
Fig. 3.
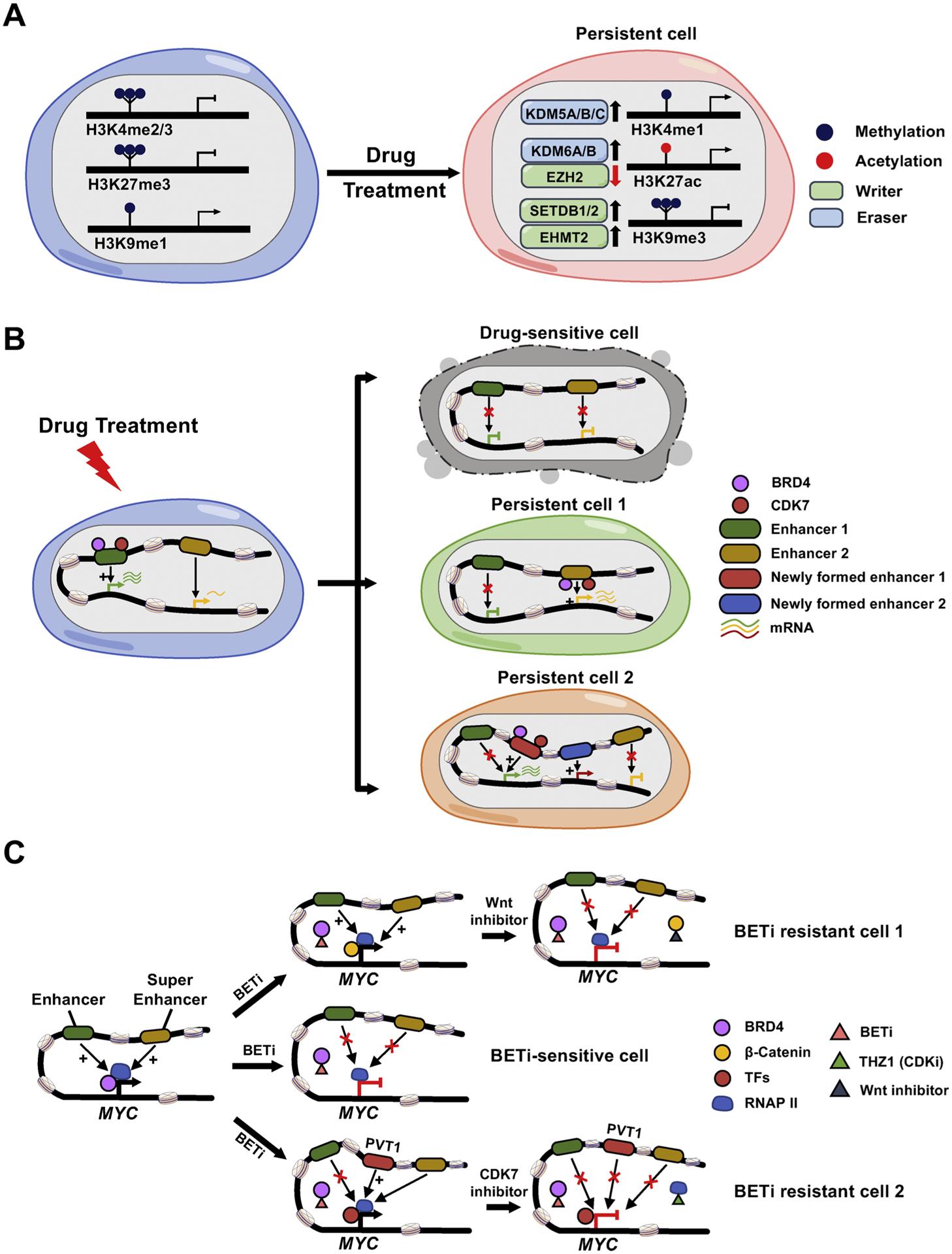
Histone modification alterations and chromatin remodeling during drug resistance development.
A. Drug treatment induces altered expression of histone modification modulators (writers and erasers), resulting in histone marks changes and the corresponding adaptive transcriptome reprogramming. B. Drug-sensitive cells are eliminated after drug treatment. Upon drug exposure, drug-sensitive cancer cells undergo cell death (top panel). However, a rare population of persistent cells withstand the initial onslaught of drug exposure, either through compensatory expression of pro-survival genes that are stimulated by drug-induced activation of previously poised enhancers (persistent cell 1; middle panel), or through enhancer switching to turn on the genes whose expressions are blocked by drug-suppressed original enhancers (persistent cell 2; bottom panel). Epigenetic regulators, such as BRD4, CDK7 and other unidentified chromatin modifying proteins, can participate in this process to mediate chromatin landscape remodeling and transcriptional reprogramming /adaptation. C. A tentative model summarizing the mechanisms of BET inhibitor (BETi) resistance in AML. MYC expression is regulated by the classic enhancer or super-enhancer in BETi-sensitive leukemia cells, which is mediated by BRD4 binding. BETi blocks the BRD4 binding to its genomic targets and subsequently inhibits the expression of MYC and suppresses cell growth (middle panel). Long-term drug treatment may restore MYC expression by distinct mechanisms: (i) activated Wnt/ β-catenin signaling pathways lead to increased binding of β-catenin to the sites that are originally occupied by BRD4 to sustain MYC expression. Targeting Wnt signaling reduces chromatin-bound β-catenin and subverts its ability to maintain the expression of MYC, resulting in the restoration of sensitivity to BETi (upper panel); (ii) PVT1 acts as a de novo BRD4 binding-independent enhancer, which can recruit other transcription factors (TFs) and RNA Polymerase II (RNAP II) to the MYC promoter and initiate MYC expression. Treatment with a CDK7 inhibitor, THZ1, reduces the Pol II occupancy to suppress the re-activated MYC transcription (lower panel).