

Abstract
Individuals mosaic for monosomy X and a cell line with Y chromosome material can have genitalia that appear phenotypical female, male or ambiguous. Those with this karyotype and typical female genitalia are diagnosed with Turner syndrome, however this definition specifically excludes those with genitalia other than typical female. There is limited information on whether medical and neurodevelopmental risks are similar among individuals with monosomy X and Y chromosome material across genital phenotypes. This multicenter retrospective study compared comorbidities and clinical management in individuals with monosomy X and Y material and male/ambiguous genitalia to those with typical female genitalia. Electronic medical records for all patients with monosomy X and Y material (n=76) at two large U.S. pediatric centers were abstracted for predetermined data and outcomes. Logistic regression was used to compare the two phenotypic groups adjusting for site and duration of follow up. The male/ambiguous genitalia group was just as likely to have congenital heart disease (RR 1.0, 95%CI [0.5–1.9]), autoimmune disease (RR 0.6 [0.2–1.3]), and neurodevelopmental disorders (RR 1.4 [0.8–1.2]) as those with female genitalia. Despite similar risks, they were less likely to receive screening and counseling. In conclusion, individuals with monosomy X and Y chromosome material have similar medical and neurodevelopmental risks relative to individuals with Turner syndrome regardless of genitalia, but there are notable differences in clinical management.
Keywords: 45,X/46,XY; Turner Syndrome; Mosaicism; monosomy X; mixed gonadal dysgenesis
INTRODUCTION
Sex chromosome mosaicism is characterized by the expression of cell lines with a different number of X and Y chromosomes within an individual (Telvi, Lebbar, Del Pino, Barbet, & Chaussain, 1999), occurring at an incidence of 1.7 per 10,000 pregnancies (Chang, Clark, & Bachman, 1990; Hook & Warburton, 2014). Mosaicism with a monosomy X cell line (45,X) in addition to either a full or partial Y chromosome (isochromosome, deletions, translocations, etc.) results in a widely variable phenotype that is understudied. Even the terminology is inconsistent with no agreed upon term to describe these individuals. Monosomy X with Y chromosome material is associated with a range of genital phenotypes including typical male, female, and ambiguous genitalia, which makes prenatal counseling, in particular, challenging (Chang et al., 1990; Farrugia et al., 2013; Huang et al., 2019). Clinically, individuals with this chromosome complement are often segregated into three distinct groups based primarily on their genitalia: typical female genitalia which is usually considered Turner syndrome; ambiguous genitalia resulting in a diagnosis of mixed gonadal dysgenesis; and typical male genitalia, which may be the most common but is the least well studied largely due to ascertainment bias (Akinsal, Baydilli, Bayramov, & Ekmekcioglu, 2018; Chang et al., 1990; Wu et al., 2017). Due to this somewhat artificial segregation, the care practices between these groups are, at least anecdotally, quite varied.
Individuals with Turner syndrome are at a higher risk for gonadal insufficiency, congenital heart and renal anomalies, acquired autoimmune conditions, poor linear growth that can be improved with growth hormone, neurocognitive deficits, and a variety of other medical and psychological conditions (Bondy, 2007). Due to the multisystem involvement associated with Turner syndrome, there are internationally endorsed Clinical Practice Guidelines (CPG) that provide detailed multidisciplinary screening and practice recommendations for individuals with Turner syndrome (Gravholt et al., 2017). However, per these CPG, the definition of Turner syndrome excludes all males and individuals with ambiguous genitalia who have a monosomy X cell line (Gravholt et al., 2017). While there are proposed recommendations for management of gender assignment and surgical intervention for individuals with monosomy X with male/ambiguous genitalia (Wu et al., 2017), there are no comprehensive clinical care guidelines for these individuals and the CPG for individuals with Turner syndrome are inconsistently applied.
Whether or not individuals with a monosomy X cell line presenting with ambiguous or normal male genitalia have the same medical and neurodevelopmental risks as females with Turner syndrome is not clear. Studies suggest that the risk of gonadal insufficiency in individuals presenting with a male phenotype is lower than individuals presenting with a female or ambiguous phenotype, however hypogonadism and infertility still affects up to 45% of males with a monosomy X cell line as well (Lindhardt Johansen et al., 2012; Martinerie et al., 2012; Toniolo & Rizzolio, 2007). There are numerous reports of poor linear growth, dysmorphic features commonly associated with Turner syndrome, congenital heart disease, and renal anomalies in individuals with male or ambiguous genitalia with a monosomy X cell line, although these are primarily case reports or descriptive studies without a comparison group making it difficult to quantify the relative risk (De Groote et al., 2013; Dumeige et al., 2018; Efthymiadou, Stefanou, & Chrysis, 2012). Less is known about the learning profiles, psychological risks, autoimmunity, or cardiometabolic disease in individuals with monosomy X presenting with male or ambiguous genitalia.
Due to the lack of evidence describing the medical and neurodevelopmental features associated with a chromosomal complement with monosomy X and Y chromosome material, it is difficult to provide counseling in the setting of a prenatal diagnosis or determine appropriate management for individuals who do not have typical female genitalia and therefore fall outside the current Turner syndrome CPG. Therefore, the objective of this multicenter observational study was to compare phenotypic features and care practices between individuals with monosomy X and Y chromosome material presenting with female genitalia (Turner syndrome) to individuals with the same genotype but with male or ambiguous genitalia. We hypothesized that the medical and neurodevelopmental risks would be similar in both groups due to the presence of a monosomy X cell line. However, individuals with male or ambiguous genitalia would be less likely to have received screenings for these risks due to the lack of CPG for this population.
METHODS
Editorial Policies and Ethical Considerations
This retrospective chart review study was conducted in accordance with the Declaration of Helsinki and approved with a waiver of consent by the local institutional review boards (COMIRB 16-1631 and IRB18-00437). De-identified data were combined for analysis.
Study Design and Participants
We conducted a retrospective study of electronic medical records (EMR) for individuals with monosomy X with Y chromosome material who were seen in outpatient clinics at Children’s Hospital Colorado or Nationwide Children’s Hospital between 2005–2019. The EMR at both institutions was queried for patients with a past medical history, problem list, and/or billing diagnosis that mapped to any of the following ICD10 codes: Q96.0 (45,X), Q96.1 (46,Xiso(Xq)), Q96.2 (45,X with abnormal sex chromosome other than isoXq), Q96.3 (mosaic 45,X/45,XX or XY), Q96.4 (45,X/other cell line(s) with abnormal sex chromosome), Q96.8 (other variants of Turner syndrome and mixed gonadal dysgenesis), Q96.9 (Turner syndrome unspecified), Q97.8 (other specified sex chromosome abnormalities, female phenotype), Q97.9 (sex chromosome abnormality, female phenotype, unspecified), Q98.6 (male with structurally abnormal sex chromosome), Q98.7 (male with sex chromosome mosaicism), Q98.8 (other specified sex chromosome abnormalities, male), Q98.9 (sex chromosome abnormalities, male, unspecified), Q99.1 (gonadal dysgenesis). Charts were individually reviewed for inclusion criteria of a karyotype (and microarray/FISH data) with a single normal X chromosome and a second sex chromosome with at least partial Y material (collectively referred to as “mX/Y”). Patients were excluded if they were not seen in an outpatient clinic capable of providing comprehensive care related to their genetic condition (i.e., genetics, endocrinology, cardiology, general pediatrics or adolescent medicine, developmental pediatrics, urology, or a multidisciplinary clinic dedicated to Turner syndrome or disorders of sex development) or had no evidence of Y material on genetic testing.
Outcomes
Data were collected and managed using REDCap (Research Electronic Data Capture) tools hosted at the University of Colorado Denver Anschutz Medical Campus (Harris et al., 2009). EMR abstraction included patient demographics, timing and indication for genetic testing, karyotype (45,X/46,XY and/or other Y variants and percentages), and presence or absence of the following medical and neurodevelopmental diagnoses: dysmorphic features associated with Turner syndrome (webbed neck, wide spaced nipples, increased carrying angle, high arched palate, posteriorly rotated ears), short stature as defined by 1) diagnosis due to short stature, 2) treatment with growth hormone, 3) last height z score < −2, or 4) final height < 10cm below mid-parental height or < 166 cm for boys, 153 cm for girls based on sex raised), gonadal function based on labs and treatment, and gonadoblastoma. Determination of predicted presence and/or number of copies of the sex-determining gene (SRY) and short-stature homeobox gene (SHOX) were determined with the CyDAS Online Package based on whether Yp11.2 and Yp11.3 were gained or lost (Hiller, Bradtke, Balz, & Rieder, 2004). Documented completion of specific clinical management recommendations outlined the Turner syndrome CPG including cardiac and renal imaging, screening for autoimmune and cardiometabolic conditions, and education/counseling on the topics of fertility implications, psychosocial comorbidities, learning differences, cardiometabolic risks/healthy lifestyle, and disclosure of the diagnosis to the patient was recorded. All abstracted data were verified and validated by a clinician (SMD) prior to analysis.
Statistical Analysis
Patients were stratified based on their phenotype into two groups based on the documented presentation of their genitalia: female or male/ambiguous. Means and standard deviations or median and interquartile ranges were provided for continuous variables and proportions reported for categorical variables. The denominator was adjusted to only include individuals for which the outcome was applicable based on age (e.g. excluded infants when comparing prevalence of learning disabilities) or completion of relevant testing (e.g. cardiac defects only assessed in those who had cardiac imaging). Binomial outcomes were compared between groups using logistical regression models that adjusted for site and duration of follow-up, with relative risk (RR) and 95% confidence intervals (CI) generated. Following this primary analysis, sensitivity analyses comparing individuals born with ambiguous versus typical male genitalia and those with prenatal vs postnatal diagnoses were conducted. Missing data were categorized as “unknown” for descriptive statistics and pairwise deletion was used for analyses. An alpha of 0.05 was considered statistically significant due to the exploratory nature of the study. Statistical analyses were performed in RStudio v 1.2.1335 and Prism GraphPad v8.4.3.
RESULTS
Demographic information for the 76 patients with mX/Y stratified by genital phenotype (female n=32; male/ambiguous n=44) are presented in Table 1 and Supplementary Table. All patients born with female genitalia were raised female; one patient subsequently identified as male later in life. All the patients with male genitalia at birth (n=15) were being raised male without evidence in the EMR of a different gender identity. Of the 29 patients with ambiguous genitalia, 86% were given a male gender assignment and raised male while the other four were raised female (three of whom had feminizing genitoplasty).
Table 1:
Cohort Demographics Stratified by Genital Phenotype
| Full cohort (n=76) |
Female Phenotype (n=32) | Male/Ambiguous Phenotype (n=44) | p-value | |
|---|---|---|---|---|
| Nationwide (Ohio) | 26 (34%) | 9 (28%) | 17 (39%) | |
| Multidisciplinary Care n (%) | 29 (38%) | 11 (34%) | 18 (41%) | 0.73 |
| Age at most recent visit (yrs) | 11.8 (6.0–15.5) | 13.0 (6.5–16.1) | 11.2 (5.7–14.2) | 0.29 |
| Duration of follow up (yrs) | 3.5 (0.8–9.8) | 1.9 (0.6–5.3) | 5.8 (0.8–11.0) | 0.018 |
| Not reported | 1 (1%) | 0 (0%) | 1 (2%) | |
| Not reported | 2 (3%) | 1 (3%) | 1 (2%) | |
| Gestational age (weeks) | 38.1 ± 2.2 | 37.9 ± 2.1 | 38.2 ± 2.3 | 0.61 |
| Birthweight (kg) | 3.0 ± 0.7 | 2.8 ± 0.8 | 3.1 ± 0.6 | 0.13 |
| Not available | 6 (8%) | 0 (0%) | 6 (14%) | |
| % 45,X Cells on Karyotype§ | 47 (20–70) | 50 (19–74) | 47 (24–66) | 0.52 |
| Other/Unknown | 3 (21%) | 0 (0%) | 3 (30%) | |
| Other/Unknown | 2 (3%) | 0 (0%) | 2 (6%) |
Continuous data are represented as mean ± SD or median (25th-75th); categorical variables are represented as n (%). P-value represents the comparison between phenotypes.
See Supplementary Table for details; genetic reports were not available for all patients. mX/Y refers to mosaic monosomy X cell line and another cell line with Y chromosome material. CHCO=Children’s Hospital Colorado; NIPT=non-invasive prenatal testing; AMA=advanced maternal age
Genetic Diagnosis
The majority of patients in both groups received a genetic diagnosis postnatally, however 8 of the 15 patients with typical male genitalia were identified prenatally incidentally through screening due to advanced maternal age or elective screening rather than a concern about the pregnancy (Table 1). Patients with male/ambiguous genitalia identified postnatally were more likely diagnosed shortly after birth secondary to congenital anomalies (including ambiguous genitalia), while females were more likely to be diagnosed later in childhood secondary to short stature (46%) or delayed puberty (21%), (Table 1). Timing and additional reasons leading to genetic evaluation, age at most recent clinical visit, and duration of care are presented in Table 1. Because duration of care was significantly different between groups, all outcomes that may be impacted by age or continuity of care were adjusted for duration of follow-up.
There were no differences in karyotype between phenotypic groups (Table 1, Figure 1, Supplementary Table). Half of the cohort had a 45,X/46,XY karyotype, with a Y isochromosome being the second most frequent (31% of female and 37% of male/ambiguous groups). Percent mosaicism of a monosomy X cell line in peripheral blood lymphocytes was also nearly identical between phenotypic groups and there was no cutoff level that could predict physical phenotype, with both phenotypic females and males having monosomy X in as low as <10% and as high as >90% of cells. Furthermore, predicted presence or absences of the SYR gene or predicted copies of the SHOX gene were not associated with genital phenotype or diagnosis of short stature. However, most individuals did not have FISH testing for these specific genes.
Figure 1.
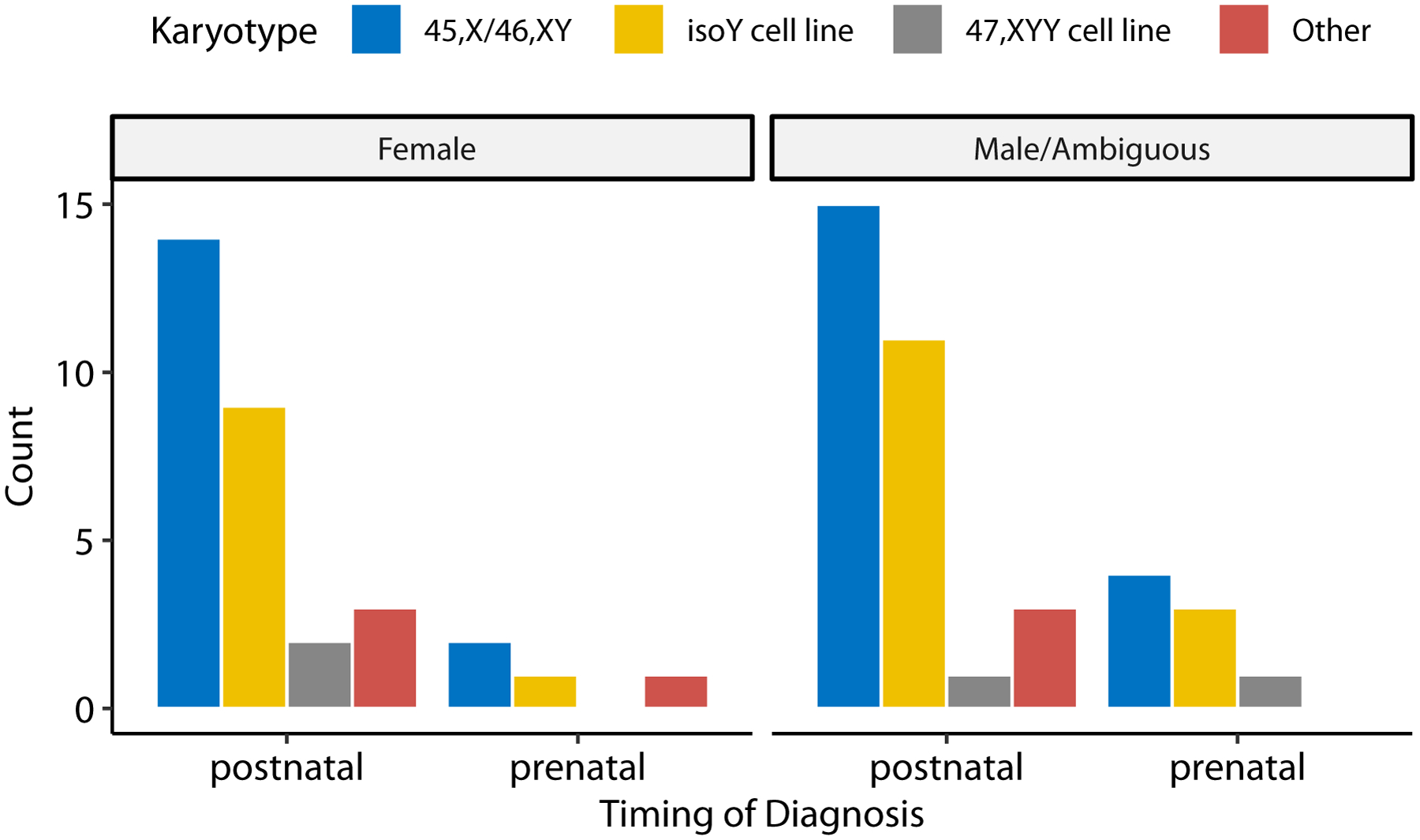
Frequency counts of different karyotypes for all patients by phenotype and timing of diagnosis.
Clinical features
Compared to females, the male/ambiguous group were at a lower risk for short stature, gonadal dysfunction, and recurrent otitis media (Table 2). However, among the individuals in the male/ambiguous group that reached a final height (n=8), the mean adult height was 160 cm ± 5 cm. A lower prevalence of dysmorphic features, renal anomalies, aortic coarctation, and musculoskeletal diagnoses in the male/ambiguous group trended toward significance (p < 0.1). All other clinical outcomes, including medical and psychological diagnoses, were present in similar prevalence in both groups (Table 2).
Table 2.
Prevalence and relative risk of Turner Syndrome-related features.
| Full cohort (n=76) | Female Phenotype (n=32) | Male/Ambiguous Phenotype (n=44) | RR^ (95%CI) | |
|---|---|---|---|---|
| Dysmorphic features | 41 (54%) | 21 (66%) | 20 (45%) | 0.7 (0.4–1.0) |
| Short stature | 50 (66%) | 26 (81%) | 24 (55%) | 0.6 (0.4–0.9)* |
| Gonadal dysfunction (≥ 11 yrs old) | 32 (76%) | 18 (95%) | 14 (61%) | 0.5 (0.4–0.9)* |
| Gonadoblastoma/dysgerminoma | 8 (16%) | 6 (24%) | 2 (8%) | 0.3 (0.1–1.6) |
| Aortic coarctation | 6 (9%) | 5 (16%) | 1 (3%) | 0.2 (0.0–1.4) |
| Aortic dilation | 7 (10%) | 2 (6%) | 5 (14%) | 2.2 (0.4–10.4) |
| Duplicated collecting system | 8 (13%) | 6 (20%) | 2 (6%) | 0.3 (0.1–1.4) |
| Recurrent otitis media | 27 (36%) | 16 (50%) | 11 (25%) | 0.4 (0.2–0.9)* |
| Strabismus/amblyopia | 6 (8%) | 1 (3%) | 5 (11%) | 3.9 (0.4–34.3) |
| Failure to thrive | 9 (12%) | 5 (16%) | 4 (9%) | 0.4 (0.1–1.7) |
| Celiac Disease | 2 (5%) | 0 | 2 (9%) | --- |
| Musculoskeletal diagnosis | 7 (9%) | 4 (13%) | 4 (9%) | 0.3 (0.1–1.1) |
| Hypertension | 5 (6%) | 4 (13%) | 1 (3%) | 0.2 (0.0–1.7) |
| Obesity | 13 (21%) | 5 (19%) | 8 (22%) | 0.9 (0.3–2.8) |
| Dyslipidemia | 17 (59%) | 7 (50%) | 10 (67%) | 1.5 (0.7–3.2) |
| Liver disease | 3 (9%) | 2 (17%) | 1 (4%) | 0.1 (0.0–1.1) |
| Seizure disorder | 7 (9%) | 4 (13%) | 3 (7%) | 0.3 (0.1–1.2) |
| Vitamin D deficiency | 15 (20%) | 7 (22%) | 8 (18%) | 0.6 (0.2–1.8) |
| Learning Disability (≥ 5 yrs old) | 9 (15%) | 3 (11%) | 6 (18%) | 1.6 (0.4–6.7) |
| Depression (≥ 5 yrs old) | 3 (5%) | 1 (4%) | 2 (6%) | 2.4 (0.5–12.3) |
| IEP/504 (≥5 yrs old) | 41 (66%) | 18 (64%) | 23 (68%) | 1.1 (0.7–1.6) |
Risk ratio (RR) and 95% confidence intervals for male/ambiguous phenotype is relative to female phenotype adjusted for site and duration of care.
p<0.05. ADHD=attention deficit hyperactivity disorder; IEP/504 = individualized education plan or 504 academic plan in school to accommodate different learning needs.
Results were similar in sensitivity analyses stratifying genitalia into three groups (Figure 2), however no individuals with a normal male phenotype were identified to have horseshoe kidney, liver disease, or gonadoblastoma. Two cases of gonadoblastoma were found in the group with ambiguous genitalia. The first was in an individual raised as female who had a bilateral gonadectomy at age 12. In the other patient, orchiectomy was performed at the age of 18 due to a dysfunctional testis, although no masses were palpable and the sonographic findings were consistent with post-surgical changes from an early orchiopexy; pathology revealed mixed intratubular germ cell neoplasm.
Figure 2.
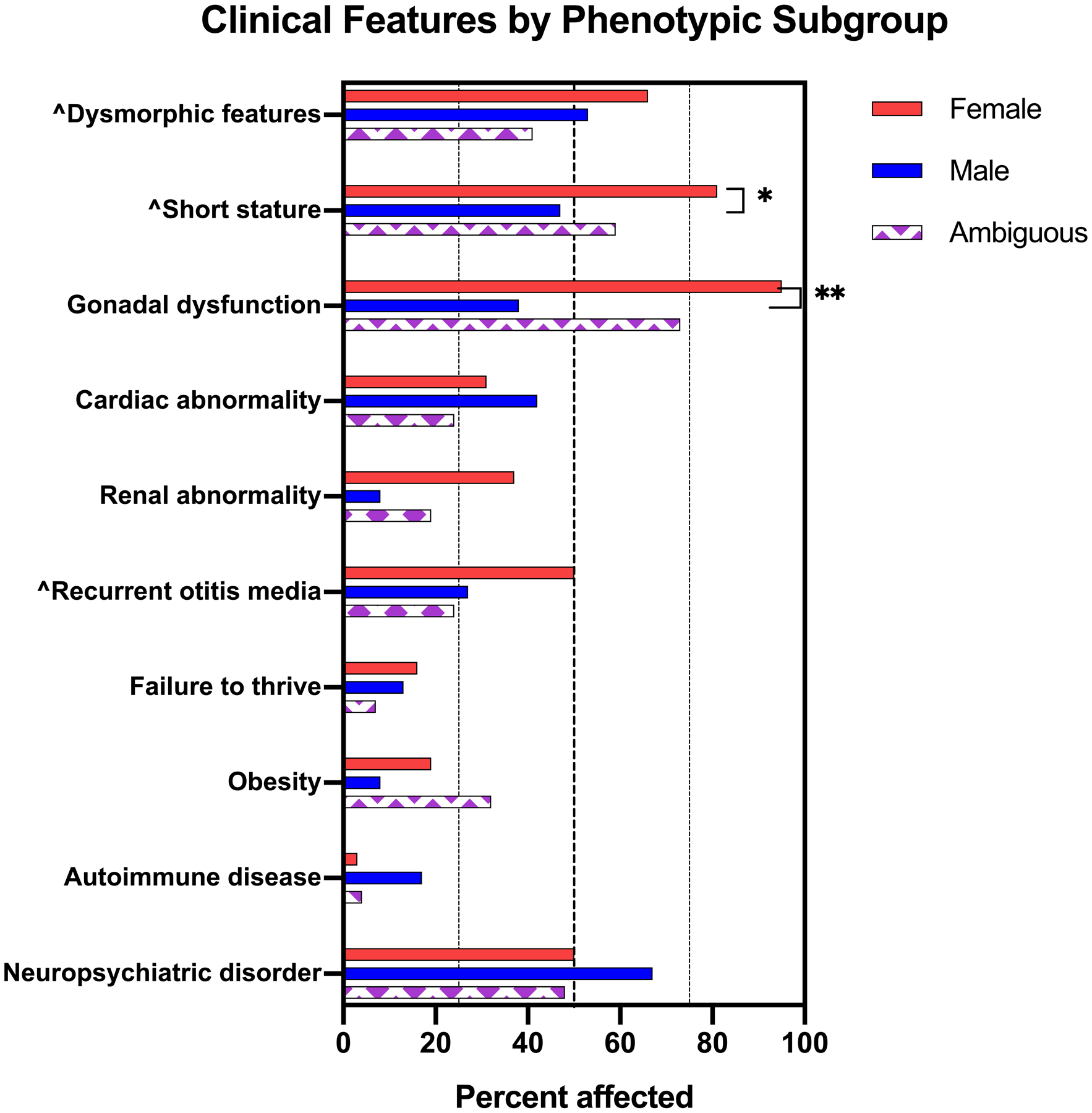
Prevalence of diagnoses compared among groups based on documented genitalia (normal female, normal male, and ambiguous). Dashed lines are at 25%, 50%, and 75%. ^Significant group differences by ANOVA at an alpha of 0.05 after adjustment for site and duration of follow up. *Significant pairwise differences after adjustment for site and duration of follow up at an alpha of 0.05 (*) or 0.01 (**).
To provide perspective for prenatal counseling for individuals with a mosaic 45,X with Y chromosome material karyotype, we explored clinical features in the 14 individuals identified prenatally compared to the group identified postnatally (Table 3). Individuals identified prenatally were much more likely to have typical genitalia compared to those identified postnatally. However, the prenatal group appeared to be at a similar risk for other clinical features including cardiac and renal anomalies, recurrent ear infections, autoimmunity, and neurodevelopmental diagnoses.
Table 3.
Exploratory sensitivity analysis stratified by diagnosis timing
| Prenatal (n=14) | Postnatal (n=62) | p-value^ | |
|---|---|---|---|
| Ambiguous | 2 (14%) | 27 (44%) | |
| Dysmorphic features | 6 (43%) | 35 (56%) | 0.39 |
| Short stature | 6 (43%) | 44 (71%) | 0.06 |
| Gonadal dysfunction (≥ 11 yrs old) | 3 (75%) | 29 (76%) | 1 |
| Gonadoblastoma/dysgerminoma | 1 (25%) | 7 (16%) | 0.52 |
| Aortic dilation | 2 (15%) | 5 (9%) | 0.61 |
| Duplicated collecting system | 3 (25%) | 5 (10%) | 0.17 |
| Recurrent otitis media | 6 (43%) | 21 (34%) | 0.55 |
| Strabismus | 0 | 6 (10%) | 0.59 |
| Failure to thrive | 1 (7%) | 7 (11%) | 1 |
| Celiac disease | 1 (17%) | 1 (3%) | 0.27 |
| Musculoskeletal diagnosis | 1 (7%) | 6 (10%) | 1 |
| Hypertension | 1 (7%) | 4 (6%) | 1 |
| Obesity | 0 | 13 (26%) | 0.06 |
| Dyslipidemia | 1 (33%) | 16 (62%) | 0.55 |
| Liver disease | 0 | 3 (12%) | 0.54 |
| Seizure disorder | 0 | 7 (11%) | 0.34 |
| Vitamin D deficiency | 3 (21%) | 12 (20%) | 1 |
| Learning Disability (≥5 yrs old) | 2 (22%) | 7 (13%) | 0.61 |
| Depression (≥ 5 yrs old) | 0 | 3 (6%) | 1 |
| IEP/504 (≥5 yrs old) | 7 (78%) | 34 (64%) | 0.71 |
Comparison of prenatal to postnatal groups using Fisher Exact without adjusting for site or duration of care due to the small prenatally identified sample, however the duration of care was similar between prenatal and postnatally diagnosed groups (mean 5.5 vs 5.0 years). ADHD=attention deficit hyperactivity disorder; IEP/504 = individualized education plan or 504 plan in school to accommodate different learning needs.
Clinical Management
Even after adjusting for site and duration of follow up, individuals in the male/ambiguous group were managed differently than the female group for numerous measures (Table 4). They were less likely to have cardiac or renal imaging; to receive counseling on risks for comorbidities; to have screening labs to assess for autoimmune or cardiometabolic conditions; and to be treated with growth hormone or antihypertensive medications. Finally, there were significant differences in the rates of gonadectomy (unilateral or bilateral), with 78% of the female genitalia group having had a gonadectomy compared to 54% of the male/ambiguous group (only 2 of those with normal male genitalia).
Table 4.
Documented clinical management stratified by phenotype
| Full cohort (n=76) | Female Phenotype (n=32) | Male/Ambiguous Phenotype (n=44) | p-value^ | |
|---|---|---|---|---|
| FISH performed | 42 (55%) | 22 (63%) | 22 (50%) | 0.37 |
| Gonadectomy (unilateral or bilateral) | 49 (65%) | 25 (78%) | 24 (55%) | 0.012 |
| Echocardiogram | 69 (91%) | 32 (100%) | 37 (84%) | 0.028 |
| Cardiac MRI | 8 (11%) | 7 (22%) | 1 (2%) | 0.013 |
| Renal ultrasound | 63 (83%) | 30 (94%) | 33 (75%) | 0.012 |
| Thyroid function testing | 62 (82%) | 28 (88%) | 34 (77%) | 0.049 |
| Thyroid function testing annually ≥2 yrs | 26 (49%) | 16 (59%) | 10 (32%) | 0.11 |
| Celiac antibody testing | 41 (54%) | 19 (59%) | 22 (50%) | 0.16 |
| Celiac antibody testing every 2 yrs ≥2 yrs | 13 (33%) | 10 (53%) | 3 (14%) | 0.040 |
| Vitamin D level testing | 26 (34%) | 14 (44%) | 12 (27%) | 0.014 |
| Lipid panel testing | 30 (39%) | 15 (47%) | 15 (39%) | 0.038 |
| Liver function testing | 44 (58%) | 22 (69%) | 22 (50%) | 0.013 |
| Liver function testing annually ≥10 yrs | 12 (36%) | 7 (47%) | 5 (28%) | 0.49 |
| Diabetes screening | 28 (37%) | 13 (41%) | 15 (34%) | 0.13 |
| Diabetes screening annually ≥10 yrs | 9 (35%) | 6 (50%) | 3 (21%) | 0.89 |
| Treatment with growth hormone | 39 (51%) | 22 (69%) | 17 (39%) | <0.001 |
| Treatment with anti-hypertensive meds | 9 (12%) | 8 (25%) | 1 (3%) | 0.001 |
| Treatment with sex steroids ≥12 yrs | 23 (62%) | 15 (83%) | 8 (42%) | 0.010 |
| Counseling on risk for delays/learning problems | 49 (65%) | 23 (72%) | 26 (60%) | 0.62 |
| Counseling on risk for mental health conditions | 20 (26%) | 12 (38%) | 8 (18%) | 0.039 |
| Counseling on diet/exercise | 39 (51%) | 17 (53%) | 22 (50%) | 0.19 |
| Counseling on risk of infertility with parents | 48 (63%) | 21 (66%) | 27 (61%) | 0.44 |
| Counseling on risk of infertility with patient ≥10 yrs | 14 (32%) | 8 (42%) | 6 (24%) | 0.074 |
| Disclosure of diagnosis to patient ≥10 yrs | 27 (61%) | 15 (79%) | 12 (48%) | 0.048 |
Comparison between female vs male/ambiguous phenotypes adjusted for site and duration of care. FISH = Fluorescence in situ hybridization; MRI = Magnetic resonance imaging
DISCUSSION
In this multicenter retrospective chart review we report on the clinical phenotype and management of 76 children with mosaic monosomy X and Y chromosome material stratified by genital phenotype given the clinical predisposition to assign a diagnosis based on physical presentation (i.e. Turner syndrome vs mixed gonadal dysgenesis). As hypothesized, individuals with male/ambiguous genitalia were just as likely as those with female genitalia to have congenital and acquired conditions commonly associated with Turner syndrome. However, there were differences in their clinical management. These findings suggest that clinical management should be based on the presence of a monosomy X cell line rather on the presentation of the genitalia.
It is well known that girls with Turner syndrome are at risk for multisystem manifestations that require multidisciplinary care (Bondy, 2007). Many features ascribed to the Turner syndrome phenotype are attributed to haploinsufficiency of genes on the sex chromosomes. Therefore, it is not surprising that the risk of these clinical findings would be similar in all individuals with a monosomy X cell line independent of their genitalia. Other studies have described the prevalence of features within cohorts of individuals with monosomy X and either mixed gonadal dysgenesis and/or male genitalia. One such study describing the clinical phenotype of 40 males with a monosomy X cell line found remarkably similar percentages as our study for congenital heart disease (26%), structural renal disease (16%), short stature (47%), and autoimmunity (20%). However, they reported fewer dysmorphic features (28%) and psychomotor delays (10%) compared to our cohort (Dumeige et al., 2018). The present study is unique in that it directly compares the risk of medical and neurodevelopmental features between individuals presenting with female versus male or ambiguous genitalia. A similar approach was used to compare cardiac pathology and found similar frequencies of cardiac pathology males and females with a monosomy X cell line with Y chromosome material (De Groote et al., 2013). In the current report we expand these outcomes in a larger sample across two hospital systems, therefore adding to our understanding of monosomy X with Y chromosome material to help guide clinical counseling and management recommendations.
Clinical presentation could not be predicted by the karyotype or level of mosaicism, which is congruent with previous studies (Chang et al., 1990; Dumeige et al., 2018; Mendez et al., 1993; Telvi et al., 1999). Approximately half of both phenotypic cohorts had a karyotype of mosaic 45,X/46,XY with 45,X/46,X,isoY as the second most common karyotype, which is not unexpected given isochromosome Y is one of the most common chromosomal abnormalities of the Y chromosome (Tuck-Muller et al., 1995; Willis, Bird, Dell’aquilla, & Jones, 2006). A novel online database developed by Liehr et al from Germany compiles published and unpublished case reports of small structurally abnormal supernumerary chromosomes (sSMC), including 383 with monosomy X and structurally abnormal Y chromosome (Liehr, 2021). Although the definition of sSMC would not include individuals with mosaic 45,X/46,XY, it could include the other ~40% in our cohort with structurally abnormal Y chromosomes (isochromosomes, rings, derivatives, etc). Within this database, 232 (~60%) are classified as female and 151 as male, with the percent of monosomy X cell lines ranging from <5% to >95% in both phenotypic classifications, congruent with the data we present here. Although clinical information on these cases is limited and variable, the karyotype does not appear to correlate with phenotypic features including puberty, infertility, short stature, or developmental delay (Liehr, 2021).
We attempted to explore whether genotype-phenotype correlations could be identified by determining the expected gene dosage of the short-stature homeobox gene (SHOX) on the distal short arm of both the X and Y chromosomes as well as the presence or absence of the sex-determining gene (SRY) on the short arm of the Y chromosome based on peripheral blood karyotype (see Supplemental Table). The predicted presence of SRY was not helpful in predicting the external genitalia, however it is important to mention that our analysis relied on primarily on prediction of the presence of the gene based on the chromosome analysis as few individuals had FISH to confirm presence or absence of SRY. There was no obvious association with the SHOX gene number and height z-score or the binary outcome of short stature in our cohort, in line with findings reported by Dumeige and colleagues (Dumeige et al., 2018). Additional research in this area is warranted especially as specific mechanisms underlying the phenotypic features of monosomy X are elucidated.
Although most cases presented in this retrospective report were postnatally diagnosed, the outcomes in the fourteen prenatal cases included are essential for comparative exploration in diagnosis timing. This comparison is imperative for guiding accurate genetic counseling in a prenatal setting given the inherent ascertainment biases introduced by postnatally diagnosed case reports. In addition, the recent advances in noninvasive prenatal screening as a standard in obstetric care (Rose et al., 2020) are anticipated to expand the proportion of aneuploidy karyotypes prenatally identified. Importantly, among the prenatally identified subgroup in this cohort, the majority had typical male genitalia. Many individuals with this karyotype and typical appearing genitalia may remain undiagnosed (Chang et al., 1990), and therefore the clinical spectrum of individuals in this study likely does not fully represent the phenotypic spectrum of the karyotype monosomy X with Y chromosome material. While no other phenotypic findings significantly differed based on timing of diagnosis (prenatal vs. postnatal) in this cohort, further studies are needed to fully inform genetic counseling for prenatally diagnosed individuals.
We found that the clinical management of individuals with male/ambiguous genitalia differed from individuals with female genitalia. Of particular concern, those in the male/ambiguous group were much less likely to receive and echocardiogram compared with those in the female group. However, among those who did receive echocardiograms, there were several cases of aortic dilation, a silent condition that can evolve over time and result in life-threatening dissection or rupture (Matura, Ho, Rosing, & Bondy, 2007). Bicuspid aortic valve and aortic dilation have been associated with haploinsufficiency of TIMP1 gene on the X chromosome, therefore monosomy X independent of the genital phenotype would be implicated (Corbitt et al., 2018). These results are congruent with De Groote and colleagues who found 4/10 boys with mX/Y had aortic dilation, (De Groote et al., 2013) and emphasizes that not only is cardiac imaging imperative in this population but it needs to be ongoing as recommended in the Turner syndrome CPG (Gravholt et al., 2017). Documentation of screening and counseling for non-cardiac conditions was insufficient for both groups, but it was especially poor for those with male/ambiguous genitalia. Interestingly, despite the minimal research available on the neurodevelopmental phenotype of individuals with mosaic monosomy X and male or ambiguous genitalia, counseling for this risk was similar between groups. However, the high prevalence of neurodevelopmental and educational concerns, including within our sub-analysis of males alone, highlights the need for future research of neuropsychological profiles and best practices for individuals with monosomy X and Y chromosome material.
Management of gender assignment and surgery has received the bulk of attention for individuals with monosomy X and Y chromosome material. Gonadectomy is currently recommended in female patients with Y chromosome material due to the risk of malignancy with likely non-functional gonads (Gravholt et al., 2017; Gravholt, Fedder, Naeraa, & Muller, 2000; Mazzanti et al., 2005). There is disagreement on the best clinical management for the gonads in individuals with ambiguous genitalia, with proposed algorithms considering the degree of virilization of the external genitalia, degree of differentiation and potential function of the gonads, presence of specific Y chromosome genes, patient age, and family preference (Cools et al., 2011; Matsumoto, Matsuyama, Matsui, Yazawa, & Matsuoka, 2020; Tam et al., 2016; Weidler, Pearson, van Leeuwen, & Garvey, 2019; Wolffenbuttel et al., 2016). Although there are no official CPG for typical or mildly undervirilized males, authors have proposed that if descended testes can be palpated and/or imaged for masses and asymmetry, a more conservative approach that avoids (or defers) gonadectomy is most appropriate (Cools et al., 2011; Weidler et al., 2019). In our cohort, clinical management was in line with these recommendations, with 83% of females undergoing bilateral gonadectomy, 76% of individuals with ambiguous genitalia undergoing unilateral or bilateral gonadectomy, and only two individuals with normal male genitalia. The prevalence of gonadoblastoma or germ cell tumor in females who had gonadectomy with available pathology was 24%, which is similar to the previously reported incidence of 15–30% (Coyle et al., 2016; Gravholt et al., 2000). Our data do not shed additional light on whether there is a malignancy risk in males with monosomy X and Y chromosome material as only two males underwent gonadectomy, and neither had evidence of atypia. However, in individuals who had at least one remaining gonad there was a paucity of documentation of surveillance for malignancy, despite the recommended management (Weidler et al., 2019).
Limitations of this study include sampling bias from pediatric specialty clinics with potential overrepresentation of individuals with ambiguous genitalia and phenotypic severity. Therefore, our conclusions are not necessarily generalizable to individuals identified in adulthood to have monosomy X and Y chromosome material. Notably, individuals incidentally identified with this chromosome complement during the prenatal period still had clinical features associated with monosomy X cell line. There is also potential for omitted or inaccurate data as an inherent bias in retrospective chart reviews. Some individuals received care outside of hospital networks, and records available for review may have been incomplete. Although this may result in inaccurate prevalence assessments, we would anticipate these errors of commission or omission would be similar across groups, and therefore, the comparison is remains valid. Additional studies in this population, including a prospective, longitudinal study of individuals with monosomy X and Y chromosome material would provide a more accurate natural history of the manifestations of the karyotype throughout the lifespan. Finally, although this is a relatively large sample for this pediatric rare disease, we were still underpowered to rule out differences between phenotypic groups. Nonetheless, this large study of children with a monosomy X cell line mosaic and Y chromosome material presents novel information regarding the phenotype that will hopefully help guide counseling, clinical management, and future research. Given our results, specific work in understanding neurodevelopment and aortopathy is particularly needed. Ultimately, development of evidence-based clinical practice guidelines for individuals with monosomy X with Y chromosme material independent of external genitalia will improve clinical care and outcomes for these patients. However, in lieu of such guidelines being available, we advocate for utilization of the Turner syndrome CPG for applicable recommendations as recommended by other authors as well (Dumeige et al., 2018).
Conclusion
The aim of this retrospective investigation of individuals with mosaic monosomy X and Y chromosome material was to describe the clinical phenotype and management within the population based on presentation of the external genitalia. The novel insights this report provides are that individuals with male/ambiguous genitalia are at a similar risk for medical and neurodevelopmental conditions as those born with normal female genitalia (i.e. Turner syndrome), however, they are less likely to receive appropriate counseling and assessments, highlighting disparities in clinical practice. These results support that all individuals with a monosomy X cell line are at risk for multisystem comorbidities commonly associated with Turner syndrome, and should receive screening, counseling, and treatment as advised by applicable recommendations in the Turner syndrome CPG until more specific consensus guidelines are adapted for this population.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the support of the Genetic Counseling Graduate Program at the University of Colorado and the providers who care for these patients at Children’s Hospital Colorado and Nationwide Children’s Hospital. This work was supported by NIH/NICHD K23HD092588, NIH/NHLBI K23 HL151868, NIH/NCATS Colorado CTSA Grant Number UL1 TR002535. Contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
NICHD K23 HD092588 (SMD), NIH/NHLBI K23 HL151868 (NJN), NIH/NCRR Colorado CTSI Grant Number UL1 TR002535
Footnotes
Author Contribution and Conflicts of Interest. Each of the co-authors acknowledges their participation in conducting the research leading to this manuscript and has agreed to its submission. None of the authors have any potential sources of conflicts of interest to disclose.
Data Sharing and Data Accessibility. The data that supports the findings of this study are available in the supplementary material of this article. Additional data are available from the corresponding author upon reasonable request.
REFERENCES
- Akinsal EC, Baydilli N, Bayramov R, & Ekmekcioglu O (2018). A Rare Cause of Male Infertility: 45,X/46,XY Mosaicism. Urol Int, 101(4), 481–485. doi: 10.1159/000484615 [DOI] [PubMed] [Google Scholar]
- Bondy CA (2007). Care of girls and women with Turner syndrome: A guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab, 92(1), 10–25. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17047017https://watermark.silverchair.com/jcem0010.pdf?token=AQECAHi208BE49Ooan9kkhW_Ercy7Dm3ZL_9Cf3qfKAc485ysgAAAj8wggI7BgkqhkiG9w0BBwagggIsMIICKAIBADCCAiEGCSqGSIb3DQEHATAeBglghkgBZQMEAS4wEQQM3w4GR4NucTcd3yCMAgEQgIIB8i0FoUPER2Yr0Ogt3FWYVk-_z5Je319ww3aB1pK7JYHezn2AHBo_MwxlkXFaJ4-L1lqX0J4jlRUyCXDVitMvEWcdQNicuWcTFYTD5sTzuiVldC5VRHdLv6u2fOMc-XDSvzT50oZrW1Lwk7tRTdUKMsPvmbovR9nCH1b1BG3NuMVGny1_w8ZbxpEsReiQQog6RY5w5kg2XFXxnOqaHHW0rcJ8Ql-yhKdF2g6nksrE6dXJhyW1oZovIrtoLDvlEcjwYQDq1cVDLB6uPPJjj0-NWHVm7YLsVsxdiPaY6sOn8ZtK2i8jjujEhf5YGShNS-F6lcvC2CVqx1IkNtloRo3Z6oOjCVOrTU15vKguWE-945r6Sf15GmKTUCPMhjrMv3wOXewDoLZCRfo0-4XPrH4M-IsXnHQmEqiNY69LPgLLoQXGzQeWfD7nNwpddEMQFBQmvqHUJC7bYrFGAJfybW6kRbxlDJj-rBGbtsbBBXppnCkqEe5bpH6-tnljS3Jv4wezkjcSWqS93oRDT4C5rW4mVMWktfp952UdxtEfUTzJqKA05BgznH4zs8yY8SmyY_4A9UNqXR1Xq0DmlDLQop34qXKRIbiZIwV85IFaTtB_r86X8sDOvU7hRAYIdsSHEUjqADk52voBLoKPEsOosWnA97BbBg [DOI] [PubMed] [Google Scholar]
- Chang HJ, Clark RD, & Bachman H (1990). The phenotype of 45,X/46,XY mosaicism: an analysis of 92 prenatally diagnosed cases. Am J Hum Genet, 46(1), 156–167. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/2294747https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1683543/pdf/ajhg00098-0160.pdf [PMC free article] [PubMed] [Google Scholar]
- Cools M, Pleskacova J, Stoop H, Hoebeke P, Van Laecke E, Drop SL, … Mosaicism Collaborative G (2011). Gonadal pathology and tumor risk in relation to clinical characteristics in patients with 45,X/46,XY mosaicism. J Clin Endocrinol Metab, 96(7), E1171–1180. doi: 10.1210/jc.2011-0232 [DOI] [PubMed] [Google Scholar]
- Corbitt H, Morris SA, Gravholt CH, Mortensen KH, Tippner-Hedges R, Silberbach M, … Gen TACRI (2018). TIMP3 and TIMP1 are risk genes for bicuspid aortic valve and aortopathy in Turner syndrome. PLoS Genet, 14(10), e1007692. doi: 10.1371/journal.pgen.1007692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle D, Kutasy B, Han Suyin K, Antao B, Lynch SA, McDermott MB, … Quinn F (2016). Gonadoblastoma in patients with 45,X/46,XY mosaicism: A 16-year experience. J Pediatr Urol, 12(5), 283.e281–283.e287. doi: 10.1016/j.jpurol.2016.02.009 [DOI] [PubMed] [Google Scholar]
- De Groote K, Cools M, De Schepper J, Craen M, Francois I, Devos D, … De Wolf D (2013). Cardiovascular pathology in males and females with 45,X/46,XY mosaicism. PLoS One, 8(2), e54977. doi: 10.1371/journal.pone.0054977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumeige L, Chatelais L, Bouvattier C, De Kerdanet M, Hyon C, Esteva B, … Martinerie L (2018). Should 45,X/46,XY boys with no or mild anomaly of external genitalia be investigated and followed up? Eur J Endocrinol, 179(3), 181–190. doi: 10.1530/EJE-18-0309 [DOI] [PubMed] [Google Scholar]
- Efthymiadou A, Stefanou EG, & Chrysis D (2012). 45,X/46,XY mosaicism: a cause of short stature in males. Hormones (Athens), 11(4), 501–504. doi: 10.14310/horm.2002.1384 [DOI] [PubMed] [Google Scholar]
- Farrugia MK, Sebire NJ, Achermann JC, Eisawi A, Duffy PG, & Mushtaq I (2013). Clinical and gonadal features and early surgical management of 45,X/46,XY and 45,X/47,XYY chromosomal mosaicism presenting with genital anomalies. J Pediatr Urol, 9(2), 139–144. doi: 10.1016/j.jpurol.2011.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, … International Turner Syndrome Consensus, G. (2017). Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol, 177(3), G1–G70. doi: 10.1530/EJE-17-0430 [DOI] [PubMed] [Google Scholar]
- Gravholt CH, Fedder J, Naeraa RW, & Muller J (2000). Occurrence of gonadoblastoma in females with Turner syndrome and Y chromosome material: a population study. J Clin Endocrinol Metab, 85(9), 3199–3202. doi: 10.1210/jcem.85.9.6800 [DOI] [PubMed] [Google Scholar]
- Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, & Conde JG (2009). Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform, 42(2), 377–381. doi: 10.1016/j.jbi.2008.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller B, Bradtke J, Balz H, & Rieder H (2004). CyDAS Online Analysis Site. Retrieved from http://www.cydas.org/OnlineAnalysis/. Retrieved 1/7/2021 http://www.cydas.org/OnlineAnalysis/
- Hook EB, & Warburton D (2014). Turner syndrome revisited: review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Hum Genet, 133(4), 417–424. doi: 10.1007/s00439-014-1420-x [DOI] [PubMed] [Google Scholar]
- Huang YC, Lee CT, Wu MZ, Liu SY, Tung YC, Ho HN, & Tsai WY (2019). The spectrum of 45,X/46,XY mosaicism in Taiwanese children: The experience of a single center. J Formos Med Assoc, 118(1 Pt 3), 450–456. doi: 10.1016/j.jfma.2018.07.003 [DOI] [PubMed] [Google Scholar]
- Liehr T (2021). Small supernumerary marker chromosomes. Retrieved from http://cs-tl.de/DB/CA/sSMC/0-Start.html. Retrieved 1/7/2021 http://cs-tl.de/DB/CA/sSMC/0-Start.html
- Lindhardt Johansen M, Hagen CP, Rajpert-De Meyts E, Kjaergaard S, Petersen BL, Skakkebaek NE, … Juul A (2012). 45,X/46,XY mosaicism: phenotypic characteristics, growth, and reproductive function--a retrospective longitudinal study. J Clin Endocrinol Metab, 97(8), E1540–1549. doi: 10.1210/jc.2012-1388 [DOI] [PubMed] [Google Scholar]
- Martinerie L, Morel Y, Gay CL, Pienkowski C, de Kerdanet M, Cabrol S, … Bouvattier C (2012). Impaired puberty, fertility, and final stature in 45,X/46,XY mixed gonadal dysgenetic patients raised as boys. Eur J Endocrinol, 166(4), 687–694. doi: 10.1530/EJE-11-0756 [DOI] [PubMed] [Google Scholar]
- Matsumoto F, Matsuyama S, Matsui F, Yazawa K, & Matsuoka K (2020). Variation of Gonadal Dysgenesis and Tumor Risk in Patients With 45,X/46,XY Mosaicism. Urology, 137, 157–160. doi: 10.1016/j.urology.2019.12.014 [DOI] [PubMed] [Google Scholar]
- Matura LA, Ho VB, Rosing DR, & Bondy CA (2007). Aortic dilatation and dissection in Turner syndrome. Circulation, 116(15), 1663–1670. doi: 10.1161/CIRCULATIONAHA.106.685487 [DOI] [PubMed] [Google Scholar]
- Mazzanti L, Cicognani A, Baldazzi L, Bergamaschi R, Scarano E, Strocchi S, … Cacciari E (2005). Gonadoblastoma in Turner syndrome and Y-chromosome-derived material. Am J Med Genet A, 135(2), 150–154. doi: 10.1002/ajmg.a.30569 [DOI] [PubMed] [Google Scholar]
- Mendez JP, Ulloa-Aguirre A, Kofman-Alfaro S, Mutchinick O, Fernandez-del-Castillo C, Reyes E, & Perez-Palacios G (1993). Mixed gonadal dysgenesis: clinical, cytogenetic, endocrinological, and histopathological findings in 16 patients. Am J Med Genet, 46(3), 263–267. doi: 10.1002/ajmg.1320460304 [DOI] [PubMed] [Google Scholar]
- Rose NC, Kaimal AJ, Dugoff L, Norton ME, American College of O, Gynecologists’ Committee on Practice, B.-O., … Society for Maternal-Fetal, M. (2020). Screening for Fetal Chromosomal Abnormalities: ACOG Practice Bulletin, Number 226. Obstet Gynecol, 136(4), e48–e69. doi: 10.1097/AOG.0000000000004084 [DOI] [PubMed] [Google Scholar]
- Tam YH, Wong YS, Pang KK, To KF, Yiu AK, Wong HY, … Lee KH (2016). Tumor risk of children with 45,X/46,XY gonadal dysgenesis in relation to their clinical presentations: Further insights into the gonadal management. J Pediatr Surg, 51(9), 1462–1466. doi: 10.1016/j.jpedsurg.2016.03.006 [DOI] [PubMed] [Google Scholar]
- Telvi L, Lebbar A, Del Pino O, Barbet JP, & Chaussain JL (1999). 45,X/46,XY mosaicism: report of 27 cases. Pediatrics, 104(2 Pt 1), 304–308. doi: 10.1542/peds.104.2.304 [DOI] [PubMed] [Google Scholar]
- Toniolo D, & Rizzolio F (2007). X chromosome and ovarian failure. Semin Reprod Med, 25(4), 264–271. doi: 10.1055/s-2007-980220 [DOI] [PubMed] [Google Scholar]
- Tuck-Muller CM, Chen H, Martinez JE, Shen CC, Li S, Kusyk C, … Wertelecki W (1995). Isodicentric Y chromosome: cytogenetic, molecular and clinical studies and review of the literature. Hum Genet, 96(1), 119–129. doi: 10.1007/BF00214200 [DOI] [PubMed] [Google Scholar]
- Weidler EM, Pearson M, van Leeuwen K, & Garvey E (2019). Clinical management in mixed gonadal dysgenesis with chromosomal mosaicism: Considerations in newborns and adolescents. Semin Pediatr Surg, 28(5), 150841. doi: 10.1016/j.sempedsurg.2019.150841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis MJ, Bird LM, Dell’aquilla M, & Jones MC (2006). Natural history of prenatally diagnosed 46,X,isodicentric Y. Prenat Diagn, 26(2), 134–137. doi: 10.1002/pd.1352 [DOI] [PubMed] [Google Scholar]
- Wolffenbuttel KP, Hersmus R, Stoop H, Biermann K, Hoebeke P, Cools M, & Looijenga LH (2016). Gonadal dysgenesis in disorders of sex development: Diagnosis and surgical management. J Pediatr Urol, 12(6), 411–416. doi: 10.1016/j.jpurol.2016.08.015 [DOI] [PubMed] [Google Scholar]
- Wu Q, Wang C, Shi H, Kong X, Ren S, & Jiang M (2017). The Clinical Manifestation and Genetic Evaluation in Patients with 45,X/46,XY Mosaicism. Sex Dev, 11(2), 64–69. doi: 10.1159/000455260 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.