimentally, it is known that the onset potential for the formation of C2H4 starts 300–400 mV more negative than the onset potential for CO evolution and that the Faradaic efficiency (F.E.) of the C2+ products shows a volcano dependence on the applied potential.7,14,15 Specific CO adsorption configurations are considered to be crucial for OC–CO dimerization,4,16 and a link between the CO coverage and the formation of C2+ products has been proposed.17,18 However, key experimental observations remained absent, and it is still a great challenge to quantify the CO coverage at the solid–liquid interface during CO2RR and give an unambiguous explanation on the potential-dependent C2+ F.E. at the molecular scale. Furthermore, a full mechanistic understanding of the C–C coupling at certain given CO coverages and adsorption configurations is still not conclusive. Therefore, an operando method is highly desirable to determine the surface coverage of CO during CO2RR.
Surface-enhanced Raman spectroscopy (SERS), detecting vibrational and rotational information in a broad spectral range with high surface sensitivity, allows to investigate the electrochemical solid–liquid interface and the interaction of surface intermediates with the active electrode.19−21 Nonetheless, the widespread application of this method is limited by the necessity of using plasmonic materials. Fortunately, nanostructured Cu materials display typical plasmonic effects that can enhance the Raman signals of surface species and improve the detection limits; thus, more attention has been paid to the use of Raman to investigate the CO adsorption and configuration during the CO2RR process.22−25 However, since the surface enhancement effect is highly related to the local nanostructure, the quantification of adsorbate surface concentrations directly from the spectral Raman intensity is hindered.
Herein, we used operando Raman spectroscopy combined with quasi-in situ Cu LMM X-ray Auger electron spectroscopy (XAES) to reveal the transformations of the electrode–liquid interface during CO2RR over Cu2O nanocube electrocatalysts. In particular, we discovered that the intensity ratio of the Cu–CO stretching to the CO rotation band is determined by the CO coverage. This observation was confirmed and explained by operando Raman experiments in CO-rich electrolytes with different CO concentrations and theoretical investigations based on Density Functional Theory (DFT). We further established a direct correlation between the C2+ product selectivity, the potential-dependent CO surface coverage, and CO adsorption configurations under reduction conditions. Our work demonstrates that operando SERS combined with DFT is an integrated methodology for investigating the electrochemical solid–liquid interface and quantifying the coverage of surface intermediates during electrocatalytic reactions.
Results and Discussion
Well-defined and surfactant-free ∼25 nm Cu2O nanocubes were used as a model system (Figures 1a, S1, S2). The X-ray diffraction pattern confirms the sole presence of Cu2O, and Rietveld refinement reveals a structural coherence length of ∼29 nm and a lattice parameter, a, of 4.267(2) Å (Figure S3). Linear combination analysis of quasi-in situ Cu LMM XAES data of the as-prepared nanocubes (Figure 1c) indicates a surface composition of about 80% Cu(I) and 20% Cu(II) species. After 1 h of CO2RR at −1.0 VRHE in 0.1 M KHCO3, the cubic morphology was partially retained, although hollow Cu structures were observed (Figures 1b, S4, S5), consistent with previous reports.26 It should be noted that the slight differences in the cube size observed in the transmission electron microscopy (TEM) images presented likely originate from the size distribution already present in the as-prepared cubes. A further plausible explanation is the redeposition of small Cu particles from dissolved Cu species in the electrolyte originating from the hollowed cubes that were observed during the CO2RR process. The surface of the Cu2O nanocubes was fully reduced to metallic Cu after CO2RR (Figure 1c), in agreement with the Cu 2p X-ray photoelectron spectroscopy data (Figure S6). For these experiments, the sample was transferred under an inert atmosphere between the electrochemical cell and the directly interfaced XAES ultrahigh vacuum analysis chamber (Figure S7).
Figure 1.

Structural and chemical characterization as well as CO2RR performance of Cu2O nanocubes. TEM images of Cu2O nanocubes in their as-prepared state (a) and after 1 h CO2RR at −1.0 VRHE (b). (c) Quasi-in situ Cu LMM XAES spectra of Cu2O nanocubes in the as-prepared state and after 1 h of CO2RR at −1.0 VRHE without air exposure. Potential-dependent F.E. of (d) ethylene, (e) ethanol (EtOH) and 1-propanol (PrOH), (f) the sum of all C2+ products, and (g) H2 and CO obtained after 1 h of CO2RR. Solid lines are guides for the eye. All electrochemical experiments were conducted in 0.1 M KHCO3, and the electrode potentials are given vs the RHE.
Figure 1d–g shows the F.E.s of CO2RR products which vary strongly with the applied potential (Figures S8 and S9). A typical volcano dependence of the F.E. on the applied potential appears for the C2+ products, which reaches a maximum of 60% at about −1.05 VRHE. The CO F.E. decreases with the potential from about −0.85 to −1.2 VRHE. The potential-dependent F.E. of H2 opposes the trend obtained for the C2+ products, with a minimum value at around −1.0 VRHE. The CH4 F.E. increases with the potential from about −1.0 to −1.2 VRHE. Similar potential-dependent F.E.s of CO2RR products have been widely reported and discussed experimentally and theoretically,7,14,15,27 but a molecular understanding is still lacking.
Operando SERS measurements were carried out to investigate the catalyst structure and surface adsorbates during CO2RR as well as their dynamics (see the experimental setup in Figure S10). Figure 2a displays the operando Raman spectra acquired on the same position of a glassy carbon electrode decorated with Cu2O nanocubes as a function of the applied potential in a CO2-saturated 0.1 M KHCO3 electrolyte. The glassy carbon substrate exhibits Raman peaks at 1313 and 1616 cm–1 for potentials ranging from the open circuit potential (OCP) to −1.2 VRHE (Figure S11). The potential-dependent change of these two peaks in Figure 2a is mainly due to the transformation of Cu2O to metallic Cu and the formation of bubbles on the electrode surface during CO2RR. Consecutive Raman spectra at OCP prove the stability of the electrode under the measurement conditions employed (Figure S12).
Figure 2.

Operando Raman spectroscopy data on Cu2O nanocubes during CO2RR. (a) Raman spectra of Cu2O nanocubes acquired during CO2RR for potentials ranging from the OCP to −1.2 VRHE. (b) Zoom-in spectra of the restricted rotation of adsorbed CO (P1) and Cu–CO stretching (P2) from −0.4 to −1.2 VRHE. (c) Potential-dependent intensity ratio of P2 to P1. The error bars were obtained as standard deviation (see the Supporting Information for details). All electrochemical experiments were conducted in 0.1 M KHCO3, and the electrode potentials are given vs the RHE.
Raman peaks at 415, 530, and 625 cm–1 belong to Cu2O.28,29 When the potential decreases to +0.3 VRHE, these peaks disappear, and a new peak at about 360 cm–1 evolves (Figure S13). The Raman shift of this band is similar to that of the Cu–CO stretching of adsorbed CO, but its assignment is still under debate.24,30,31 Our data shows that this band shifts to lower wavenumbers as the potential decreases (Figure S14), just opposite to the change of the Cu–CO stretching band (Figure 2b). The red shift of the wavenumber and the relative high onset potential preliminarily rule out its assignment to the Cu–CO stretching band. Interestingly, the 360 cm–1 peak is accompanied by another peak at 706 cm–1 from +0.3 to 0.0 VRHE, which has been assigned to surface hydroxyl species.31,32 Concurrently, a strong signal of carbonate species at 1077 cm–1 is detected, in line with previous experimental studies and DFT benchmarks (Table S1).24,30,52,53 In order to provide a reasonable assignment, systematic control experiments were carried out. During the backward scan from CO2RR conditions to the OCP (Figures S15 and S16), with only metallic Cu, no peaks at such wavenumber were observed in the similar potential range, which means that this peak is not related to reaction intermediates of CO2RR but related to the reduction of copper oxide species. Control experiments in Ar-saturated KHCO3 showed the same peaks at a similar potential (Figure S17), indicating that this band is independent of the CO2 electrolyte saturation. Without carbonate ions in the electrolyte, these bands disappeared as seen in Ar-saturated NaClO4 (Figure S18). Thus, we assign this band at 360 cm–1 to the surface copper carbonate species formed during the reduction of copper oxide species in the presence of carbonate electrolyte ions (KHCO3) and hydroxyl species,33 in agreement with Raman spectra of malachite and azurite,34 and DFT vibrational frequencies of carbonate on Cu (Table S1).
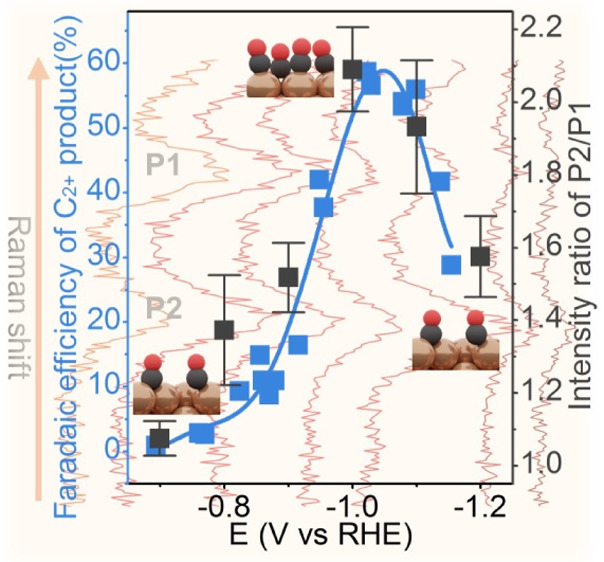
At about −0.1 VRHE, the electrode with the surface copper carbonate species is reduced to metallic Cu, consistent with the cyclic voltammogram (Figure S19). Furthermore, during the reduction process, no significant CO signal is detected. A peak around 2000 cm–1 is observed; however, this feature disappears at about −0.4 VRHE. The same vibrational fingerprint is detected at a similar potential in a control experiment using Ar-saturated KHCO3 as electrolyte (Figure S17). In the literature, this peak was previously attributed to H adsorbed on Cu.35 From −0.5 VRHE, the presence of adsorbed CO is demonstrated by the Raman peaks located at 280, 355–370, and 1970–2110 cm–1, corresponding to the restricted rotation of adsorbed CO (P1), Cu–CO stretching (P2), and C–O stretching, respectively (Figure 2a,b).36−38 In some reports, the broad C–O stretching band was attributed to different CO adsorption configurations or sites, including bridge-bonded and atop-bonded CO or CO adsorbed on terrace and defect sites.4,53,39 Moreover, the P1 and P2 bands, which reflect the interaction between reaction intermediates and the Cu electrode surface, change regularly with the potential. P2 displays a blue shift in the peak frequencies as the electrode potential decreases (Figure 2b). A similar potential-dependent phenomenon has been reported for carbon monoxide chemisorbed on a platinum surface, and it was attributed to the electrochemical Stark effect.40 Because the influence of dipole–dipole coupling interactions on the Pt–CO vibration has been proven negligible, the Raman shift of the Pt–CO band can be considered an indicator for potential-induced changes in the bonding strength and bond length of Pt–CO.40 The same approach might be applied to our system, since Cu exhibits lower binding strength to CO as compared to Pt, and therefore, it is plausible that the blue shift of the Cu–CO vibration frequencies observed reflect a stronger Cu–CO bond at more negative potentials. At −0.5 VRHE, the spectral intensity of P1 is significantly stronger than that of P2, and as the potential shifts negatively, P2 becomes gradually stronger than P1. Figure 2c displays the potential dependence of the intensity ratio of P2 and P1, which exhibits a volcano-type profile increasing from P2/P1 values of 0.7 to 1.4 as the potential decreases from −0.6 to −0.9 VRHE and reaching a maximum at −1.0 VRHE and then decreasing quickly (Figures S20 and S21).
Most importantly, the intensity ratio of these two peaks as a function of the applied potential follows a similar trend as the CO2RR F.E. of the C2+ products. Through carefully reviewing and reanalyzing the Raman data in previous literature, we found that higher P2/P1 Raman peak ratios were associated with higher F.E.s for C2+ products in a variety of different catalysts, for example, CuAg nanowires versus Cu nanowires.22 Previous studies conducted with synchrotron radiation in the far-infrared range suggested that the intensities of these two peaks could be related to the surface coverage of CO on Cu in vacuum, although the Fano-like infrared peak made it difficult to precisely extract the ratio.41 Thus, a reasonable assumption is that our data reflect the CO coverage at the solid–liquid interface during the CO2RR process. Such a ratio may also reveal information on the electrochemical double layer, including the local pH, coadsorption, or solution environment.
To gain further insight into the evolution of the P2/P1 Raman peak ratio, we performed operando measurements in the presence of CO as well as DFT vibrational analysis for different CO coverages on the Cu(100) surface (Figure 3. First, we determined the optimal electrode potential to follow the CO adsorption on Cu in the CO-saturated KHCO3 electrolyte. We identified a potential window between about −0.32 VRHE and −0.62 VRHE in which adsorbed CO can be detected and the P2/P1 peak ratio also increases with decreasing potential (Figure S22). The CO bands disappeared at −0.72 VRHE, indicating the electrochemical conversion of CO. We also verified the stability of CO at different potentials in time-dependent CORR Raman data, with the P2/P1 intensity peak ratio decreasing due to the consumption of CO at −0.72 VRHE and below (Figure S23). To minimize the conversion of CO, we carried out operando Raman measurements at −0.52 VRHE in electrolytes with different CO concentrations (Figures S24 and S25). The volume fraction of CO-saturated KHCO3 in the electrolyte is used to describe the CO concentration. In principle, the CO surface coverage can be adjusted by controlling the CO concentration in the electrolyte, and their relationship is usually described by the Langmuir’s equation. As shown in Figure 3a, the P2/P1 intensity ratio increases with increasing CO concentration, following Langmuir equation (Figure 3b). Similar results were observed in the CO-containing NaClO4 solution (Figures S26–S30). These data demonstrate that the P2/P1 Raman peak ratio is a valid measure of the surface coverage of CO.
Figure 3.
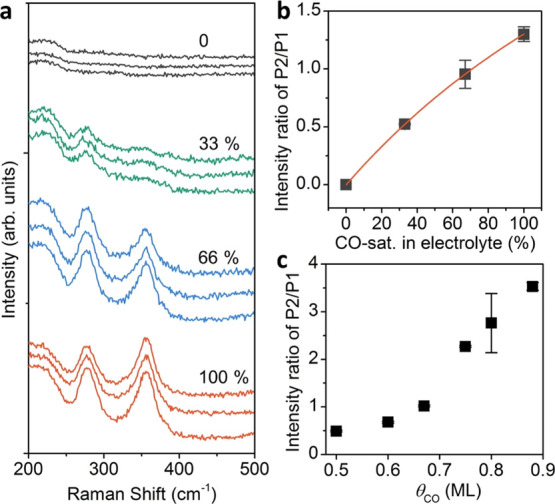
Raman spectra of adsorbed CO on Cu with different CO concentrations in the 0.1 M KHCO3 electrolyte. (a) Operando Raman spectra of Cu2O nanocubes in 0.1 M KHCO3 electrolyte with different CO concentrations at −0.52 VRHE. We mixed the CO-saturated KHCO3 with the Ar-saturated KHCO3 to prepare the CO-rich KHCO3 with different CO concentrations. The percentage represents the volume fraction of CO-saturated KHCO3 in the electrolyte from 0 to 100%. The experiments with different CO concentrations are repeated three times. (b) Intensity ratio of the P2/P1 Raman peaks as a function of the CO concentration. The red line shows the fitting result based on a Langmuir equation. (c) Theoretical benchmark of the P2/P1 intensity ratio vs CO surface coverage, θCO (Tables S4–S6).
We then performed DFT simulations of CO vibrational frequencies on Cu(100) for different CO surface coverages and adsorption configurations (0.11–0.88 ML) to link the experimental Raman spectral features to the relevant CO surface coverage (see Computational Details in the Supporting Information). Cu(100) terraces have been reported on OD-Cu under reduction conditions,6 and this facet is suggested to be the most active toward ethylene production.42 At relatively high surface coverages, θCO ≥ 0.6 ML, CO adsorbs in a mix of atop and bridge configurations,16,43 an experimental observation which is correctly reproduced by our DFT results (Table S2). To prove the robustness of our computational setup, we benchmarked the CO binding energy as a function of Hubbard’s Ueff,44 Cu slab thicknesses, and applied electric field (Figures S31 and S32, Tables S3). A four-layer Cu(100) model without Hubbard correction on C(O) 2p orbitals provided excellent agreement with the experimental results for CO binding energy for coverages expected under CO2 reduction conditions, θCO ≥ 0.5 ML (Table S2).43,44
Figure 3c shows the calculated P2/P1 peak ratio as a function of the CO coverage for θCO ≥ 0.5 ML (Tables S4–S6) which we extracted from DFT-derived Raman spectra (Figure S33). A clear increase of this ratio is observed with increasing CO coverage, and the values obtained theoretically are comparable with those experimentally determined. The increase of the P2/P1 ratio is correlated with a larger population of weakly bound COatop at high surface coverage, for which C=O rotation (P1) is increasingly reduced. Furthermore, by comparing the theoretical (Figure 3c) and experimental data (Figure 2c), we can associate the intensity ratio detected experimentally under CO2RR conditions at −0.6 and −1.0 VRHE (P2/P1 = 0.8 and 2.1) with a DFT CO surface coverage of 0.60 to 0.75 monolayer (ML), respectively (Table S6). Thus, we confirmed that the intensity ratio of the frustrated Cu–CO rotation and the Cu–CO stretching is not only a valid measure of the CO coverage but also linked to a predominant CO binding motif to the Cu surface.
To reveal the catalytic role of the CO binding configuration in the formation of C2+ products, we calculated C–C coupling on Cu(100) from CO precursor pairs bound in atop and bridge configuration (see Computational Details in the Supporting Information). Our theoretical and experimental spectroscopic results show a direct correlation between high surface coverage of weakly bound COatop at the catalyst surface (0.75 ML, determined via DFT, Figure 3c) and C2+ production (detected experimentally, Figure 1f). In the literature, COatop hydrogenation was reported more favorable than the reduction of bridge-bound CO,16 and a lower activation barrier for CO–CO dimerization was calculated for a COatop–CObridge precursor rather than CObridge–CObridge.4 C–C activation barrier is a key descriptor for assessing C2+ selectivity since CO–CO coupling to form the OCCO– dimer is considered the rate-determining step for CO2 electroreduction to C2+ products.46 Thus, we assessed the thermodynamics and kinetics of this process from nine different coupling configurations at θ*CO = 0.11 ML (Figures 4a and S34) to rationalize the facile COatop reduction to C2+ at −1.05 VRHE (Figure 1f). In general, our calculations show that the formation of the OCCO– dimer from COatop–COatop(CObridge) precursors (red and blue data points in Figure 4b) has an activation barrier around 0.9–1.0 eV, ∼0.4 eV lower than its formation from the lower-coverage CObridge–CObridge precursor (1.4 eV, black data points in Figure 4b). This means that C2+ products are more easily formed if at least one COatop participates in the critical CO–CO dimerization step.
Figure 4.

C–C coupling geometries depending on the CO adsorption configuration. (a) Adsorption geometry for different COatop–COatop (left), CObridge–COatop (center), and CObridge–CObridge (right) precursors. (b) Activation barriers Ea for C–C coupling from different COatop–COatop (red), CObridge–COatop (blue), and CObridge–CObridge (black) precursors vs rebonding energy, Erebond = E*CO(1)-ts + E*CO(2)-ts (eqs 1 and 2).45E*CO(1)-ts (E*CO(2)) is the adsorption energy of one CO molecule in the CO–CO transition state (ts) geometry once the other CO is excluded. The number labels relate each data point in panel (b) to the configuration of its initial state in panel (a).
Since standard Bronsted–Evans–Polanyi-type linear scaling relationships between the Gibbs free energy47 and the activation energy of C–C coupling cannot account for this 0.4 eV difference between atop and bridge adsorption configurations, we employed the Hammer’s decomposition scheme for the activation energies.45,48 Hammer’s formalism states that the activation energy for dissociation (coupling) processes correlates with Erebond, the rebonding energy required to bind products (dissociation) or rebind precursors (coupling) from the transition-state configuration. This correlation involves as well a geometric offset, Eint, which describes the interaction (repulsion) between precursors. Thus, the activation barriers for C–C coupling from the nine different COx–COy initial configurations scale with the energy to rebind the CO precursors (Figure 4b, eq 1), here calculated as adsorption energies of each CO molecule in the absence of the coupling partner (eq 2).
| 1 |
| 2 |
Adsorption configurations which account for less endothermic rebonding energies (Erebond ∼ +0.5 eV, Figure 4b), such as bridge–bridge, can rebind CO molecules from the transition state, thus impeding a successful C–C coupling. Instead, atop–atop(bridge) configurations show poor interaction with the transition state (very endothermic Erebond ∼ +1.0 eV, Figure 4b), which hinders CO–CO dissociation and enables the formation of the final state, *OCCO–. The promoting effect of atop–atop(bridge) adsorption configurations is further confirmed by the structural and electronic features of the transition-state geometry. C–O distance, dC–O(2) = 1.26 Å, and OCCO Bader charge, qCO–CO = −0.9 |e–| are close to the characteristics of *OCCO– (dC–O(2) = 1.39 Å, qOCCO = −1.0 |e–|, Tables S7 and S8), thus suggesting a fast evolution toward the dimer. Finally, the interaction energy, Eint, accounts for a positive offset, 1.75 ± 0.13 eV, due to the repulsion between the CO fragments (Figure 4b).
As a final remark, both, experimental results and theoretical insights confirmed that CO coverage is the key in enabling C–C coupling on Cu and providing a molecular-level understanding of the change of the F.E. of C2+ products versus CO at different potentials. High CO coverage at an appropriate potential (−1.05 VRHE) implies large surface population of C–C selective COatop (Table S2) and reduces the occurrence of the competing hydrogen evolution reaction (minimum F.E. at about −1.0 VRHE). Instead, lower CO coverages at more positive or negative potentials affect CO2 reduction toward ethylene negatively since COatop can convert to more static configurations (e.g., CObridge),16,53 either inert for CO2 reduction,16 or precursors for methane formation, in agreement with our evidence of high CH4 F.E. at −1.15 VRHE (Figure S8).49
Furthermore, we applied operando Raman spectroscopy to track the temporal evolution of the surface CO coverage on Cu during the CO2RR. Figure 5 shows time-dependent Raman spectra with a resolution of 5 s acquired at different applied potentials, −0.6 VRHE, −0.8 VRHE, and −1.0 VRHE (Figure 5a,b,c respectively), corresponding to the potential of CO formation, the initial potential of CO conversion, and the optimal potential of C–C coupling during CO2RR, respectively. No significant surface adsorption of CO was detected in the first 5 s due to the reduction of Cu2O, and Figure 5d shows the data after 20 s (potentials were applied after 15 s). At −0.6 VRHE, CO generated by CO2RR adsorbs on the Cu surface and reaches a steady state within 150 s. The time-dependent curve mainly reflects the balance of the CO generation with CO desorption and adsorption. The surface concentration of CO increases rapidly due to the large number of accessible Cu active sites at the beginning of the reaction and then reaches equilibrium as the number of available active surface sites decreases and the adsorption and desorption are balanced. At −0.8 VRHE, the CO coverage increases faster than at −0.6 VRHE to reach a similar equilibrium of the peak ratio at about 0.8, which means faster CO formation rate. However, there is a second-wave increase of the CO surface coverage after ∼180 s. The most reasonable explanation is the existence of another adsorption site or configuration of CO on the electrode surface. With the saturation of the first type of adsorption site, CO gradually adsorbs on another site with lower adsorption free energy. DFT simulations confirmed these experimental results (Table S2): for θCO ≥ 0.6 ML, COatop population increases and CO binding energy decreases to 0.2–0.3 eV, fingerprint of weakly bound COatop, with the C=O rotation band (P1) less intense (Table S4). Thus, the time-dependent curve of the peak ratio shows a process of equilibrium in two phases. At a more negative potential (−1.0 VRHE), the optimal potential for C–C coupling during CO2RR, the CO coverage increases more quickly and reaches a higher equilibrium coverage, which determines a higher P2/P1 ratio. Although the complex CO2RR mechanism and the electrode structure made it difficult to give a kinetics equation, we provide an effective strategy to investigate the dynamics of CO on the Cu surface during the CO2RR.
Figure 5.

Time-dependent operando Raman spectra acquired on Cu2O nanocubes at different potentials during CO2RR. (a) −0.6 VRHE. (b) −0.8 VRHE. (c) −1.0 VRHE. (d) Time dependence of the peak ratio at different applied potentials vs RHE.
Conclusions
In conclusion, operando Raman spectroscopy and DFT modeling were used to reveal the change of the electrode structure and the composition and dynamics of the surface intermediates during CO2RR on Cu2O nanocubes. During the CO2RR, a Raman band at about 360 cm–1 appeared between +0.3 and 0.0 VRHE, which was assigned to surface copper carbonate species formed from the KHCO3 electrolyte. We also revealed that the ratio of the Cu–CO stretching (P2) and Cu–CO rotation (P1) bands changes with the applied potential and is strongly related to the CO coverage, allowing us to track the dynamics of the CO surface coverage on Cu during CO2RR. Interestingly, a clear correlation exists between the P2/P1 ratio and the F.E. of the C2+ products on the applied potential. Our experiments and theoretical insights allowed us to conclude that the degree and ease of the C–C coupling is determined by the CO surface coverage, which in turn influences the preferred CO adsorption configuration. At a high surface coverage, CO adsorbs in a mix of atop and bridge sites, and both COatop–COatop and COatop–CObridge couplings are thermodynamically and kinetically more favorable than CObridge–CObridge due to both electronic and structural effects. Weakly bound atop configurations show no interaction with CO in the transition state, thus limiting CO–CO dissociation and boosting the evolution toward the final state OCCO–. Overall, we were able to provide molecular-level insight into the correlation between the CO coverage and the potential-dependent C2+ F.E. Finally, it was illustrated that operando Raman is an effective method to investigate the electrochemical solid–liquid interface and the interaction of surface intermediates with the electrode during an electrocatalytic reaction.
Acknowledgments
C.Z. thanks the Alexander von Humboldt Foundation (AvH) for supporting him with an AvH postdoctoral research grant. This work was also partially funded by the European Research Council (ERC-725915, OPERANDOCAT) as well as the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—project no. 406944504—SPP 2080 and Germany’s Excellence Strategy—EXC 2008—390540038—UniSysCat. F.D., R.G.-M., and N.L. thank the financial support from the Spanish Ministry of Science and Innovation (RTI2018-101394-B-I00). We further acknowledge the support by the IMPRS Functional Interfaces Physics and Chemistry and BSC-RES for providing generous computational resources.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c01478.
Datasets generated during the current study are available in the ioChem-BD database50 at DOI: 10.19061/iochem-bd-1-192. Synthesis of Cu2O cubes and electrodes, ex situ and quasi-in situ characterization of samples, operando Raman experiments, CO2RR testing, and computational details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- De Luna P.; Hahn C.; Higgins D.; Jaffer S. A.; Jaramillo T. F.; Sargent E. H. What Would It Take for Renewably Powered Electrosynthesis to Displace Petrochemical Processes?. Science 2019, 364, eaav3506 10.1126/science.aav3506. [DOI] [PubMed] [Google Scholar]
- Gao D.; Arán-Ais R. M.; Jeon H. S.; Roldan Cuenya B. Rational Catalyst and Electrolyte Design for CO2 Electroreduction towards Multicarbon Products. Nat. Catal. 2019, 2, 198–210. 10.1038/s41929-019-0235-5. [DOI] [Google Scholar]
- Wang X.; de Araújo J. F.; Bagger A.; Schmies H.; Kühl S.; Rossmeisl J.; Strasser P.; Strasser P. Mechanistic Reaction Pathways of Enhanced Ethylene Yields during Electroreduction of CO2–CO Co-Feeds on Cu and Cu-Tandem Electrocatalysts. Nat. Nanotechnol. 2019, 14, 1063–1070. 10.1038/s41565-019-0551-6. [DOI] [PubMed] [Google Scholar]
- Li F.; Thevenon A.; Rosas-Hernández A.; Wang Z.; Li Y.; Gabardo C. M.; Ozden A.; Dinh C. T.; Li J.; Wang Y.; Edwards J. P.; Xu Y.; McCallum C.; Tao L.; Liang Z.-Q.; Luo M.; Wang X.; Li H.; O’Brien C. P.; Tan C.-S.; Nam D.-H.; Quintero-Bermudez R.; Zhuang T.-T.; Li Y. C.; Han Z.; Britt R. D.; Sinton D.; Agapie T.; Peters J. C.; Sargent E. H. Molecular Tuning of CO2-to-Ethylene Conversion. Nature 2020, 577, 509–513. 10.1038/s41586-019-1782-2. [DOI] [PubMed] [Google Scholar]
- Ma W.; Xie S.; Liu T.; Fan Q.; Ye J.; Sun F.; Jiang Z.; Zhang Q.; Cheng J.; Wang Y. Electrocatalytic Reduction of CO2 to Ethylene and Ethanol through Hydrogen-Assisted C–C Coupling over Fluorine-Modified Copper. Nat. Catal. 2020, 3, 478–487. 10.1038/s41929-020-0450-0. [DOI] [Google Scholar]
- Arán-Ais R. M.; Scholten F.; Kunze S.; Rizo R.; Roldan Cuenya B. The Role of in Situ Generated Morphological Motifs and Cu(I) Species in C2+ Product Selectivity during CO2 Pulsed Electroreduction. Nat. Energy 2020, 5, 317–325. 10.1038/s41560-020-0594-9. [DOI] [Google Scholar]
- Hori Y.; Murata A.; Takahashi R. Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at a Copper Electrode in Aqueous Solution. J. Chem. Soc., Faraday Trans. 1989, 85, 2309–2326. 10.1039/f19898502309. [DOI] [Google Scholar]
- Li C. W.; Ciston J.; Kanan M. W. Electroreduction of Carbon Monoxide to Liquid Fuel on Oxide-Derived Nanocrystalline Copper. Nature 2014, 508, 504. 10.1038/nature13249. [DOI] [PubMed] [Google Scholar]
- Nitopi S.; Bertheussen E.; Scott S. B.; Liu X.; Engstfeld A. K.; Horch S.; Seger B.; Stephens I. E. L.; Chan K.; Hahn C.; Nørskov J. K.; Jaramillo T. F.; Chorkendorff I. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. 10.1021/acs.chemrev.8b00705. [DOI] [PubMed] [Google Scholar]
- Kim D.; Kley C. S.; Li Y.; Yang P. Copper Nanoparticle Ensembles for Selective Electroreduction of CO2 to C2–C3 Products. Proc. Nat. Acad. Sci. 2017, 114, 10560–10565. 10.1073/pnas.1711493114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh C.-T.; Burdyny T.; Kibria M. G.; Seifitokaldani A.; Gabardo C. M.; García de Arquer F. P.; Kiani A.; Edwards J. P.; De Luna P.; Bushuyev O. S.; Zou C.; Quintero-Bermudez R.; Pang Y.; Sinton D.; Sargent E. H. CO2 Electroreduction to Ethylene via Hydroxide-Mediated Copper Catalysis at an Abrupt Interface. Science 2018, 360, 783–787. 10.1126/science.aas9100. [DOI] [PubMed] [Google Scholar]
- Dattila F.; García-Muelas R.; López N. Active and Selective Ensembles in Oxide-Derived Copper Catalysts for CO2 Reduction. ACS Energy Lett. 2020, 5, 3176–3184. 10.1021/acsenergylett.0c01777. [DOI] [Google Scholar]
- Montoya J. H.; Shi C.; Chan K.; Nørskov J. K. Theoretical Insights into a CO Dimerization Mechanism in CO2 Electroreduction. J. Phys. Chem. Lett. 2015, 6, 2032–2037. 10.1021/acs.jpclett.5b00722. [DOI] [PubMed] [Google Scholar]
- Kortlever R.; Shen J.; Schouten K. J. P.; Calle-Vallejo F.; Koper M. T. M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. 10.1021/acs.jpclett.5b01559. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Handoko A. D.; Hirunsit P.; Yeo B. S. Electrochemical Reduction of CO2 Using Copper Single-Crystal Surfaces: Effects of CO* Coverage on the Selective Formation of Ethylene. ACS Catal. 2017, 7, 1749–1756. 10.1021/acscatal.6b03147. [DOI] [Google Scholar]
- Grosse P.; Gao D.; Scholten F.; Sinev I.; Mistry H. Dynamic Changes in the Structure, Chemical State and Catalytic Selectivity of Cu Nanocubes during CO2 Electroreduction: Size and Support Effects. Angew. Chem., Int. Ed. 2018, 57, 6192–6197. 10.1002/anie.201802083. [DOI] [PubMed] [Google Scholar]
- Gunathunge C. M.; Ovalle V. J.; Li Y.; Janik M. J.; Waegele M. M. Existence of an Electrochemically Inert CO Population on Cu Electrodes in Alkaline pH. ACS Catal. 2018, 8, 7507–7516. 10.1021/acscatal.8b01552. [DOI] [Google Scholar]
- Li J.; Wang Z.; McCallum C.; Xu Y.; Li F.; Wang Y.; Gabardo C. M.; Dinh C.-T.; Zhuang T.-T.; Wang L.; Howe J. Y.; Ren Y.; Sargent E. H.; Sinton D. Constraining CO Coverage on Copper Promotes High-efficiency Ethylene Electroproduction. Nat. Catal. 2019, 2, 1124–1131. 10.1038/s41929-019-0380-x. [DOI] [Google Scholar]
- Sandberg R. B.; Montoya J. H.; Chan K.; Nørskov J. K. CO-CO Coupling on Cu facets: Coverage, Strain and Field Effects. Surf. Sci. 2016, 654, 56–62. 10.1016/j.susc.2016.08.006. [DOI] [Google Scholar]
- Handoko A. D.; Wei F.; Jenndy B. S.; Yeo B. S.; Seh Z. W. Understanding Heterogeneous Electrocatalytic Carbon Dioxide Reduction through Operando Techniques. Nat. Catal. 2018, 1, 922–934. 10.1038/s41929-018-0182-6. [DOI] [Google Scholar]
- Tian Z.-Q.; Ren B. Adsorption and Reaction at Electrochemical Interfaces as Probed by Surface-Enhanced Raman Spectroscopy. Annu. Rev. Phys. Chem. 2004, 55, 197–229. 10.1146/annurev.physchem.54.011002.103833. [DOI] [PubMed] [Google Scholar]
- Dong J.-C.; Zhang X.-G.; Briega-Martos V.; Jin X.; Yang J.; Chen S.; Yang Z.-L.; Wu D.-Y.; Feliu J. M.; Williams C. T.; Tian Z.-Q.; Li J.-F. In Situ Raman Spectroscopic Evidence for Oxygen Reduction Reaction Intermediates at Platinum Single-Crystal Surfaces. Nat. Energy 2019, 4, 60–67. 10.1038/s41560-018-0292-z. [DOI] [Google Scholar]
- Gao J.; Zhang H.; Guo X.; Luo J.; Zakeeruddin S. M.; Ren D.; Grätzel M. Selective C–C Coupling in Carbon Dioxide Electroreduction via Efficient Spillover of Intermediates As Supported by Operando Raman Spectroscopy. J. Am. Chem. Soc. 2019, 141, 18704–18714. 10.1021/jacs.9b07415. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Chang X.; Malkani A. S.; Yang X.; Thompson L.; Jiao F.; Xu B. Speciation of Cu Surfaces during the Electrochemical CO Reduction Reaction. J. Am. Chem. Soc. 2020, 142, 9735–9743. 10.1021/jacs.0c02354. [DOI] [PubMed] [Google Scholar]
- Chernyshova I. V.; Somasundaran P.; Ponnurangam S. On the Origin of the Elusive First Intermediate of CO2 Electroreduction. Proc. Nat. Acad. Sci. 2018, 115, E9261–E9270. 10.1073/pnas.1802256115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Wang Z.; García de Arquer F. P.; Dinh C.-T.; Ozden A.; Li Y. C.; Nam D.-H.; Li J.; Liu Y.-S.; Wicks J.; Chen Z.; Chi M.; Chen B.; Wang Y.; Tam J.; Howe J. Y.; Proppe A.; Todorović P.; Li F.; Zhuang T.-T.; Gabardo C. M.; Kirmani A. R.; McCallum C.; Hung S.-F.; Lum Y.; Luo M.; Min Y.; Xu A.; O’Brien C. P.; Stephen B.; Sun B.; Ip A. H.; Richter L. J.; Kelley S. O.; Sinton D.; Sargent E. H. Efficient Electrically Powered CO2-to-Ethanol via Suppression of Deoxygenation. Nat. Energy 2020, 5, 478–486. 10.1038/s41560-020-0607-8. [DOI] [Google Scholar]
- Gao D.; Zegkinoglou I.; Divins N. J.; Scholten F.; Sinev I.; Grosse P.; Roldan Cuenya B. Plasma-Activated Copper Nanocube Catalysts for Efficient Carbon Dioxide Electroreduction to Hydrocarbons and Alcohols. ACS Nano 2017, 11, 4825–4831. 10.1021/acsnano.7b01257. [DOI] [PubMed] [Google Scholar]
- Peterson A. A.; Abild-Pedersen F.; Studt F.; Rossmeisl J.; Nørskov J. K. How Copper Catalyzes the Electroreduction of Carbon Dioxide into Hydrocarbon Fuels. Energy Environ. Sci. 2010, 3, 1311–1315. 10.1039/c0ee00071j. [DOI] [Google Scholar]
- Mao Y.; He J.; Sun X.; Li W.; Lu X.; Gan J.; Liu Z.; Gong L.; Chen J.; Liu P.; Tong Y. Electrochemical Synthesis of Hierarchical Cu2O Stars with Enhanced Photoelectrochemical Properties. Electrochim. Acta 2012, 62, 1–7. 10.1016/j.electacta.2011.10.106. [DOI] [Google Scholar]
- Ren D.; Ang B. S.-H.; Yeo B. S. Tuning the Selectivity of Carbon Dioxide Electroreduction toward Ethanol on Oxide-Derived CuxZn catalysts. ACS Catal. 2016, 6, 8239–8247. 10.1021/acscatal.6b02162. [DOI] [Google Scholar]
- Jiang S.; Klingan K.; Pasquini C.; Dau H. New Aspects of Operando Raman Spectroscopy Applied to Electrochemical CO2 Reduction on Cu Foams. J. Chem. Phys. 2019, 150, 041718. 10.1063/1.5054109. [DOI] [PubMed] [Google Scholar]
- He M.; Li C.; Zhang H.; Chang X.; Chen J. G.; Goddard W. A. Oxygen Induced Promotion of Electrochemical Reduction of CO2 via Co-Electrolysis. Nat. Commun. 2020, 11, 3844. 10.1038/s41467-020-17690-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodappa N.; Su M.; Zhao Y.; Le J.-B.; Yang W.-M.; Radjenovic P.; Dong J.-C.; Cheng J.; Tian Z.-Q.; Li J.-F. Early Stages of Electrochemical Oxidation of Cu(111) and Polycrystalline Cu Surfaces Revealed by in Situ Raman Spectroscopy. J. Am. Chem. Soc. 2019, 141, 12192–12196. 10.1021/jacs.9b04638. [DOI] [PubMed] [Google Scholar]
- Moradzaman M.; Mul G. In Situ Raman Study of Potential-Dependent Surface Adsorbed Carbonate, CO, OH, and C Species on Cu Electrodes during Electrochemical Reduction of CO2. ChemElectroChem 2021, 8, 1478–1485. 10.1002/celc.202001598. [DOI] [Google Scholar]
- An H.; Wu L.; Mandemaker L. D. B.; de Ruiter J.; Wijten J. H.; Janssens J.; van der Stam W.; Weckhuysen B. M.. Sub-Second Time-Resolved Surface Enhanced Raman Spectroscopy Reveals Dynamic CO Intermediates during Electrochemical CO2 Reduction on Copper. Angew. Chem., Int. Ed. 2021, 10.1002/anie.202104114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-Vélez J.-J.; Chuang C.-H.; Gao D.; Zhu Q.; Ivanov D.; Jeon H. S.; Arrigo R.; Mom R. V.; Stotz E.; Wu H.-L.; Jones T. E.; Roldan Cuenya B.; Knop-Gericke A.; Schlögl R. On the Activity/Selectivity and Phase Stability of Thermally Grown Copper Oxides during the Electrocatalytic Reduction of CO2. ACS Catal. 2020, 10, 11510–11518. 10.1021/acscatal.0c03484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost R. L.; Martens W. N.; Rintoul L.; Mahmutagic E.; Kloprogge J. T. Raman Spectroscopic Study of Azurite and Malachite at 298 and 77 K. J. Raman Spectrosc. 2002, 33, 252–259. 10.1002/jrs.848. [DOI] [Google Scholar]
- Heyes J.; Dunwell M.; Xu B. CO2 Reduction on Cu at Low Overpotentials with Surface-Enhanced in Situ Spectroscopy. J. Phys. Chem. C 2016, 120, 17334–17341. 10.1021/acs.jpcc.6b03065. [DOI] [Google Scholar]
- Smith B. D.; Irish D. E.; Kedzierzawski P.; Augustynski J. A Surface Enhanced Roman Scattering Study of the Intermediate and Poisoning Species Formed during the Electrochemical Reduction of CO2 on Copper. J. Electrochem. Soc. 1997, 144, 4288–4296. 10.1149/1.1838180. [DOI] [Google Scholar]
- Gunathunge C. M.; Li X.; Li J.; Hicks R. P.; Ovalle V. J.; Waegele M. M. Spectroscopic Observation of Reversible Surface Reconstruction of Copper Electrodes under CO2 Reduction. J. Phys. Chem. C 2017, 121, 12337–12344. 10.1021/acs.jpcc.7b03910. [DOI] [Google Scholar]
- Akemann W.; Otto A. The Effect of Atomic Scale Surface Disorder on Bonding and Activation of Adsorbates: Vibrational Properties of CO and CO2 on Copper. Surf. Sci. 1993, 287-288, 104–109. 10.1016/0039-6028(93)90751-5. [DOI] [Google Scholar]
- Gunathunge C. M.; Li J.; Li X.; Hong J. J.; Waegele M. M. Revealing the Predominant Surface Facets of Rough Cu Electrodes under Electrochemical Conditions. ACS Catal. 2020, 10, 6908–6923. 10.1021/acscatal.9b05532. [DOI] [Google Scholar]
- Zhang P.; Cai J.; Chen Y.-X.; Tang Z.-Q.; Chen D.; Yang J.; Wu D.-Y.; Ren B.; Tian Z.-Q. Potential-Dependent Chemisorption of Carbon Monoxide at a Gold Core–Platinum Shell Nanoparticle Electrode: A Combined Study by Electrochemical in Situ Surface-Enhanced Raman Spectroscopy and Density Functional Theory. J. Phys. Chem. C 2010, 114, 403–411. 10.1021/jp908478m. [DOI] [Google Scholar]
- Hirschmugl C. J.; Williams G. P.; Hoffmann F. M.; Chabal Y. J. Adsorbate-Substrate Resonant Interactions Observed for CO on Cu (100) in the Far Infrared. Phys. Rev. Lett. 1990, 65, 480. 10.1103/physrevlett.65.480. [DOI] [PubMed] [Google Scholar]
- Bagger A.; Ju W.; Varela A. S.; Strasser P.; Rossmeisl J. Electrochemical CO2 Reduction: Classifying Cu Facets. ACS Catal. 2019, 9, 7894–7899. 10.1021/acscatal.9b01899. [DOI] [Google Scholar]
- Vollmer S.; Witte G.; Wöll C. Determination of Site Specific Adsorption Energies of CO on Copper. Catal. Lett. 2001, 77, 97–101. 10.1023/a:1012755616064. [DOI] [Google Scholar]
- Gajdoš M.; Hafner J. CO Adsorption on Cu(111) and Cu(001) Surfaces: Improving Site Preference in DFT Calculations. Surf. Sci. 2005, 590, 117–126. 10.1016/j.susc.2005.04.047. [DOI] [Google Scholar]
- Hammer B. Bond Activation at Monatomic Steps: NO Dissociation at Corrugated Ru(0001). Phys. Rev. Lett. 1999, 83, 3681. 10.1103/physrevlett.83.3681. [DOI] [Google Scholar]
- Birdja Y. Y.; Pérez-Gallent E.; Figueiredo M. C.; Göttle A. J.; Calle-Vallejo F.; Koper M. T. M. Advances and Challenges in Understanding the Electrocatalytic Conversion of Carbon Dioxide to Fuels. Nat. Energy 2019, 4, 732–745. 10.1038/s41560-019-0450-y. [DOI] [Google Scholar]
- Pérez-Ramírez J.; López N. Strategies to Break Linear-Scaling Relationships. Nat. Catal. 2019, 2, 971–976. 10.1038/s41929-019-0376-6. [DOI] [Google Scholar]
- Liu Z.-P.; Hu P. General Rules for Predicting Where a Catalytic Reaction Should Occur on Metal Surfaces: A Density Functional Theory Study of C–H and C–O Bond Breaking/Making on Flat, Stepped, and Kinked Metal Surfaces. J. Am. Chem. Soc. 2003, 125, 1958–1967. 10.1021/ja0207551. [DOI] [PubMed] [Google Scholar]
- Bai H.; Cheng T.; Li S.; Zhou Z.; Yang H.; Li J.; Xie M.; Ye J.; Ji Y.; Li Y. Controllable CO Adsorption Determines Ethylene and Methane Productions from CO2 Electroreduction. Sci. Bull. 2021, 66, 62. 10.1016/j.scib.2020.06.023. [DOI] [PubMed] [Google Scholar]
- Álvarez-Moreno M.; de Graaf C.; López N.; Maseras F.; Poblet J. M.; Bo C. Managing the Computational Chemistry Big Data Problem: The IoChem-BD Platform. J. Chem. Inf. Model. 2015, 55, 95–103. 10.1021/ci500593j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.