

Abstract
Human macrophage galactose-type lectin (hMGL, HML, CD301, CLEC10A), a C-type lectin expressed by dendritic cells and macrophages, is a receptor for N-acetylgalactosamine α-linked to serine/threonine residues (Tn antigen, CD175) and its α2,6-sialylated derivative (sTn, CD175s). Because these two epitopes are among malignant cell glycan displays, particularly when presented by mucin-1 (MUC1), assessing the influence of the site and frequency of glycosylation on lectin recognition will identify determinants governing this interplay. Thus, chemical synthesis of the tandem-repeat O-glycan acceptor region of MUC1 and site-specific threonine glycosylation in all permutations were carried out. Isothermal titration calorimetry (ITC) analysis of the binding of hMGL to this library of MUC1 glycopeptides revealed an enthalpy-driven process and an affinity enhancement of an order of magnitude with an increasing glycan count from 6–8 μM for monoglycosylated peptides to 0.6 μM for triglycosylated peptide. ITC measurements performed in D2O permitted further exploration of the solvation dynamics during binding. A shift in enthalpy–entropy compensation and contact position-specific effects with the likely involvement of the peptide surroundings were detected. KinITC analysis revealed a prolonged lifetime of the lectin–glycan complex with increasing glycan valency and with a change in the solvent to D2O.
The presence of glycans is increasingly interpreted as a means of conveying molecular signals for many (patho)physiological processes “read” by tissue receptors (lectins).1−6 This already holds true for the smallest sugar epitope, a single saccharide, the Tn antigen, i.e., N-acetylgalactosamine (GalNAc), O-linked in the α-anomeric bond to Ser/Thr residues, which is abundantly present in mucins such as MUC1.7−11 A mutation in the molecular chaperone Cosmc that is responsible for ensuring acquisition of the optimal activity of T-synthase, the essential enzyme for Tn elongation by β1,3-galactosylation, leads to cancer-associated overexpression of Tn antigen.12,13 Interestingly, the profile of the occupancy of acceptor sites in glycoproteins can vary, and it is tempting to interpret this as establishing switches for affinity regulation when interacting with tissue lectins. Strategically teaming up glycopeptide synthesis with calorimetric analysis of the molecular rendezvous between the ligand and a physiologically relevant tissue lectin is a means to provide answers about the significance of site-specific glycosylation and glycan presentation in clusters. In fact, in the case of mucin-type O-glycosylation, a diversity of N-acetylgalactosaminyltransferases gives rise to regions with varying degrees of sugar density; this phenomenon is not yet fully understood in functional terms.14,15 Toward this end, we here focused on a GalNAc receptor expressed by macrophages and dendritic cells, the human macrophage galactose-type lectin (hMGL, UniProtKB Q8IUN9).
hMGL belongs to the C-type family of lectins, which recruits Ca2+ for coordination bonding of the ligand.16,17 Structurally, this immune cell lectin is closely related to the hepatic asialoglycoprotein receptor (ASGPR), with specificity directed toward the Tn antigen18−21 and its α2,6-sialylated derivative.22,23 The relevance of glycan density with a set of six mono- to hexaglycosylated peptides generated on the platform of the synthetic 11-mer core sequence PTTTPITTTTK20 and the role of the neighboring peptide backbone portions21 in recognition by hMGL were described previously. Similarly, evidence of an emerging relevance of the degree of occupancies in mucin (fragments) obtained with leguminous lectins was reported.24−26 These studies of plant lectins prompted us to perform a detailed analysis of ligand binding with synthetic bioinspired glycopeptides for a human lectin. We first prepared the complete panel of Thr-modified glycopeptides of the MUC1 tandem-repeat motif. In this context, it is noteworthy that comparative analysis of staining profiles of GalNAc-specific lectins from plants (DBA and SBA) or snails (HPA) with that of hMGL in mucin-rich murine jejunum had disclosed differences so that extrapolations between lectins of the same nominal monosaccharide specificity should be done with caution.27
Having made the glycopeptides with site-specific glycosylation in systematic permutations to yield mono- to triglycosylated products, available in the first part of our study, we then performed a systematic ITC analysis. It includes examining the effect of glycan site variations at constant valency. To gain further insights into the binding process, we assessed binding thermodynamics in both H2O and D2O as a means to probe hydration contribution(s); this was studied for the first time with a human C-type lectin, so far exclusively done on leguminous lectins, especially concanavalin A.28,29 Our applications of ITC and KinITC shed light on differential thermodynamics and kinetics of association among the MUC1 glycopeptides and the carbohydrate recognition domain (CRD) of hMGL that are dependent on the specific site and density of glycosylation, as well as the type of solvent environment.
Materials and Methods
Reagents and General Procedures
All reagents and solvents were purchased from commercial sources (Thermo Fisher Scientific or Sigma-Aldrich) and used without further purification. Spectra/Por-Float-A-Lyzer dialysis units and Slide-A-Lyzer dialysis cassettes were purchased from Thermo Fisher Scientific. d-Galactal (1), Fmoc-l-threonine (Fmoc-Thr-OH), N,N′-diisopropyl-carbodiimide (DIC), pentafluorophenol (Pfp), and hydroxybenzotriazole (HOBt) were purchased from CHEM-IMPEX. Hydrogen peroxide (30%, H2O2), anhydrous silver perchlorate (AgClO4), molecular sieves (4 Å, 1.6 and/or 3.2 mm), N-acetyl-d-galactosamine (GalNAc), and deuterium oxide (D2O) were from Sigma-Aldrich. Methyl N-acetyl-α-d-galactosamine (α-Me-GalNAc), methyl N-acetyl-β-d-galactosamine (β-Me-GalNAc), and methyl β-glucopyranoside (β-Me-Glu) were purchased from Toronto Research Chemicals. The standard Fmoc-protected amino acids and O-(1H-6-chlorobenzotriazol1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) were obtained from Protein Technologies, and Tentagel S RAM Fmoc resin was purchased from Advanced ChemTech. Filter devices used in centrifugation were obtained from Pall Corp.
Reactions were monitored using TLC on 200 μm thick silica gel F-254-coated aluminum plates (Silicycle, Inc.) with detection by ultraviolet (UV) light (λ = 254 nm) and/or by charring with a 10% solution of sulfuric acid in ethanol. Column chromatography was carried out using Silicycle silica gel F60 (230–400 mesh). 1H and 13C NMR spectra were obtained on Varian Mercury 400 MHz or Bruker Advanced III 400 MHz spectrometers. Signals are reported in terms of their chemical shifts (δ in parts per million) relative to CDCl3 (1H, δ 7.26; 13C, δ 77.16). Coupling constants (J values) are reported in hertz. All spectra were analyzed using ACD/LABORATORIES software.
Synthesis of 2-Azido Tn Antigen Building Block Nα-(9H-Fluoren-9-yl)-methoxycarbonyl-O-(3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-d-galactopyranosyl)-l-threonine Pfp Ester (4)
Tri-O-acetylated d-galactal (2) was prepared from d-galactal (1) by using acetic anhydride and pyridine as a catalyst, according to the well-established protocol.30 3,4,6-Tri-O-acetyl-2-azido-2-desoxy-α-d-galactopyranosyl chloride (3) was synthesized according to the method of Plattner et al.31 and used in the next glycosylation step without further purification. Compound 3 (1.89 g, 5.40 mmol) and Nα-Fmoc-Thr-OPfp (1.31 g, 2.58 mmol) were dissolved in a mixture of anhydrous DCM (25 mL) and toluene (20 mL), and then the mixture was stirred with activated 4 Å molecular sieves (2.0 g) at −30 °C for 20 min under an argon atmosphere. A solution of AgClO4 (780 mg, 3.76 mmol) in anhydrous toluene (5 mL) was gradually added to the reaction flask over 15 min. The reaction proceeded at room temperature (rt) for 24 h or until compound 3 was completely consumed, as confirmed by TLC. The reaction mixture was diluted with DCM (75 mL), and the suspension was filtered through a Celite and washed with DI water (200 mL). The organic layer was dried with sodium sulfate (Na2SO4) and concentrated in vacuo before fractionation by column chromatography purification [4:1 (v/v) toluene/EtOAc] that gave a light-yellow viscous oil (4). The final 2-azido Fmoc-protected O-glycosylated Thr (4) was precipitated in cold petroleum ether affording a white powder, obtained in 71.7% (1.56 g, diastereochemically pure α-anomer) yield and 11.9% (0.26 g, β-anomer) yield. The HPLC chromatogram, MALDI-TOF mass spectrum, and NMR spectrum (1H and 13C) are shown in Figures S1–S4.
Synthesis of Tn Antigen Building Block Nα-(9H-Fluoren-9-yl)-methoxycarbonyl-O-(3,4,6-tri-O-acetyl-2-acetamido-2-deoxy-α-d-galactopyranosyl)-l-threonine Pfp Ester (5)
To a solution of glacial acetic acid (2.7 mL), acetic anhydride (25 mL), and tetrahydrofuran (THF) (25 mL) was added compound 4 (α-anomer, 1.17 g, 1.43 mmol), and the mixture was allowed to cool to 0 °C for 15 min before activated zinc powder (1.86 g, 28.44 mmol) was stirred into the mixture and continued to spin overnight at rt. Freshly activated zinc was prepared by successive rapid washings with 3% diluted hydrochloric acid (four times), distilled water (four times), EtOH (four times), and dry diethyl ether (four times) and dried under vacuum overnight. The suspension was quenched with THF (60 mL) and filtered through Celite, and the resulting product was rinsed with EtOAc. The solvents were evaporated azeotropically with toluene before column chromatography purification [1:1 (v/v) toluene/EtOAc]. Recrystallization with cold petroleum ether afforded the white precipitate, Tn antigen (5), obtained in 78.1% (0.93 g, diastereochemically pure α-anomer) yield. The HPLC chromatogram, MALDI-TOF mass spectrum, and NMR spectrum (1H and 13C) are provided in Figures S5–S8.
Synthesis of MUC1 Tandem-Repeat (Glyco)peptides
Syntheses of MUC1 peptides were completed using the solid phase peptide synthesis (SPPS) approach on Tentagel S RAM Fmoc resin (0.1 mmol scale). Automated peptide synthesis was carried out using a Protein Technologies, Inc., PS3 peptide synthesizer. The Fmoc amino acids and coupling reagents, HCTU and 1-HOBt, were used in 4-fold molar excess. The Pfp ester of the glycoamino acid (5) was coupled manually in a 1.5-fold molar excess in the presence of DIPEA (230 μL, 1.32 mmol). Piperidine at a concentration of 20% in DMF was used for Fmoc deprotection reactions. Removal of amino acid side chain protecting groups and release of the peptides from the resin were performed concurrently using a water/thioanisole/trifluoroacetic acid (TFA) cocktail [2.5:2.5:95 (v/v/v)] while samples were being agitated for 3 h. Peptides were precipitated with cold methyl tert-butyl ether (MTBE) and centrifugated at 4000 rpm for 10 min at 4 °C. The precipitate was suspended in fresh MTBE, centrifugated, and decanted three additional times, successively. Peptides were deacetylated by agitation in a 0.1 M solution of NaOH for 15 min. The pH was adjusted to 4 by dropwise addition of 6 M HCl. The lyophilized crude deacetylated peptide was screened by analytical RP-HPLC and analyzed by MALDI-TOF MS. The crude peptides were purified by a semipreparative RP-HPLC 1260 Infinity Agilent Technologies system using a Grace Vydac monomeric C18 column [250 mm × 22 mm (inside diameter)] with a 10 mm core–shell particle size (120 Å pore size) as a stationary phase. For elution, a linear gradient with eluent A (0.1% TFA in water) and eluent B (0.08% TFA in acetonitrile) was used at a flow rate of 10 mL/min. Homogeneous fractions were combined, and their purity was evaluated by analytical RP-HPLC on a model 1260 Infinity Agilent Technologies system using a Phenomenex Aeris Peptide XB-C18 column [150 mm × 4.6 mm (inside diameter)] with a 3.6 μm core–shell particle size (100 Å pore size) as a stationary phase. A linear elution gradient (0.5% B at 0 min, 0.5% B at 2 min, and 30% B at 30 min) with eluent A (0.1% TFA in water) and eluent B (0.08% TFA in acetonitrile) was used at a flow rate of 0.8 mL/min. Absorption signals were detected with a UV diode array detector at a wavelength of 214 nm. The collected fractions were analyzed by MALDI-TOF MS (Applied Biosystems Voyager-De Pro) using α-cyano-4-hydroxycinnamic acid (α-CHCA) as the matrix. HPLC and MALDI-TOF MS analyses of the purified glycosylated peptides are provided in Figures S10–S25.
CD of (Glyco)peptides
The CD spectra were recorded by a JASCO J-810 spectropolarimeter (Jasco, Eaton, MD) and processed using Spectragryph software.32 Spectra were recorded in the wavelength range of 180–260 nm at 25 °C using a scan speed of 200 nm/min at a 0.1 nm bandwidth in a rectangular quartz cell with a path length of 0.1 cm. The optimum absorbance was obtained with a peptide concentration of 0.13 mg/mL. Molar concentrations of the peptidyl solutions were determined by analytical RP-HPLC; in turn, molar ellipticity values were normalized. CD measurements are presented in [θ] in degrees square centimeter per decimole. Peptides were dissolved in deionized water or heavy water as specified, and the resulting spectra are given in Figure S26.
Lectin Preparation
The lectin’s extracellular domain that includes the stalk and three noncovalently associated CRDs (Q8IUN9) was obtained by recombinant production. Protein was isolated from inclusion bodies by steps that involve solubilization, refolding, and purification. The obtained product was then ascertained for purity as described previously.21,27 To obtain the full-length cDNA sequence of the extracellular domain of hMGL, specific oligonucleotides were designed on the basis of the published sequence in the GSDB, DDBJ, EMBL, and NCBI databases with accession number D50532.19 The PCR-based amplification was directed using sense primer 5′-CCGGATCCTGGTGACCCTGAGAAC-3′ and antisense primer 5′-CCGGATCCGGGTGGTCCCACCAA-3′ with an internal BamHI restriction site (underlined). The PCR product was separated by agarose gel electrophoresis, eluted from the respective gel slice by using a gel extraction kit (Invitek, Berlin, Germany), digested by BamHI, and finally ligated without a start codon for the in-frame insertion into the BamHI-linearized pET-3d vector (Novagen). This process resulted in an N-terminal elongation of the expressed protein sequence of 11 amino acids of the T7 tag (underlined) and one amino acid of the vector backbone before the BamHI restriction side (MASMTGGQQMG R). For plasmid-directed protein expression, Escherichia coli strain BL21 (DE3) cells were transformed with the constructed pET3d-hMGL plasmid and cultured at 37 °C until an optical density of 0.6–0.8 was reached. Optimal protein expression was obtained with terrific broth (TB) medium at 30 °C for 4 h in the presence of isopropyl β-d-thiogalactopyranoside at a final concentration of 0.1 mM. To recover expressed proteins that formed inclusion bodies, cells were harvested by centrifugation (7000 × g for 10 min) and washed with 50 mM Tris-HCI buffer (pH 8.0) containing 0.15 M NaCl. Cells were frozen for 1 h, resuspended in Tris-buffered saline (TBS) containing 1 mM phenylmethanesulfonyl fluoride (PMSF), and lysed on ice by three cycles of sonication for 1 min each. The bacterial lysate was centrifuged at 39000g and 4 °C for 20 min. The preparation of inclusion bodies and refolding of the active proteins had been described previously.19,33 In brief, pellets containing the insoluble inclusion bodies were recovered and washed by centrifugation at 14000 rpm for 20 min at 4 °C with 60 mL of TBS containing 0.1% Triton X-100 and 10 mM ethylenediaminetetraacetic acid (EDTA), followed by a wash with 30 mL of H2O, and the suspension was finally centrifuged at 39000 × g for 20 min at 4 °C. Washed pellets were solubilized with 100 mL of 2 M ammonium hydroxide under stirring for 6 h at rt and then dialyzed against 20 mM MOPS buffer (pH 7.0) containing 20 mM CaCl2 and 0.5 M NaCl at 4 °C. Soluble recombinant hMGL was purified by affinity chromatography on a column of homemade lactose-Sepharose 4B, as described previously.21,27
Lectin Purity and Structural Status
Gel electrophoresis and analysis were performed using one- and two-dimensional separation. Isoelectric focusing was conducted on a ZOOMIPGRunner system (Invitrogen, Carlsbad, CA) with ZOOM strips (pI 4–7 and 3–10). Focusing conditions for the pI 4–7 strips were 175 V for 15 min, 175–2000 V for 45 min, and 2000 V for 105 min. For the pI 3–10 strips, conditions were 175 V for 15 min, 175–2000 V for 45 min, and 2000 V for 30 min. Strips were treated with 125 mM iodoacetamide for protein alkylation. SDS–PAGE under reducing conditions was carried out in an XCell SureLock electrophoresis chamber (Invitrogen) on a NuPAGE Novex 4% to 12% Bis-Tris ZOOM gel at 200 V.
To prepare the protein for gel filtration, lyophilized protein was dissolved in running phosphate-buffered saline (PBS) and 50 μL aliquots were chromatographed on a Superose-12HR10/30 column using an ÄKTApurifier 10 system with a flow rate of 0.5 mL/min at 4 °C. Protein elution was recorded at 280 nm. The column was calibrated with the following molecular weight markers: blue dextran (Mr > 2000 kDa), aldolase (Mr = 158 kDa), albumin (Mr = 67 kDa), ovalbumin (Mr = 44 kDa), chymotrypsinogen (Mr = 25 kDa), and vitamin B12 (Mr = 1.35 kDa).
Matrix-assisted laser desorption ionization (MALDI) time-of-flight (TOF) mass spectrometry (MS) was applied for molecular weight determination and peptide mass fingerprinting using trypsin and chymotrypsin in independent experiments, as described for lectin analysis previously.34−36 Briefly, each protein sample was dissolved in water to reach a final concentration of 4 μg/μL; for molecular mass determination with double-layer sinapinic acid as the matrix, the protein-containing sample was further diluted with 0.1% TFA [1:5 (v/v)]. For peptide fingerprinting, 5 μg of protein was separately digested with either 50 ng of trypsin or 50 ng of chymotrypsin. Digest mixtures were desalted with reversed phase ZipTipC-18 (Merck, Darmstadt, Germany), and samples spotted on a MALDI target plate with α-CHCA as a matrix for analysis from Bruker Daltonik (Bremen, Germany). Spotted samples were dried at ambient temperature prior to mass spectrometric analysis. MALDI mass spectra were collected on a RapifleX Tissue Typer instrument (Bruker Daltonik). FlexControl (version 3.4) was used for instrument control, and FlexAnalysis (version 3.4) for processing the data of the spectra. Annotated spectra were further analyzed with BioTools 3.2 (Bruker Daltonik). Enzyme specificity was set to that of trypsin or chymotrypsin, allowing also for cleavage N-terminally to proline residues and up to two missed cleavage sites, or to that of chymotrypsin allowing up to three missed cleavage sites. Carbamidomethylation (CAM) was set as the fixed modification, whereas oxidation of methionine (M) and N-terminal acetylation were considered as variable modifications.
ITC Experiments
Microcalorimetric measurements were performed using a MicroCal PEAQ-ITC calorimeter (Malvern) for GalNAc and (glyco)peptides and iTC200 (Microcal) for α-Me-GalNAc, β-Me-GalNAc, Thr-Tn, and β-Me-Glc. The ligand-containing solution was injected in 2 μL aliquots by a computer-controlled microsyringe at 150 s intervals between injections into the solution of lectin buffer (calorimetric cell volume of 200 μL) at 25 °C for a total of 19 injections while stirring at 750 rpm. Solutions of the monosaccharide and its derivatives were diluted to concentrations of 1.50 mM, while the solutions of (glyco)peptides were diluted to concentrations of 0.25–0.50 mM based on analytical RP-HPLC. Lectin concentrations were 11–50 μM as confirmed by measurements using a BioTek Epoch microplate spectrophotometer at λ = 280 nm, using the monomer ε = 22.9 value for a 1% (w/v) solution. The concentrations of the ligand and lectin for each experiment are provided in the Supporting Information along with the corresponding thermograms (Figures S28–S30). Solutions of both the ligand and the lectin (extracellular domain) were prepared in 10 mM HEPES sodium salt, 50 mM NaCl, and 2 mM CaCl2 (pH 7.4) in both deionized H2O and D2O (heavy water, >99.8 atom % D), as shown previously to be practical for human galectins-1 and -3.37 Thermodynamic analysis was performed using MicroCal PEAQ-ITC and Origin analysis software based on a one-set-of-sites model, and with the fitted offset parameter applied to each titration following the company’s guideline and previous applications.38−40 Thermograms, integrated heat values, and signature plot analysis of the thermodynamic bindings are provided in Figures S28–S30. Additionally, experimental ITC data were processed using NITPIC version 1.2.7 (biophysics.swmed.edu/MBR/software.html),41,42 fit into a 1:1 binding model using SEDPHAT version 15.2b (sedfitsedphat.nibib.nih.gov), and the output was documented graphically using GUSSI version 1.4.2 (biophysics.swmed.edu/MBR/software.html) (Figure S31).
Kinetic Parameters
The application of the KinITC software43 led to estimations of the off and on rate constants [koff and kon, respectively (Figures S32 and S33)]. The commercially available ITC analysis software AFFINImeter version 1.2.3 (affinimeter.com/site/download/affinimeter-itc-windows/) was used in our analysis.44 Dissociation and association rate constants were obtained from the time-dependent peak shapes determined experimentally by ITC.
Results and Discussion
Synthesis of Fmoc-Protected O-Glycosylated Thr-Based Building Blocks
The main challenge in the synthesis of the Tn antigen building block stems from the low yield and the achievement of stereoselective formation of the 1,2-cis-α-glycoside [4 (Scheme 1; for analytical data, see Figures S1–S8)]. A one-pot azidochlorination method31 was used to obtain the fully protected 2-azido-2-deoxy-galactopyranosyl chloride donor [3 (Scheme 1); 97% crude yield]. The use of the azidochlorinated carbohydrate moiety enables stereochemically controlled generation of the α-glycosidic bond due to the nonparticipating nature of the C-2 azido group and the ability of the chloride to act as the leaving group at the anomeric carbon.45 With access to sufficient quantities of the chloroazide donor (3), a modified Koenigs–Knorr reaction ensued upon incorporation of the Pfp ester of the Fmoc-protected Thr acceptor. To minimize formation of the β-anomer, the reaction was conducted in noncoordinating co-solvents, and the Fmoc-Thr-OPfp acceptor was used in a limiting amount (0.75 equiv). This strategy exploited the dual-purpose Pfp group, both as a C-terminal acid-protecting group and an activating group during SPPS. Pure α-glycoside was obtained by gradient elution with toluene and ethyl acetate (EtOAc) in silica gel [4 (Scheme 1); 71.7% α-anomer yield]. The assumption of formation of the α-O-glycosidic linkage was confirmed by measuring the characteristic anomeric doublet at 5.18 ppm (at 4.5 ppm expected for the β-proton) and 99.1 ppm for the anomeric carbon (Figures S3 and S4). Using a one-pot reductive acetylation approach in the presence of activated zinc powder, the O-glycosylated Thr azide (4) was converted into the acetamide. This step affords the desired Fmoc-protected Pfp ester of O-glycosylated Thr [5 (Scheme 1); 78.1% yield] after purification by silica gel chromatography. The anomeric purity of the building blocks (4 and 5) was confirmed by analytical RP-HPLC and 1H and 13C NMR spectroscopy, and the mass by MALDI-TOF MS (Figures S1–S8).
Scheme 1. Synthetic Procedures for Fmoc-Thr(Tn) Building Block 5 for Use in SPPS.

Reagents and conditions: (a) acetic anhydride, pyridine, DCM, rt, 16 h; (b) FeCl3, NaN3, H2O2, ACN, −45 °C, 6 h; (c) DIC, Pfp-OH, EtOAc, −15 °C, 4 h; (d) AgClO4, anhydrous DCM/toluene [1:1 (v/v)], −30 °C, 16 h; (e) zinc powder, acetic acid/acetic anhydride/THF [1:6:6 (v/v/v)], rt, 18 h.
MUC1-Based (Glyco)peptide Synthesis and Characterization
The three Thr residues in the MUC1 tandem repeat are acceptors for O-glycans. The peptide was used as a platform for the site-specific introduction of α-linked GalNAc to achieve all Thr-linked permutations for a systematic study. The panel of seven glycopeptides covering the range from single-site to fully glycosylated scaffolds was prepared by SPPS [7–13 (Table 1 and Figure S9)], together with the glycan-free control peptide (6). Fmoc-protected amino acids with HCTU/HOBt activation in equimolar quantities (4-fold excess) were used in an iterative process on an automated peptide synthesizer (Figure S9). The coupling of the orthogonally protected α-linked GalNAc building block (5) in a 1.5 equiv excess led to the three monoglycosylated peptides (7–9), the three diglycosylated peptides (10–12), and the single triglycosylated peptide (13) (Table 1 and Scheme 1). After completion of the (glyco)peptide chain synthesis, they were cleaved from the resin under acidic conditions (water/thioanisole/TFA), followed by complete removal of the O-acetyl (protecting) groups under basic conditions (0.1 M aqueous NaOH). The preparations were processed by RP-HPLC, and their purity was ascertained by analytical RP-HPLC and MALDI-TOF MS (Table 1 and Figures S10–S25).
Table 1. SPPS of MUC1 (Glyco)peptides and Their Characterization by MALDI-TOF MS and RP-HPLC.
| MALDI-TOF MS [M +
H]+a |
RP-HPLC | ||||
|---|---|---|---|---|---|
| entry | amino acid sequenceb | no. of sugars | expected (Da) | observed (Da) | tRc (min) |
| 6 | H-HGVTSAPDTRPAPGSTAPPA-NH2 | 0 | 1884.93 | 1885.65 | 17.42 |
| 7 | H-HGVT*SAPDTRPAPGSTAPPA-NH2 | 1 | 2089.25 | 2088.73 | 16.34 |
| 8 | H-HGVTSAPDT*RPAPGSTAPPA-NH2 | 1 | 2089.25 | 2088.49 | 16.85 |
| 9 | H-HGVTSAPDTRPAPGST*APPA-NH2 | 1 | 2089.25 | 2088.44 | 16.57 |
| 10 | H-HGVTSAPDT*RPAPGST*APPA-NH2 | 2 | 2292.45 | 2290.19 | 15.93 |
| 11 | H-HGVT*SAPDTRPAPGST*APPA-NH2 | 2 | 2292.45 | 2291.47 | 15.38 |
| 12 | H-HGVT*SAPDT*RPAPGSTAPPA-NH2 | 2 | 2292.45 | 2290.39 | 15.62 |
| 13 | H-HGVT*SAPDT*RPAPGST*APPA-NH2 | 3 | 2495.28 | 2493.69 | 15.11 |
The matrix used was α-cyano-4-hydroxycinnamic acid.
T* is a Thr O-linked Tn monosaccharide.
HPLC analysis conditions can be found in the Figures S10–S25.
As expected, upon performance of RP-HPLC, the control peptide (6) displayed the longest retention time (tR). The addition of a single (hydrophilic) glycan moiety at either Thr4, Thr9, or Thr16 (7, 8, or 9, respectively) resulted in a decrease in tR by 1 min (on average), followed by a further 1 min decrease for the diglycosylated peptides (10–12). The triglycosylated peptide (13) eluted from the column first, almost 3 min earlier than the nonglycosylated peptide (6) (Table 1). It is noteworthy that a separation among mono- or diglycosylated peptides that differ in the position of the glycan attachment site was detected (Table 1). Under these conditions, the tertiary structure of a peptide is assumed to be disrupted during RP-HPLC, either by the solvents used for elution and/or due to interaction of the peptide with the hydrophobic stationary material.46
With the (glyco)peptides (6–13) in hand, their secondary structure could be investigated by circular dichroism (CD) spectroscopy. No significant differences in the overall conformations were observed in either H2O or buffered D2O (Figure S26). A random coil with polyproline II (PPII) helical elements present was observed, as indicated by the spectral molar ellipticity ([θ]) minima at 198 nm and the increased maximum ([θ]max) intensity at 222 nm.47 It is noteworthy that the extent of the presence of structured elements positively correlates to GalNAc density (Figure S26). These findings correlate well with the corresponding decrease in tR on RP-HPLC (Table 1). This panel of glycopeptides could next be used as ligands for the endogenous lectin.
hMGL Lectin Purification and Characterization
The protein’s purity was assessed by one- and two-dimensional gel electrophoresis analyses (Figure 1A–C) using human galectin-3 (Figure 1A) and galectin-1 (Figure 1C) as internal standards due to the similar properties of the mass/isoelectric point as well as by mass spectrometry of intact and proteolytically cleaved material (Figure S27A–D). Gel filtration analysis (elution profiles shown in Figure 1D) revealed the typical asymmetrical peak so that, in line with previous work, these results “are consistent with the presence of a dissociating trimer”.48
Figure 1.
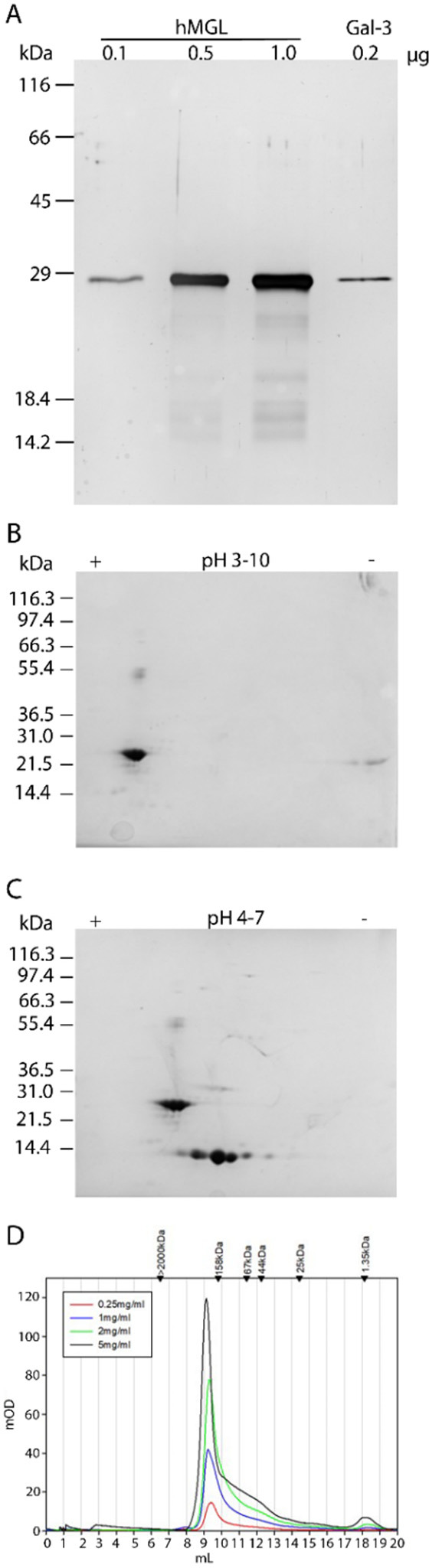
Characterization of hMGL by SDS–PAGE, two-dimensional (2D) gel electrophoresis, and gel filtration. (A) SDS–PAGE (15% polyacrylamide) of 0.1, 0.5, and 1.0 μg of hMGL as well as 0.2 μg of galectin-3 (protein with a similar mass and constitution with stalk and CRD). (B) 2D gel electrophoresis of 20 μg of hMGL in the pI range of 3–10. (C) 2D electrophoresis of a mixture of 20 μg of hMGL and 20 μg of galectin-1 in the pI range of 4–7. (D) Gel filtration of hMGL at the indicated concentrations (applied volume of 50 μL in each case).
ITC Titrations in H2O
As an internal reference for the work with the glycopeptides, ITC titrations were first performed in H2O with free GalNAc (Figure 2A), its α/β-methyl derivatives, and the Thr-GalNAc conjugate. In each case, titration reached full saturation with an n value consistently around 1, when n is normalized to the active cell concentration of the dissociating trimer. The KD values were in the range of 10 μM (Figure S29). Expectedly, there was no indication of a detectable affinity for methyl β-glucopyranoside used as a negative (specificity) control with hMGL. The α-methyl anomer (present in mucins) yielded an affinity that was 2-fold higher than that of its β-anomer (present in the disaccharide LacdiNAc) presented by N-glycan chains of cognate glycoproteins49 and the GalNAcα1-O-Thr conjugate (Figure S29). Binding of the monosaccharide, its methyl derivatives, and the conjugate with Thr was enthalpy driven in each case, a common feature of association of glycan to a lectin.
Figure 2.

Selected ITC binding curves, including thermographic profiles for interaction of (A) GalNAc (1.50 mM) with hMGL (29.6 μM), (B) MUC1-Thr16 (0.50 mM) with hMGL (22 μM), (C) MUC1-Thr4,16 (0.50 mM) with hMGL (20 μM), and (D) MUC1-Thr4,9,16 (0.25 mM) with hMGL (20 μM) in buffered H2O determined by ITC. Isotherms and thermograms were reproduced in GUSSI version 1.4.2 from the data obtained by MicroCal PEAQ-ITC software.
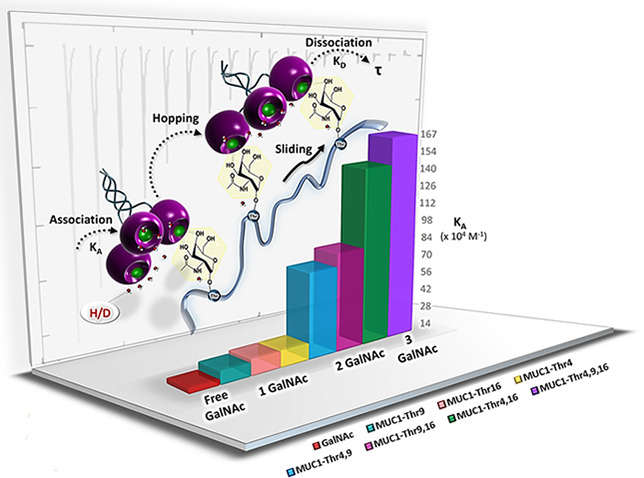
A panel of synthetic MUC1 glycopeptides were tested as ligands for hMGL. The derived stoichiometry (Table 2) revealed the functional valence of MUC1 glycopeptides, defined as the inverse of the obtained n value (1/n). For example, the n value for MUC1-Thr4,9,16 is determined to be 0.32, equating to a maximum of three glycans binding to the trimeric hMGL protein under saturation conditions.
Table 2. ITC Data for Association of hMGL with GalNAc and MUC1-Based Glycopeptides in Buffered H2O and D2Oa.
|
KA
(×104 M–1) |
ΔG (kcal mol–1) |
ΔH (kcal mol–1) |
Δ−(TΔS)
(kcal mol–1) |
n |
KD (μM) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ligand | entry | H2O | D2O | H2O | D2O | H2O | D2O | H2O | D2O | H2O | D2O | H2O | D2O |
| GalNAc | 5.64 | 13.79 | –6.49 | –7.01 | –11.30 | –6.81 | 4.81 | –0.20 | 0.99 | 0.97 | 17.70 | 7.25 | |
| MUC1-Thr4 | 7 | 14.66 | 26.88 | –7.05 | –7.41 | –11.40 | –12.50 | 4.38 | 5.13 | 0.99 | 1.00 | 6.82 | 3.72 |
| MUC1-Thr9 | 8 | 11.87 | 10.84 | –6.93 | –6.87 | –10.50 | –14.10 | 3.53 | 7.22 | 1.01 | 1.01 | 8.42 | 9.22 |
| MUC1-Thr16 | 9 | 14.47 | 16.75 | –7.05 | –7.13 | –10.00 | –14.18 | 2.95 | 7.63 | 1.03 | 1.01 | 6.91 | 5.97 |
| MUC1-Thr9,16 | 10 | 81.30 | 263.15 | –8.07 | –8.76 | –24.00 | –19.20 | 16.00 | 10.40 | 0.46 | 0.49 | 1.23 | 0.38 |
| MUC1-Thr4,16 | 11 | 144.92 | 263.15 | –8.41 | –8.76 | –22.80 | –24.60 | 14.30 | 15.80 | 0.48 | 0.50 | 0.69 | 0.38 |
| MUC1-Thr4,9 | 12 | 70.92 | 144.50 | –7.98 | –8.41 | –20.70 | –15.60 | 12.70 | 7.22 | 0.50 | 0.50 | 1.41 | 0.69 |
| MUC1-Thr4,9,16 | 13 | 166.67 | 338.98 | –8.49 | –8.91 | –30.30 | –28.50 | 21.80 | 19.50 | 0.32 | 0.32 | 0.60 | 0.29 |
Errors in ΔH ranged between ±0.01 and 1.12 kcal mol–1 and between ±0.01 and 0.61 μM/experiment for KD. The error values and concentrations of the ligand and lectin for each experiment are provided in the Supporting Information along with the corresponding thermograms (Figures S28–S30).
When proceeding to performing thermodynamic analysis of binding the glycopeptide to hMGL, we determined the glycan-free peptide lacks affinity for the lectin. The presence of a GalNAc residue was essential for binding (Table 2, Figure 2B–D, and Figure S29). The affinity increased from a KD value of approximately 16–17 μM for free GalNAc and the GalNAcα1-O-Thr conjugate to 7–8 μM for the monoglycosylated peptides. Evidently, the peptide backbone presence matters but the nature of the microenvironment of each GalNAc residue (at Thr4, Thr9, or Thr16) has only a minor influence under these conditions.
Moving from monovalent to bivalent and trivalent glycopeptides, the affinity increased markedly to KD values of ≤1 μM. The magnitude of this effect was calculated by the affinity enhancement factor β using eq 1.50
| 1 |
The highest enhancement value (β) was obtained for the triglycosylated MUC1-Thr4,9,16 peptide (13) (Table 3). The three individual binding steps exhibit negative cooperativity evidenced by cooperativity coefficient (α) values of <1 (Table 3). Upon characterization of the thermodynamics of glycopeptide binding with stepwise increases in valency from one to three, proportional enhancement of ΔH, increased negative cooperativity, and increased entropic penalties were observed (Tables 2 and 3).
Table 3. Enhancement Factors (β) and Cooperativity Coefficients (α) for Multivalent MUC1 Glycopeptidesa.
| ligand | entry | β | α |
|---|---|---|---|
| MUC1-Thr9,16 | 10 | 6.17 | 0.57 |
| MUC1-Thr4,16 | 11 | 9.94 | 0.59 |
| MUC1-Thr4,9 | 12 | 5.34 | 0.57 |
| MUC1-Thr4,9,16 | 13 | 12.20 | 0.40 |
β enhancement factor values are calculated using the equation β = KAmulti/KA. β measures the increase in the affinity of each ligating unit obtained by its multivalent presentation. Cooperativity values (α) are calculated using the equation ΔGmul,n = αnΔGmono. An α of >1 indicates positive cooperativity in the multivalent interaction, and an α of <1 indicates negative cooperativity in the multivalent interaction.50 The ΔGmono values were calculated by averaging the respective ΔG values of the glycan regions being evaluated.
In terms of drawing an analogy, the spatial presentation of each GalNAc moiety of a single glycopeptide can be viewed to resemble that of terminal contact sites of a triantennary N-glycan (Gal/GalNAc) binding to the CRDs of the trimeric ASGPR.51 The approximate distances between amino acids Thr4 and Thr9 (15.5 Å), Thr9 and Thr16 (21.7 Å), and Thr4 and Thr16 (37.2 Å) calculated from known PPII amino acid distances52 mimic that of the triantennary N-glycan, showing that glycoclusters can avidly interact with hMGL, as also revealed by measuring respective IC50 values.53
We further analyzed our ITC data by the KinITC method.43 Dissociation and association rate constants (koff and kon, respectively) were derived from the ITC titration curve data. The applicability of the method has recently been documented experimentally by running surface plasmon resonance (SPR) experiments in parallel to ITC to study the thermodynamics and kinetics of binding of mannoside to the fimbrial adhesin FimH.54 In our study, a fit of the kon and koff rates was not possible across the entire set of data (Table 4). Previously published data using SPR had already disclosed this problem for the case of Tn, sTn, and Neu5Gc-Tn.22 The occurrence of this phenomenon may suggest that the free sugar and the monovalent glycopeptides MUC1-Thr4 and MUC1-Thr9 have relatively high dissociation rates (koff), so that ligand–hMGL complexes will have very short lifetimes (τ), defined as the reciprocal of koff. Fittingly, we were able to record a kon rate of 0.096 × 106 M–1 s–1 for binding of hMGL to the monoglycosylated MUC1-Thr16 peptide (Table 4 and Figure S32).
Table 4. Kinetic Parameters and Lifetime Calculations of GalNAc and Glycopeptides in Buffered H2O When Binding to hMGLa.
| ligand | entry | kon (×106 M–1 s–1) | koff (s–1) | τ (s) |
|---|---|---|---|---|
| GalNAc | – | – | – | |
| MUC1-Thr4 | 7 | – | – | – |
| MUC1-Thr9 | 8 | – | – | – |
| MUC1-Thr16 | 9 | 0.096 | 0.600 | 1.67 |
| MUC1-Thr9,16 | 10 | 0.032 | 0.049 | 20.41 |
| MUC1-Thr4,16 | 11 | 0.058 | 0.051 | 19.61 |
| MUC1-Thr4,9 | 12 | 0.016 | 0.060 | 16.67 |
| MUC1-Thr4,9,16 | 13 | 0.026 | 0.019 | 52.63 |
Kinetic information was calculated from experimental ITC data using AFFINImeter KinITC software.44 Lifetimes were calculated using the equation: lifetime (τ) = 1/koff. The standard errors of koff and kon values and the equilibration time curves can be found in Figure S32.
In this context, upon examination of a member of another class of C-type lectins, association rate constants in the range of 105–106 M–1 s–1 have been reported for selectin binding to in vivo ligands, i.e., L-selectin to P-selectin glycoprotein ligand-1 (PSGL-1, CD162).55 Also of interest for comparison, the dissociation rate koff of 0.060 s–1 for the MUC1-Thr16 glycopeptide–hMGL complex is similar to koff values obtained for binding using optical tweezers56 and indicates a longer lifetime (τ = 1.67 s) in comparison to those of GalNAc and the two other monoglycosylated peptides. In our experimental series, the triglycosylated MUC1 peptide (13) exhibited the lowest dissociation rate (koff = 0.019 s–1) and the longest lifetime of all complexes (τ = 52.63 s) (Table 4).
In overview, the lifetimes ranged from 1.67 s for the monoglycosylated (7–9) and 16.67–20.41 s for diglycosylated (10–12) to the mentioned value of 52.63 s for the triglycosylated MUC1 peptide (13). To better analyze the solvent contribution to thermodynamics, the measurements in H2O were supplemented by work in D2O, especially important in resolving cases of extended ligand structures (peptide portion), beyond the direct contact site contributing to binding.
ITC Titrations in D2O
The thermodynamic parameters in D2O are listed in Table 2 and shown in Figure S30. They were then further used to calculate ΔΔH, Δ−(TΔS), and ΔΔG values (Table 5). Equation 2 was applied to examine the occurrence of deuterium isotope effects (DIEs). Considering a 10% change in the energy for a hydrogen bond in D2O versus H2O, this energy difference helps estimate the contribution of solvent reorganization to the observed enthalpy of binding.28,29,57
| 2 |
Compensation between ΔΔH and Δ−(TΔS) afforded a small net change in ΔΔG (Table 5). The enthalpy of binding for the free glycan to hMGL was less favorable in D2O (ΔΔH = −4.49 kcal mol–1). This suggests a significant contribution of solvent reorganization to the enthalpy of binding. Such a scenario had previously been seen for the leguminous lectin concanavalin A, in which Ca2+ does not directly contact the sugar but assists the amino acids in acquiring the suited orientations for mannose binding.28,29 The data are the first results of a human tissue lectin employing Ca2+ by engaging in coordination bonds for ligand binding.
Table 5. DIE Parameters of Association for GalNAc and Glycopeptides in H2O versus D2O (DIE = H2O – D2O) with hMGL.
| ligand | entry | ΔΔG (kcal mol–1) | ΔΔH (kcal mol–1) | Δ−(TΔS) (kcal mol–1) |
|---|---|---|---|---|
| GalNAc | 0.52 | –4.49 | 5.01 | |
| MUC1-Thr4 | 7 | 0.36 | 1.10 | –0.75 |
| MUC1-Thr9 | 8 | –0.06 | 3.60 | –3.69 |
| MUC1-Thr16 | 9 | 0.08 | 4.18 | –4.68 |
| MUC1-Thr9,16 | 10 | 0.69 | –4.80 | 5.60 |
| MUC1-Thr4,16 | 11 | 0.35 | 1.80 | –1.50 |
| MUC1-Thr4,9 | 12 | 0.43 | –5.10 | 5.48 |
| MUC1-Thr4,9,16 | 13 | 0.42 | –1.80 | 2.30 |
When proceeding from using the free sugar to applying glycopeptides, we found the H/D exchange in solvent for the monoglycosylated peptides (7–9) resulted in more favorable enthalpic contributions, accompanied by compensating higher entropic penalties. Evidently, the presence of the peptide backbone and the site of glycan presentation affected solvation differently. The nature of the microenvironment matters. The enthalpy component of MUC1-Thr16 (9) was most affected by the isotope substitution (ΔΔH = 4.18 kcal mol–1) (Table 5). Thermodynamic parameters of binding the diglycosylated MUC1-Thr4,16 glycopeptide (11) complement the trend seen with the monoglycosylated MUC1 peptides (Table 5). The ΔΔG values for the MUC1 glycopeptides (7–13) do not significantly change in H2O or D2O due to the equally compensating behavior observed in −TΔS values (Table 5). The observed variations in the enthalpy component of the DIE further suggest that not only the entire peptide backbone but also the glycan density and the nature of the glycan vicinity impact solvation when in contact with hMGL.
When processing the data to obtain kinetic values, we found an overall increase in the lifetime for lectin–glycopeptide complexes in D2O (Figure S33) over that in H2O (Table 4). Such an increased lifetime may reflect a slower proton transfer rate between the ligand bound to the lectin than when free in the solvent.58 This is most evident in the case of the diglycosylated glycopeptide MUC1-Thr4,16 (11).
Overall, the enthalpy–entropy compensation (EEC) plot in D2O displays a linear relationship with a slope near unity (Figure 3B). The data imply that solvent reorganization is a key factor of binding when coordination bonds are involved, and this also has been shown to be effective for antibody–antigen binding.59 In comparison to the EEC in water (Figure 3A), similar values near unity between the two isosteres are indicative of comparable binding mechanisms operative in both systems. Furthermore, the (compensation) plot of Δ−(TΔS) (H2O–D2O) against ΔΔH (H2O–D2O) shows an excellent fit to the regression line with a near unity slope (−0.92) and a very small intercept (0.20 kcal mol–1) with a correlation coefficient of 0.997 (Figure 3C). These results suggest that the discrepancy in conformational/structural changes taking place in the two solvents is likely minor, and that the (de)hydration processes appear to be a notable source of variety for thermodynamic parameters in this system.
Figure 3.

Left and middle plots showing enthalpy–entropy compensation for hMGL–glycopeptide interaction in (A) H2O and (B) D2O. The enthalpy and entropy from each of the individual experiments for the glycopeptides binding with hMGL are plotted. Numbered points indicate individual measurements (see Table 2 for point identification). A linear fit to all of the data points yielded lines with slopes of −1.077 and −1.131 for the glycopeptides in H2O and D2O, respectively. Panel C shows transfer between the two solvents. Enthalpy–entropy compensation for the transfer from H2O to D2O yielded a line with a slope of −0.92.
Conclusions
Synthesis of the MUC1 glycopeptide panel, based on the tandem-repeat motif, allowed for the thermodynamics of their interactions with hMGL to be determined in a systematic manner. An increased affinity, reaching KD values below 1 μM with negative cooperativity and with enthalpy–entropy compensation, was determined upon bi- and triglycosylation. KinITC analysis of the ITC data revealed notable decreases in koff rates with increases in valency. Lifetimes of the hMGL-ligand complexes reached 52.63 s for the triglycosylated MUC1 peptide (13) in comparison to 1.67 s for the monoglycosylated (at Thr16) peptide. This is in line with a “bind and jump” mechanism originally proposed for binding of proteins to DNA and leguminous lectin binding to mucin or fragments thereof.60 The noted decrease in the affinity of free (monomeric) CRD and the gradient change in affinity in correlation with glycan density measured by fluorescence polarization equilibrium binding assays20 relative to trimeric hMGL also argue along this line and call for calorimetric analysis with the CRD, in both H2O and D2O. Interestingly, in comparison to interaction analysis with leguminous lectins in D2O,28 the enthalpy contribution on DIE for MUC1 glycopeptides as ligands varied. These results suggest that features of the peptide backbone likely come into play and impact solvation beyond the cognate sugar for hMGL. Testing sTn as a ligand will broaden the analysis of this aspect. Of relevance in this context, extending the length of a canonical disaccharide ligand to two tetrasaccharides had recently revealed major solvent rearrangements to occur in the case of human galectins-1 and -3.37 Finally, testing these glycopeptides as antigens will allow comparative thermodynamic studies with antibodies to be performed.
Acknowledgments
The authors are thankful to Stoyan Milev (Malvern Panalytical) and Juan Sabin and Adrian Velazquez-Campoy (Affinimeter) for valuable discussions on data analysis software used in this study.
Glossary
Abbreviations
- MUC
mucin
- Tn
T antigen nouvelle
- sTn
sialyl-T antigen nouvelle
- hMGL
human macrophage galactose-type lectin
- PPII
polyproline II
- ITC
isothermal titration calorimetry
- CD
circular dichroism
- CRD
carbohydrate recognition domain
- SPPS
solid phase peptide synthesis
- Fmoc
fluorenyl methoxycarbonyl
- Pfp
pentafluorophenyl
- EtOAc
ethyl acetate
- EtOH
ethanol
- KD
dissociation constant
- D2O
deuterium oxide
- 1H
hydrogen-1
- 13C
carbon-13
- α-CHCA
α-cyano-4-hydroxycinnamic acid
- CDCl3
deuterochloroform
- δ
chemical shift
- J
spin–spin splitting
- RP
reverse phase
- DCM
methylene chloride
- AgClO4
silver perchlorate
- DI
deionized
- Na2SO4
sodium sulfate
- NaHCO3
sodium bicarbonate
- HOBt
hydroxybenzotriazole
- HCTU
O-(1H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- NMM
N-methylmorpholine
- DMF
dimethylformamide
- DIPEA
N,N-diisopropylethylamine
- NaOH
sodium hydroxide
- HCl
hydrochloric acid
- n
number of binding sites
- MTBE
methyl tert-butyl ether
- λ
wavelength
- H2O2
hydrogen peroxide
- DIPEA
N,N-diisopropylethylamine
- DIC
diisopropylcarbodiimide
- NaN3
sodium azide
- FeCl3
iron(III) chloride hexahydrate
- Ac2O
acetic anhydride
- NaCl
sodium chloride
- CaCl2
calcium chloride
- CuSO4
copper sulfate
- Pyr
pyridine
- AcOH
acetic acid
- OH
hydroxyl
- H/D
hydrogen/deuterium exchange
- DIE
deuterium isotope effect
- NMR
nuclear magnetic resonance
- RP-HPLC
reverse phase high-performance liquid chromatography
- MALDI-TOF MS
matrix-assisted laser desorption ionization time-of-flight mass spectroscopy
- koff
dissociation rate
- kon
association rate
- EEC
enthalpy–entropy compensation
- ΔΔH
ΔHH2O – ΔHD2O
- Δ−(TΔS)
–(TΔS)H2O – (TΔS)D2O
- ΔΔG
ΔGH2O – ΔGD2O
- ASGPR
asialoglycoprotein receptor
- FimH
fimbrial adhesin
- SPR
surface plasmon resonance
- PSGL-1
P-selectin glycoprotein ligand-1
- Neu5Gc
N-glycol(o)ylneuraminic acid
- β
affinity enhancement factor
- LacdiNAc
GalNAcβ1,4GlcNAc, N-[(3R,4R,5S,6R)-5-[(2S,3R,4R,5R,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-2,4-dihydroxy-6-(hydroxymethyl)oxan-3-yl]acetamide
- τ
lifetime
- DBA
Dolichos biflorus agglutinin
- SBA
soybean agglutinin
- HPA
Helix pomatia agglutinin
- EDTA
ethylenediaminetetraacetic acid
- cDNA
complementary DNA
- PCR
polymerase chain reaction
- TB
terrific broth
- IPTG
isopropyl β-d-thiogalactopyranoside
- TBS
Tris-buffered saline
- PMSF
phenylmethanesulfonyl fluoride
- NH4OH
ammonium hydroxide
- MOPS
3-(N-morpholino)propanesulfonic acid
- pI
isoelectric point
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- PBS
phosphate-buffered saline
- Mr
relative molar mass
- CAM
carbamidomethylation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00942.
RP-HPLC chromatograms, MALDI-TOF mass spectra, NMR spectra, CD spectra, ITC titration profiles, Affinimeter/KinITC data, and NITPIC/SEDPHAT data (PDF)
Accession Codes
hMGL, UniProtKB Q8IUN9.
Author Present Address
∥ F.G.F.: Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139.
Author Present Address
⊥ M.C.R.B.: Department of Biochemistry and Molecular Biology, Complex Carbohydrate Research Center, University of Georgia, 120 E. Green St., Athens, GA 30602.
Author Contributions
D.M.B. conducted the synthesis of the building blocks, synthesis of glycopeptides, and their CD analysis. E.R.M. assisted in building block synthesis. F.G.F., D.M.B., and M.C.R.B. conducted and analyzed the ITC experiments. D.M.B. analyzed the kinetic data. A.-K.L. and H.K. conducted lectin preparation. M.M. and J.K. performed purity assessments and gel filtration. M.C. and H.-J.G. supervised experiments and validated results. D.M.B., F.G.F., H.-J.G., and M.C. contributed in writing the manuscript.
This research was supported by start-up funds, Florida Atlantic University (FAU), to M.C., National Institutes of Health (NIH) Grant CA242351 to M.C., a FAU Graduate Research and Inquiry Program (GRIP) grant to D.M.B., and FAU OURI Undergraduate Research grants to F.G.F. and E.R.M. Support by COST Action CA18103 (InnoGly; to H.-J.G.) is also gratefully acknowledged.
The authors declare no competing financial interest.
Notes
All data generated or analyzed during this study are included in this published article (and its Supporting Information).
Supplementary Material
References
- Kaltner H.; Abad-Rodríguez J.; Corfield A. P.; Kopitz J.; Gabius H.-J. (2019) The sugar code: letters and vocabulary, writers, editors and readers and biosignificance of functional glycan-lectin pairing. Biochem. J. 476, 2623–2655. 10.1042/BCJ20170853. [DOI] [PubMed] [Google Scholar]
- Manning J. C.; Romero A.; Habermann F. A.; García Caballero G.; Kaltner H.; Gabius H.-J. (2017) Lectins: a primer for histochemists and cell biologists. Histochem. Cell Biol. 147, 199–222. 10.1007/s00418-016-1524-6. [DOI] [PubMed] [Google Scholar]
- Corfield A. (2017) Eukaryotic protein glycosylation: a primer for histochemists and cell biologists. Histochem. Cell Biol. 147, 119–147. 10.1007/s00418-016-1526-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabius H.-J.; Roth J. (2017) An introduction to the sugar code. Histochem. Cell Biol. 147, 111–117. 10.1007/s00418-016-1521-9. [DOI] [PubMed] [Google Scholar]
- Cummings R. D. (2019) Stuck on sugars - how carbohydrates regulate cell adhesion, recognition, and signaling. Glycoconjugate J. 36, 241–257. 10.1007/s10719-019-09876-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckwith D. M.; Cudic M. (2020) Tumor-associated O-glycans of MUC1: carriers of the glyco-code and targets for cancer vaccine design. Semin. Immunol. 47, 101389. 10.1016/j.smim.2020.101389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh S.; Gnanapragassam V. S.; Jain M.; Rachagani S.; Ponnusamy M. P.; Batra S. K. (2015) Pathobiological implications of mucin glycans in cancer: sweet poison and novel targets. Biochim. Biophys. Acta 1856, 211–225. 10.1016/j.bbcan.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corfield A. P. (2015) Mucins: a biologically relevant glycan barrier in mucosal protection. Biochim. Biophys. Acta, Gen. Subj. 1850, 236–252. 10.1016/j.bbagen.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Ju T.; Otto V. I.; Cummings R. D. (2011) The Tn antigen-structural simplicity and biological complexity. Angew. Chem., Int. Ed. 50, 1770–1791. 10.1002/anie.201002313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervoni G. E.; Cheng J. J.; Stackhouse K. A.; Heimburg-Molinaro J.; Cummings R. D. (2020) O-glycan recognition and function in mice and human cancers. Biochem. J. 477, 1541–1564. 10.1042/BCJ20180103. [DOI] [PubMed] [Google Scholar]
- Stowell S. R.; Ju T.; Cummings R. D. (2015) Protein glycosylation in cancer. Annu. Rev. Pathol.: Mech. Dis. 10, 473–510. 10.1146/annurev-pathol-012414-040438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Ju T.; Ding X.; Xia B.; Wang W.; Xia L.; He M.; Cummings R. D. (2010) Cosmc is an essential chaperone for correct protein O-glycosylation. Proc. Natl. Acad. Sci. U. S. A. 107, 9228–9233. 10.1073/pnas.0914004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju T.; Lanneau G. S.; Gautam T.; Wang Y.; Xia B.; Stowell S. R.; Willard M. T.; Wang W.; Xia J. Y.; Zuna R. E.; Laszik Z.; Benbrook D. M.; Hanigan M. H.; Cummings R. D. (2008) Human tumor antigens Tn and sialyl Tn arise from mutations in Cosmc. Cancer Res. 68, 1636–1646. 10.1158/0008-5472.CAN-07-2345. [DOI] [PubMed] [Google Scholar]
- Bennett E. P.; Mandel U.; Clausen H.; Gerken T. A.; Fritz T. A.; Tabak L. A. (2012) Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology 22, 736–756. 10.1093/glycob/cwr182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman J.; Guan Y.; Perrine C. L.; Gerken T. A.; Tabak L. A. (2012) UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyltransferases: completion of the family tree. Glycobiology 22, 768–777. 10.1093/glycob/cwr183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drickamer K.; Taylor M. E. (2015) Recent insights into structures and functions of C-type lectins in the immune system. Curr. Opin. Struct. Biol. 34, 26–34. 10.1016/j.sbi.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabius H.-J.; Kaltner H.; Kopitz J.; André S. (2015) The glycobiology of the CD system: a dictionary for translating marker designations into glycan/lectin structure and function. Trends Biochem. Sci. 40, 360–376. 10.1016/j.tibs.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Ozaki K.; Lee R. T.; Lee Y. C.; Kawasaki T. (1995) The differences in structural specificity for recognition and binding between asialoglycoprotein receptors of liver and macrophages. Glycoconjugate J. 12, 268–274. 10.1007/BF00731329. [DOI] [PubMed] [Google Scholar]
- Suzuki N.; Yamamoto K.; Toyoshima S.; Osawa T.; Irimura T. (1996) Molecular cloning and expression of cDNA encoding human macrophage C-type lectin. Its unique carbohydrate binding specificity for Tn antigen. J. Immunol. 156, 128–135. [PubMed] [Google Scholar]
- Iida S.; Yamamoto K.; Irimura T. (1999) Interaction of human macrophage C-type lectin with O-linked N-acetylgalactosamine residues on mucin glycopeptides. J. Biol. Chem. 274, 10697–10705. 10.1074/jbc.274.16.10697. [DOI] [PubMed] [Google Scholar]
- Marcelo F.; Garcia-Martin F.; Matsushita T.; Sardinha J.; Coelho H.; Oude-Vrielink A.; Koller C.; André S.; Cabrita E. J.; Gabius H.-J.; Nishimura S.; Jiménez-Barbero J.; Cañada F. J. (2014) Delineating binding modes of Gal/GalNAc and structural elements of the molecular recognition of tumor-associated mucin glycopeptides by the human macrophage galactose-type lectin. Chem. - Eur. J. 20, 16147–16155. 10.1002/chem.201404566. [DOI] [PubMed] [Google Scholar]
- Mortezai N.; Behnken H. N.; Kurze A. K.; Ludewig P.; Buck F.; Meyer B.; Wagener C. (2013) Tumor-associated Neu5Ac-Tn and Neu5Gc-Tn antigens bind to C-type lectin CLEC10A (CD301, MGL). Glycobiology 23, 844–852. 10.1093/glycob/cwt021. [DOI] [PubMed] [Google Scholar]
- Artigas G.; Monteiro J. T.; Hinou H.; Nishimura S. I.; Lepenies B.; Garcia-Martin F. (2017) Glycopeptides as targets for dendritic cells: exploring MUC1 glycopeptides binding profile toward macrophage galactose-type lectin (MGL) orthologs. J. Med. Chem. 60, 9012–9021. 10.1021/acs.jmedchem.7b01242. [DOI] [PubMed] [Google Scholar]
- Dam T. K.; Gerken T. A.; Cavada B. S.; Nascimento K. S.; Moura T. R.; Brewer C. F. (2007) Binding studies of α-GalNAc-specific lectins to the α-GalNAc (Tn-antigen) form of porcine submaxillary mucin and its smaller fragments. J. Biol. Chem. 282, 28256–28263. 10.1074/jbc.M704677200. [DOI] [PubMed] [Google Scholar]
- Dam T. K.; Gerken T. A.; Brewer C. F. (2009) Thermodynamics of multivalent carbohydrate-lectin cross-linking interactions: importance of entropy in the bind and jump mechanism. Biochemistry 48, 3822–3827. 10.1021/bi9002919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh Y.; Rodriguez Benavente M. C.; Al-Huniti M. H.; Beckwith D.; Ayyalasomayajula R.; Patino E.; Miranda W. S.; Wade A.; Cudic M. (2020) Positional scanning MUC1 glycopeptide library reveals the importance of PDTR epitope glycosylation for lectin binding. J. Org. Chem. 85, 1434–1445. 10.1021/acs.joc.9b02396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltner H.; Manning J. C.; García Caballero G.; Di Salvo C.; Gabba A.; Romero-Hernández L. L.; Knospe C.; Wu D.; Daly H. C.; O’Shea D. F.; Gabius H.-J.; Murphy P. V. (2018) Revealing biomedically relevant cell and lectin type-dependent structure-activity profiles for glycoclusters by using tissue sections as an assay platform. RSC Adv. 8, 28716–28735. 10.1039/C8RA05382K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam T. K.; Oscarson S.; Sacchettini J. C.; Brewer C. F. (1998) Differential solvation of “core” trimannoside complexes of the Dioclea grandiflora lectin and concanavalin A detected by primary solvent isotope effects in isothermal titration microcalorimetry. J. Biol. Chem. 273, 32826–32832. 10.1074/jbc.273.49.32826. [DOI] [PubMed] [Google Scholar]
- Chervenak M. C.; Toone E. J. (1994) A direct measure of the contribution of solvent reorganization to the enthalpy of binding. J. Am. Chem. Soc. 116, 10533–10539. 10.1021/ja00102a021. [DOI] [Google Scholar]
- Wang T.; Demchenko A. V. (2019) Synthesis of carbohydrate building blocks via regioselective uniform protection/deprotection strategies. Org. Biomol. Chem. 17, 4934–4950. 10.1039/C9OB00573K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner C.; Hofener M.; Sewald N. (2011) One-pot azidochlorination of glycals. Org. Lett. 13, 545–547. 10.1021/ol102750h. [DOI] [PubMed] [Google Scholar]
- Menges F. (2020) Spectragryph - optical spectroscopy software, ver. 1.2 (http://www.effemm2.de/spectragryph/).
- Sato M.; Kawakami K.; Osawa T.; Toyoshima S. (1992) Molecular cloning and expression of cDNA encoding a galactose/ N-acetylgalactosamine-specific lectin on mouse tumoricidal macrophages. J. Biochem. 111, 331–336. 10.1093/oxfordjournals.jbchem.a123758. [DOI] [PubMed] [Google Scholar]
- Kopitz J.; Vértesy S.; André S.; Fiedler S.; Schnölzer M.; Gabius H.-J. (2014) Human chimera-type galectin-3: Defining the critical tail length for high-affinity glycoprotein/cell surface binding and functional competition with galectin-1 in neuroblastoma cell growth regulation. Biochimie 104, 90–99. 10.1016/j.biochi.2014.05.010. [DOI] [PubMed] [Google Scholar]
- García Caballero G.; Flores-Ibarra A.; Michalak M.; Khasbiullina N.; Bovin N. V.; André S.; Manning J. C.; Vértesy S.; Ruiz F. M.; Kaltner H.; Kopitz J.; Romero A.; Gabius H.-J. (2016) Galectin-related protein: An integral member of the network of chicken galectins. 1. From strong sequence conservation of the gene confined to vertebrates to biochemical characteristics of the chicken protein and its crystal structure. Biochim. Biophys. Acta, Gen. Subj. 1860, 2285–2297. 10.1016/j.bbagen.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak M.; Warnken U.; André S.; Schnölzer M.; Gabius H.-J.; Kopitz J. (2016) Detection of proteome changes in human colon cancer induced by cell surface binding of growth-inhibitory human galectin-4 using quantitative SILAC-based proteomics. J. Proteome Res. 15, 4412–4422. 10.1021/acs.jproteome.6b00473. [DOI] [PubMed] [Google Scholar]
- Diercks T.; Medrano F. J.; FitzGerald F. G.; Beckwith D.; Pedersen M. J.; Reihill M.; Ludwig A.-K.; Romero A.; Oscarson S.; Cudic M.; Gabius H.-J. (2021) Galectin-glycan interaction: guideline for monitoring by 77Se NMR spectroscopy and solvent (H2O/D2O) impact on binding. Chem. - Eur. J. 27, 316–325. 10.1002/chem.202003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITC data analysis in Origin. Tutorial Guide, ver. 7 (2004) MicroCal.
- Kutzner T. J.; Gabba A.; FitzGerald F. G.; Shilova N. V.; García Caballero G.; Ludwig A.-K.; Manning J. C.; Knospe C.; Kaltner H.; Sinowatz F.; Murphy P. V.; Cudic M.; Bovin N. V.; Gabius H.-J. (2019) How altering the modular architecture affects aspects of lectin activity: case study on human galectin-1. Glycobiology 29, 593–607. 10.1093/glycob/cwz034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García Caballero G.; Beckwith D.; Shilova N. V.; Gabba A.; Kutzner T. J.; Ludwig A.-K.; Manning J. C.; Kaltner H.; Sinowatz F.; Cudic M.; Bovin N. V.; Murphy P. V.; Gabius H.-J. (2020) Influence of protein (human galectin-3) design on aspects of lectin activity. Histochem. Cell Biol. 154, 135–153. 10.1007/s00418-020-01859-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S.; Vargas C.; Zhao H.; Piszczek G.; Brautigam C. A.; Schuck P. (2012) High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 84, 5066–5073. 10.1021/ac3007522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann T. H.; Brautigam C. A. (2015) High-precision, automated integration of multiple isothermal titration calorimetric thermograms: new features of NITPIC. Methods 76, 87–98. 10.1016/j.ymeth.2014.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnouf D.; Ennifar E.; Guedich S.; Puffer B.; Hoffmann G.; Bec G.; Disdier F.; Baltzinger M.; Dumas P. (2012) KinITC: a new method for obtaining joint thermodynamic and kinetic data by isothermal titration calorimetry. J. Am. Chem. Soc. 134, 559–565. 10.1021/ja209057d. [DOI] [PubMed] [Google Scholar]
- Dumas P.; Ennifar E.; Da Veiga C.; Bec G.; Palau W.; Di Primo C.; Piñeiro A.; Sabin J.; Muñoz E.; Rial J. (2016) Extending ITC to kinetics with kinITC. Methods Enzymol. 567, 157–180. 10.1016/bs.mie.2015.08.026. [DOI] [PubMed] [Google Scholar]
- Nigudkar S. S.; Demchenko A. V. (2015) Stereocontrolled 1,2-cis glycosylation as the driving force of progress in synthetic carbohydrate chemistry. Chem. Sci. 6, 2687–2704. 10.1039/C5SC00280J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripet B.; Cepeniene D.; Kovacs J. M.; Mant C. T.; Krokhin O. V.; Hodges R. S. (2007) Requirements for prediction of peptide retention time in reversed-phase high-performance liquid chromatography: hydrophilicity/hydrophobicity of side-chains at the N- and C-termini of peptides are dramatically affected by the end-groups and location. J. Chromatogr. A 1141, 212–225. 10.1016/j.chroma.2006.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes J. L.; Miles A. J.; Whitmore L.; Wallace B. A. (2014) Distinct circular dichroism spectroscopic signatures of polyproline II and unordered secondary structures: applications in secondary structure analyses. Protein Sci. 23, 1765–1772. 10.1002/pro.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jégouzo S. A.; Quintero-Martínez A.; Ouyang X.; dos Santos Á.; Taylor M. E.; Drickamer K. (2013) Organization of the extracellular portion of the macrophage galactose receptor: A trimeric cluster of simple binding sites for N-acetylgalactosamine. Glycobiology 23, 853–864. 10.1093/glycob/cwt022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirro M.; Rombouts Y.; Stella A.; Neyrolles O.; Burlet-Schiltz O.; van Vliet S. J.; de Ru A. H.; Mohammed Y.; Wuhrer M.; van Veelen P. A.; Hensbergen P. J. (2020) Characterization of macrophage galactose-type lectin (MGL) ligands in colorectal cancer cell lines. Biochim. Biophys. Acta, Gen. Subj. 1864, 129513. 10.1016/j.bbagen.2020.129513. [DOI] [PubMed] [Google Scholar]
- Reynolds M.; Pérez S. (2011) Thermodynamics and chemical characterization of protein-carbohydrate interactions: the multivalency issue. C. R. Chim. 14, 74–95. 10.1016/j.crci.2010.05.020. [DOI] [Google Scholar]
- Lee R. T.; Lee Y. C. (2000) Affinity enhancement by multivalent lectin-carbohydrate interaction. Glycoconjugate J. 17, 543–551. 10.1023/A:1011070425430. [DOI] [PubMed] [Google Scholar]
- Adzhubei A. A.; Sternberg M. J. (1993) Left-handed polyproline II helices commonly occur in globular proteins. J. Mol. Biol. 229, 472–493. 10.1006/jmbi.1993.1047. [DOI] [PubMed] [Google Scholar]
- André S.; O’Sullivan S.; Koller C.; Murphy P. V.; Gabius H.-J. (2015) Bi- to tetravalent glycoclusters presenting GlcNAc/GalNAc as inhibitors: from plant agglutinins to human macrophage galactose-type lectin (CD301) and galectins. Org. Biomol. Chem. 13, 4190–4203. 10.1039/C5OB00048C. [DOI] [PubMed] [Google Scholar]
- Zihlmann P.; Silbermann M.; Sharpe T.; Jiang X.; Muhlethaler T.; Jakob R. P.; Rabbani S.; Sager C. P.; Frei P.; Pang L.; Maier T.; Ernst B. (2018) KinITC-one method supports both thermodynamic and kinetic SARs as exemplified on FimH antagonists. Chem. - Eur. J. 24, 13049–13057. 10.1002/chem.201802599. [DOI] [PubMed] [Google Scholar]
- Wild M. K.; Huang M. C.; Schulze-Horsel U.; van der Merwe P. A.; Vestweber D. (2001) Affinity, kinetics, and thermodynamics of E-selectin binding to E-selectin ligand-1. J. Biol. Chem. 276, 31602–31612. 10.1074/jbc.M104844200. [DOI] [PubMed] [Google Scholar]
- Hadjialirezaei S.; Picco G.; Beatson R.; Burchell J.; Stokke B. T.; Sletmoen M. (2017) Interactions between the breast cancer-associated MUC1 mucins and C-type lectin characterized by optical tweezers. PLoS One 12, e0175323 10.1371/journal.pone.0175323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toone E. J. (1994) Structure and energetics of protein-carbohydrate complexes. Curr. Opin. Struct. Biol. 4, 719–728. 10.1016/S0959-440X(94)90170-8. [DOI] [Google Scholar]
- Cioni P.; Strambini G. B. (2002) Effect of heavy water on protein flexibility. Biophys. J. 82, 3246–3253. 10.1016/S0006-3495(02)75666-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam T. K.; Torres M.; Brewer C. F.; Casadevall A. (2008) Isothermal titration calorimetry reveals differential binding thermodynamics of variable region-identical antibodies differing in constant region for a univalent ligand. J. Biol. Chem. 283, 31366–31370. 10.1074/jbc.M806473200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam T. K.; Gerken T. A.; Brewer C. F. (2009) Thermodynamics of multivalent carbohydrate-lectin cross-linking interactions: importance of entropy in the bind and jump mechanism. Biochemistry 48, 3822–3827. 10.1021/bi9002919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.