

Graphical abstract

Keywords: COVID-19, SARS-CoV-2, NiRAN, Oleuropein
Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA-dependent RNA polymerase (RdRp) is a promising target for antiviral drugs. In this study, a chemical library (n = 300) was screened against the nidovirus RdRp-associated nucleotidyltransferase (NiRAN) domain. Blind docking was performed using a selection of 30 compounds and nine ligands were chosen based on their docking scores, safety profile, and availability. Using cluster analysis on a 10 microsecond molecular dynamics simulation trajectory (from D.E. Shaw Research), the compounds were docked to the different conformations. On the basis of our modelling studies, oleuropein was identified as a potential lead compound.
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) belongs to the betacoronavirus genus and is the infectious agent that causes coronavirus disease 2019 (COVID-19) [1]. To date, there are seven coronaviruses that can infect humans and they are divided into two groups [1]. The common human coronaviruses (hCoVs) generally cause a mild to moderate respiratory infection and include hCoV-229E, hCoV-NL63, hCoV-OC43, and hCoV-HKUI [1]. Additionally, three hCoVs have been reported to cause severe disease and they include severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV-2 [1]. These three coronaviruses have the capacity to cause lower respiratory infections, which can result in acute lung injury (ALI), acute respiratory distress syndrome (ARDS), and multiorgan failure [2]. The long-term consequences of infection continue to be investigated. The high transmissibility of SARS-CoV-2 and its emerging variants, has resulted in strict public health measures being implemented and a significant amount of attention has been placed on developing vaccines and investigating potential antiviral drugs.
Betacoronaviruses are enveloped viruses that consist of a positive-sense, single stranded RNA genome [1]. The SARS-CoV-2 genome consists of a replicase complex that is formed by the open reading frames, ORF1a and ORF1b [3]. These two ORFs encode the polyproteins pp1a and pp1ab, which are cleaved to produce the non-structural proteins (nsp1-16) [3]. The SARS-CoV-2 genome is also comprised of structural and accessory genes. The four major structural proteins are the spike protein (S), nucleocapsid protein (N), envelope protein (E), and membrane glycoprotein (M) [3]. The receptor binding domain of the spike protein attaches to the host cell receptor angiotensin-converting enzyme 2 (ACE2) and this interaction mediates SARS-CoV-2 infection [4].
Non-structural protein 12 (nsp12), which is also known as RNA-dependent RNA polymerase (RdRp), is a crucial component of the replication-transcription complex and catalyses the synthesis of RNA from RNA templates [5]. The RdRp interacts with proteins such as nsp7, nsp8, nsp9 and nsp13 to facilitate virus replication and transcription [5]. The structure of the SARS-CoV-2 RdRp has been determined and is comprised of a right-hand RdRp domain, a nidovirus RdRp-associated nucleotidyltransferase domain (NiRAN), an interface domain and an N-terminal β-hairpin [6]. The RdRp domain consists of the fingers, palm and thumb subdomains [6].
The SARS-CoV-2 RdRp has also been identified as an ideal target for antiviral drugs. Remdesivir and favipiravir are examples of prodrugs that are being tested for their ability to inhibit the RdRp, as they act as nucleoside analogues and are incorporated into the growing RNA chain [7]. This results in the termination of RNA synthesis. Remdesivir was the first drug to be approved by the U.S. Food and Drug Administration (FDA), despite the contradicting findings from the Solidarity Trial conducted by the World Health Organization [7]. The NiRAN domain has also been of interest and it is conserved in the Nidovirales [8]. This domain was first discovered in the RdRp of the equine arteritis virus (EAV) and it was hypothesised to have RNA ligase activity, nucleotidyltransferase activity, and protein priming function [8]. The potential kinase or phosphotransferase activity of the NiRAN domain is also being investigated and more recently, its interaction with nsp9 has been explored [9].
In addition to developing drugs that inhibit the catalytic activity of the RdRp through covalently binding to the RNA template, the NiRAN domain could be a potential target site for therapeutic agents [10]. Drug repurposing will continue to play an integral role in combating infectious diseases and a number of studies have utilised computational methods to identify potential lead compounds from existing drugs [11]. In this study, molecular modelling tools were used to screen a library of 300 compounds against the NiRAN domain of the SARS-CoV-2 RdRp. This consisted of pharmacological compounds and natural compounds, with antiviral, antioxidant, and anti-inflammatory properties. As aforementioned, the NiRAN domain has nucleotidylating activity and adenosine diphosphate (ADP), uridine-5′-triphosphate (UTP), and guanosine-5′-triphosphate (GTP) were used as the control ligands. Based on the results, the library was narrowed down to 30 compounds. The potential lead ligands were subsequently identified through performing blind docking on the RdRp structures and molecular docking on several conformations of the RdRp from a 10 μs trajectory [12].
2. Materials and methods
2.1. Structures of the proteins and ligands
The cryo-electron microscopy (cryo-EM) structures of the SARS-CoV-2 RdRp were obtained from the RCSB Protein Data Bank (PDB ID: 6 M71 and 6XEZ) [6], [13], [14]. The RdRp chain was isolated from the replication-transcription complex, the waters and ligands were removed, and the relevant ions were retained. This included the zinc (Zn2+) ions in both structures. Adenosine diphosphate was the ligand present in the NiRAN domain of the 6XEZ cryo-EM structure and this was used as a control [13]. Likewise, UTP and GTP were used as control compounds. The chemical structures of ADP, UTP, GTP, and 300 ligands were obtained from the National Centre for Biotechnology Information (NCBI) PubChem Database [15]. If unavailable, the chemical structures were obtained from the ChEMBL Database [16]. The library of 300 ligands consisted of 220 phenolic compounds and 13 fatty acids from OliveNetTM [17]. A number of compounds (n = 63) with antimicrobial and anti-inflammatory properties were also utilised.
2.2. Preparation of the proteins and ligands
The cryo-EM structures of the RdRp protein were imported into Maestro and were prepared using the Protein Preparation Wizard of the Schrödinger Suite (version 2020–4) [18]. Similarly, the compounds were imported into Maestro and were prepared using the LigPrep tool. The default settings were utilised and the optimised potentials for liquid simulations (OPLS3e) force field was selected [19]. A receptor grid that was 20 × 20 × 20 Å in size was generated around the conserved residues of the NiRAN domain and they were K73, E83, R116, L119, T120, T123, T206, D208, N209, Y217, D218, G220, D221, and S236 [20]. The Glide Receptor Grid Generation protocol was used for this step.
2.3. Molecular docking using the Schrödinger Suite
The 300 ligands and controls were initially screened using the Glide Ligand Docking protocol. The Glide standard precision (SP) mode was selected for this process. The SP mode allows for compounds to be docked in a timely manner and is more accurate than the high throughput screening option. The promising ligand poses were refined using the OPLS3e force field and this was followed by post-docking minimisation.
Based on the results, 30 compounds were examined further. The ligands were docked to the NiRAN domain using the quantum-mechanics-polarised ligand docking (QPLD) protocol of the Schrödinger Suite for improved docking accuracy [21]. The compounds were initially docked using Glide and energy calculations were then performed on the protein–ligand complexes generated using ab initio quantum mechanics (QM) methods. The ligands were re-docked using the charges that were predicted by the QSite software and the poses were ranked in the final stage. The extra precision (XP) mode was chosen for the initial docking and redocking steps, and the QM level was set to accurate for the Jaguar component [22], [23], [24]. The GlideScore (kcal/mol) was recorded and the protein–ligand interactions were visualised using the Ligand Interaction Diagram tool.
2.4. Blind docking and binding site prediction
The P2Rank software package is a template-free tool that predicts ligand-binding sites based on machine learning and the SARS-CoV-2 RdRp cryo-EM structures were analysed using this program [25]. Blind docking was also performed with the selection of 30 compounds. The goal of blind docking was to investigate whether the ligands would preferentially bind to the NiRAN domain or any other site in the protein, which may potentially include an allosteric binding site. The structures of the proteins and compounds were imported into PyRx and they were prepared as macromolecules and ligands, respectively [26]. The protein was set as rigid, while all torsions of the ligands were activated. The receptor grid was generated around the entire protein and the exhaustiveness was increased to 2048. AutoDock Vina was used to perform the blind docking calculations and the jobs were run on Galileo, which is a cloud computing service (Hypernet Labs), and the Spartan High Performance Computing (HPC) system [27], [28], [29].
2.5. Cluster analysis of MD simulation trajectory
A 10 µs molecular dynamics (MD) simulation trajectory of the SARS-CoV-2 nsp7-nsp8-nsp12 RNA polymerase complex (PDB ID: 6 M71) was obtained from the D.E. Shaw Research group and analysed using the Gromacs 2018.2 software package with plug-ins for Visual Molecular Dynamics 1.9.3 [12], [30], [31], [32]. Nsp12 was isolated from the protein complex and analysed using root mean square deviation (RMSD) and root mean square fluctuation (RMSF) analysis tools included in Gromacs 2018.2. The Gromacs clustering tool gmx cluster was utilised to calculate clusters of similar structures based on RMSD of the protein. The gromos clustering algorithm, as described by Daura et al. [33], was applied. Cluster analysis was performed on the partially disordered N-terminal region of the protein (residues 30–120) for the entire trajectory, where the time interval between frames was 1.2 ns. Using an RMSD cut-off of 0.3 nm to define two structures as neighbours, 15 clusters were obtained. Representative protein structures for the top six clusters were extracted from the trajectory based on the median frame of each group for molecular docking of compounds to the NiRAN domain of RdRp.
2.6. Protein structure alignment
The Protein Structure Alignment tool in Maestro was used to align the NiRAN domain of the conformations that were representative of each cluster, using the cluster 1 structure as the reference. The NiRAN domain of the 6XEZ cryo-EM structure was also aligned to the conformation corresponding to cluster 1 for comparison. The RMSD values of the aligned amino acids were recorded.
3. Results and discussion
3.1. Molecular docking of 300 compounds to the NiRAN domain
In a study that was performed on the RdRp of the EAV, the nucleotidylation activity of the NiRAN domain was observed when UTP and GTP were present as substrates [8]. In a recent paper by Slanina et al. it was demonstrated that the coronavirus NiRAN domains could transfer nucleoside monophosphates to nsp9 and that there was relatively low specificity for a particular NTP substrate [9]. Residues K73, R116, T123, D126, D218 and F219 of the SARS-CoV-2 RdRp have previously been predicted to be essential for the enzymatic activity of the NiRAN domain, and multiple sequence alignment of coronavirus RdRp sequences has revealed that there are a number of conserved residues (Fig. 1 ) [20].
Fig. 1.

Structure of the SARS-CoV-2 RdRp. The cryo-EM structure of the apo RdRp (A) and the RdRp chain in complex with RNA (B) can be seen. The key active site residues in the NiRAN domain are labelled (C). The orientation of the ADP (red) present within the cryo-EM structure of the RdRp (PDB ID: 6XEZ) has also been compared to the docked ADP (blue) (D).
Using the Glide Ligand Docking protocol, 300 compounds were screened against the NiRAN domain of the SARS-CoV-2 RdRp (PDB ID: 6 M71) (Table S1). Lucidumoside C was the flavonoid compound that had the weakest binding affinity and the GlideScore was −0.4 kcal/mol. Delphinidin was predicted to be the strongest binding ligand and the GlideScore was found to be −7.2 kcal/mol. In addition to the library of 300 ligands, ADP, UTP and GTP were used as the control compounds. The GlideScores of these compounds were −6.3, −6.3, and −6.0 kcal/mol, respectively.
Based on this initial screen, 30 compounds with a broad range of binding affinities were selected for further analysis. This allowed for comparison of a range of ligands to ensure that there was a reasonable agreement between the ranking according to binding affinities. The commercial availability, approval by the FDA, and known side effects of these compounds were also taken into consideration. They included protease inhibitors, antibiotics, kinase inhibitors, nucleoside analogues, dietary compounds, and compounds with antioxidant and anti-inflammatory properties. For improved docking accuracy and further refinement, the 30 ligands and control compounds were subsequently docked to the NiRAN domain using the QPLD protocol. The GlideScores ranged from −3.6 to −10.8 kcal/mol and the chemical structures of these ligands are provided in Table 1 . In order to evaluate whether the controls and selected compounds would preferentially bind to the active site of the NiRAN domain, blind docking was performed on the cryo-EM structure of the SARS-CoV-2 RdRp. For the 6 M71 structure, 20 ligands had poses within the NiRAN domain (Table S2).
Table 1.
The selection of 30 compounds that were docked to the NiRAN domain are provided, along with their GlideScores (kcal/mol) (PDB ID: 6 M71). The control compounds are also included.
| Description | Ligand | GlideScore (kcal/mol) | Structure |
|---|---|---|---|
| Protease inhibitors | Indinavir: FDA approved | −7.4 |  |
| Saquinavir: FDA approved | −6.5 | 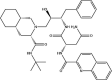 |
|
| Nelfinavir: FDA approved | −5.7 | 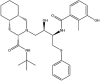 |
|
| Ritonavir: FDA approved | −5.1 |  |
|
| Lopinavir: FDA approved | −4.9 | 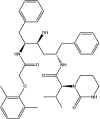 |
|
| Darunavir: FDA approved | −4.4 | 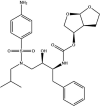 |
|
| Antibiotics | Amikacin: FDA approved | −10.2 |  |
| Tobramycin: FDA approved | −10.1 |  |
|
| Ceftriaxone: FDA approved | −6.0 |  |
|
| Ertapenem: FDA approved | −5.5 |  |
|
| Cefotaxime: FDA approved | −4.2 |  |
|
| Ceftazidime: FDA approved | −3.8 |  |
|
| Cefuroxime: FDA approved | −3.6 |  |
|
| Kinase inhibitors | Sunitinib: FDA approved | −5.0 |  |
| Ibrutinib: FDA approved | −4.4 |  |
|
| Zanubrutinib: FDA approved | −4.3 |  |
|
| Sorafenib: FDA approved | −4.2 |  |
|
| Acalabrutinib: FDA approved | −3.9 |  |
|
| Dietary compounds: Antioxidant and anti-inflammatory activities | Hellicoside: Phenolic compound | −10.8 |  |
| Rutin: Phenolic compound | −9.8 |  |
|
| Oleuropein: Phenolic compound | −8.8 | 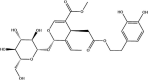 |
|
| Epicatechin gallate: Polyphenol | −8.0 | 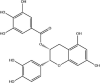 |
|
| Epigallocatechin gallate: Polyphenol | −7.6 |  |
|
| Cyanidin-3-O-glucoside: Phenolic compound | −7.2 | 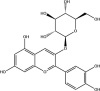 |
|
| Hypericin: Orphan drug designation for the synthetic form of hypericin (SGX301) | −5.8 |  |
|
| Curcumin: Polyphenol | −5.5 | 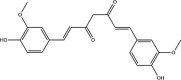 |
|
| RdRp inhibitors | Remdesivir: FDA approved | −6.5 | 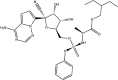 |
| Anti-inflammatory agents | SRT1720: Experimental drug – sirtuin activator | −5.2 |  |
| Sulfasalazine: FDA approved | −4.1 |  |
|
| SRT2104: Experimental drug – sirtuin activator | −4.0 |  |
|
| Controls | UTP | −10.0 | 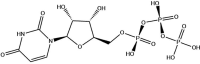 |
| GTP | −7.2 | 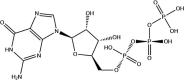 |
|
| ADP | −7.0 | 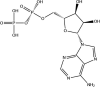 |
In a study by Dwivedy et al. the NiRAN domain was found to assume a kinase-like fold and is thought that this region may have pseudokinase or phosphotransferase activity [20]. The motif search also predicted the presence of kinase-like motifs and to explore this further, they docked broad specificity kinase inhibitors to the active site of the NiRAN domain [20]. Sunitinib and sorafenib were predicted to interact with aspartate residues, while SU6656 formed a hydrogen bond with K73 [20]. Through using an ADP-Glo Kinase assay kit, Dwivedy et al. were also able to provide evidence that the SARS-CoV-2 RdRp had intrinsic kinase/phosphotransferase like activity and that the kinase inhibitors significantly reduced its kinase-like activity [20]. Sunitinib, ibrutinib, zanubrutinib, sorafenib and acalabrutinib were the kinase inhibitors examined in the current study and when examining the protein–ligand interactions, it was apparent that the ligands also formed hydrogen bonds with negatively charged aspartate residues in the NiRAN domain (Table S3). It is important to note that kinase inhibitors may provide clinical benefit by exerting dual antiviral and anti-inflammatory effects, and the potential side-effects should also be taken into consideration [34].
Furthermore, the antiretroviral protease inhibitors that are used to treat patients with human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS) have been of interest [35]. Lopinavir, for example, is an inhibitor of the SARS-CoV main protease (Mpro) and in vitro studies have shown that this drug has inhibitory activity against SARS-CoV, SARS-CoV-2, and MERS-CoV [36]. Lopinavir is commonly used in combination with ritonavir, and these inhibitors have been tested in patients admitted to hospital with COVID-19 [37]. Broad-spectrum antibiotics have also played a role in the drug repurposing process, as they may be used for the treatment of co-infections, and their mechanisms of action require further elucidation [38].
Several dietary compounds were also part of the 30 ligands to be selected, with rutin, hellicoside, oleuropein, and cyanidin-3-O-glucoside being phenolic compounds from the OliveNetTM database [17]. Curcumin, which is the major constituent of turmeric, as well as the catechins (epicatechin gallate and epigallocatechin gallate) are also classified as polyphenols [39]. Hypericin is classified as an anthraquinone derivative and is found in St. John’s Wort [40]. Over one-third of new molecular entities that are approved by the FDA are natural products and their derivatives, and numerous studies have focused on screening large libraries of phytochemicals against coronavirus proteins to identify potential lead compounds [41]. Various natural compounds have been examined for their ability to target the spike protein, Mpro, papain-like protease (PLpro), and RdRp, and further research is required to validate their antiviral effects and pharmacokinetic properties. Curcumin, piperine, demethoxycurcumin, glycyrrhizic acid, rutin, nicotiflorin, epigallocatechin-3-gallate, and theaflavin are natural compounds that have been identified as potential antiviral drugs against the SARS-CoV-2 RdRp based on in silico analysis [42], [43], [44].
3.2. Structural analysis of 10 µs MD simulation trajectory of RdRp protein complex
Due to the NiRAN domain being a flexible region of the RdRp, cluster analysis was performed on a 10 μs MD simulation trajectory of the RdRp protein complex that was made available by the D.E Shaw Research group. Ensemble docking uses MD simulations to generate conformations of the protein for docking calculations, aiming to reproduce the selection of ligands for specific protein conformations that form more thermodynamically favourable protein/ligand complexes [45]. Thus, the aim was to select a subset of conformations where a representative protein structure for each cluster could be used for further docking.
The average RMSD of nsp12 protein backbone was 0.47 nm over the duration of the trajectory. There was a slight fluctuation in backbone RMSD at approximately 3 µs before stabilising after 4.2 µs (Fig. 2 A). RMSF analysis (Fig. 2B) indicated that this may be attributed to flexibility in the partially disordered residues 30 – 120 encompassing the N-terminal region of nsp12. The most prominent peaks in this region are at residues D61 and D107 located on the outer loops of the protein with RMSF values of 0.90 nm. It is noted that due to the highly flexible nature of the N-terminal residues, the structure of this region was previously unable to be resolved in SARS-CoV nsp12 [46]. As this was the most flexible region of the protein in proximity to the proposed binding site, this region was selected for cluster analysis.
Fig. 2.

Analysis of 10 µs MD simulation trajectory of the SARS-CoV-2 nsp7-nsp8-nsp12 RNA polymerase complex. A) Root mean square deviation (RMSD) of nsp12 backbone after fitting to backbone for 10 µs trajectory. B) Root mean square fluctuation (RMSF) of nsp12 backbone. C) Cluster analysis of flexible N-terminal region of NiRAN domain in SARS-CoV-2 nsp12, where the six most prevalent clusters are highlighted in representative structures. The cluster number over time throughout the trajectory is represented as a heat map.
Cluster analysis was performed to identify the most prevalent conformations in the trajectory for further screening of compounds. Cut-off values were varied between 0.1 and 0.5 nm in increments of 0.1 nm, with clustering analysis performed for each of these values. Based on the distribution of structures captured by each group, a cut-off distance of 0.3 nm for the N-terminal protein was selected. 8334 frames of the trajectory were divided into 15 clusters. Representative structures from the six most prevalent structures were used as starting structures for docking with lead compounds. The majority (59.2%) of frames were assigned to cluster 1, followed by 20.7% to cluster 2. The remaining clusters captured: 6.9% (cluster 3), 5.5% (cluster 4), 4.6% (cluster 5), 1.4% (cluster 6) of frames. Clusters 7 to 15 each captured less than 0.5% of frames, and were thus excluded from analysis.
From the 10 µs trajectory of the SARS-CoV-2 nsp12, this N-terminal region is initially partially disordered, becoming folded into a stable ordered structure resembling the N-lobe fold of protein kinases in agreement with the same complex determined in the presence of a reducing agent [6], [12]. From the heatmap shown in Fig. 2C, the N-terminal residues at the beginning of the trajectory are in conformations consistent with clusters 4, 2, and 6 until approximately 3 µs. From this time point, the protein becomes stable in conformations corresponding to cluster 1, which becomes the most common structure for the remainder of the trajectory. Conformations assigned to cluster 5 emerge at approximately 5 µs, while conformations corresponding to cluster 3 occur 8 µs into the trajectory. It is inferred from this analysis that clusters 1, 5, and 3 may represent conformations of the stable ordered N-terminal region of nsp12. However, it is acknowledged that further analysis will be required to characterise this. For the purpose of the present manuscript, representative structures for each cluster were utilised for molecular docking.
3.3. Molecular docking to the representative conformations of the RdRp identified from cluster analysis
The selected 30 compounds and control ligands were docked to the NiRAN domain of the representative structure for cluster 1 (Table S4). Interestingly, the phenolic compounds rutin, oleuropein, and hellicoside from the OliveNetTM database were the top three ligands with the strongest binding affinities. The GlideScores of these ligands were −10.9, −10.0, and −9.9 kcal/mol, respectively. Rutin, oleuropein, and hellicoside predominantly formed hydrogen bonds with the residues of the NiRAN domain, and hellicoside also formed a π-π cation with the amino acid R116. The control compounds ADP, UTP, and GTP had GlideScores between −7.4 and −9.0 kcal/mol (Fig. 3 ).
Fig. 3.

Molecular docking results of nine compounds and the control ligands. A) Oleuropein, cefotaxime, hypericin, indinavir, sulfasalazine, nelfinavir, sunitinib, lopinavir and ritonavir were docked to the NiRAN domain of the conformation representative of cluster 1 from the 10 μs MD simulation trajectory. B) The docking results of ADP, GTP, and UTP can be seen for the NiRAN domain. The residues that were involved in intermolecular bonds with the ligands are labelled (hydrogen bonds: bold font, salt bridge: italics, π-π interaction: regular font, π-π cation: regular font and underline, hydrogen bonds and salt bridges: bold font and italics, salt bridge and π-π cation: regular font, underline, and italics). The GlideScores (kcal/mol) are provided. Polar residues are coloured blue, positively charged residues are coloured purple, negatively charged residues are coloured red, and hydrophobic residues are coloured green.
Blind docking was performed on the representative structure for cluster 1 using the control compounds and selection of 30 ligands (Table S5). Several poses of ADP (n = 3), UTP (n = 6), and GTP (n = 7) were predicted to be positioned in the NiRAN domain (Fig. 4 ). Conversely, the ligands sunitinib, tobramycin, hellicoside, rutin, SRT1720, and hypericin were predicted to bind away from this region and had no poses within the NiRAN site.
Fig. 4.

Blind docking results of nine compounds and the control ligands for the conformation representative of cluster 1 from the 10 μs MD simulation trajectory. A) The blind docking results for hypericin, oleuropein, lopinavir, sunitinib, cefotaxime, ritonavir, sulfasalazine, indinavir and nelfinavir are shown. B) The blind docking results for ADP, GTP, and UTP are also depicted. The number of poses that were present in the NiRAN domain are provided for each ligand. The NiRAN domain is coloured tan and RdRp chain is coloured silver.
In addition to the control compounds, nine ligands with a range of binding affinities were selected and were docked to the conformations of the NiRAN domain that were assigned to clusters 2 to 6 for comparison (Table S6). They were indinavir, ritonavir, nelfinavir, sulfasalazine, lopinavir, hypericin, oleuropein, cefotaxime, and sunitinib. There were differences in the GlideScores for each cluster, as well as the intermolecular bonds that were formed between the ligands and the protein residues. The amino acids that participated in hydrogen bond interactions (maximum distance of 2.8 Å) with each ligand are described in Table 2 .
Table 2.
Hydrogen bond interactions that were formed between the ligands and each conformation representative of the clusters identified from the 10 μs MD simulation trajectory.
| Ligands | Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | Cluster 6 |
|---|---|---|---|---|---|---|
| Indinavir |
|
|
|
|
|
|
| Ritonavir |
|
|
|
No H-bonds |
|
|
| Nelfinavir |
|
|
|
|
|
|
| Sulfasalazine |
|
|
|
|
|
|
| Lopinavir |
|
|
|
|
|
|
| Hypericin |
|
|
|
|
|
|
| Oleuropein |
|
|
|
|
|
|
| Cefotaxime |
|
|
|
|
|
|
| Sunitinib | No H-bonds |
|
|
|
|
|
| GTP |
|
|
|
|
|
|
| UTP |
|
No H-bonds |
|
|
|
|
| ADP |
|
No H-bonds |
|
|
|
|
When examining the intermolecular bonds that were present between the ligands and the protein structures for each cluster, it was evident that several residues formed part of the NiRAN domain and N-terminal β-hairpin structure. To compare this region in the representative conformations assigned to the clusters, protein structure alignment was performed using the cluster 1 structure as the reference. The RMSD value for cluster 2 was 2.315 Å, 1.970 Å for cluster 3, 2.313 Å for cluster 4, 1.903 Å for cluster 5, and 2.117 Å for cluster 6.
The RMSD values of the amino acids in the NiRAN domain and nearby β-hairpin were also evaluated. Greater RMSD values were observed for the residues in the conformations corresponding to clusters 2, 4 and 6. The larger RMSD values were mainly associated with residues K50 to Y69, and K103 to P112. The conformations corresponding to clusters 3 and 5 were found to be more similar to cluster 1. As aforementioned, cluster 1, 3 and 5 were prominent towards the end of the 10 μs MD simulation trajectory and may represent conformations of the stable ordered N-terminal region of the RdRp. Moreover, differences were observed in the RMSD values for several residues that formed intermolecular bonds with the ligands and this was more noticeable in the conformations corresponding to clusters 2, 4 and 6 (Table S7). The conformational changes that occur in this region over the course of the trajectory and the flexibility of some of the residues may consequently be contributing to the differences seen in the binding affinities of the compounds and intermolecular bonds that are formed.
Oleuropein was found to consistently bind strongly to the conformations corresponding to each cluster and was selected as a potential lead compound. The GlideScore for the cluster 1 structure was −10.0 kcal/mol and oleuropein predominantly formed hydrogen bonds with the protein residues including N209, D208, T206, V204, D221, and N52. Due to there being missing residues in the NiRAN domain of the 6 M71 structure that was originally obtained from the RCSB PDB, oleuropein and the control compounds were also docked to the RdRp chain of the cryo-EM replication-transcription complex that was determined by Chen et al (PDB ID: 6XEZ) [13]. When comparing the NiRAN domain of the 6XEZ cryo-EM structure to the conformation that was representative of cluster 1, the RMSD was found to be 1.945 Å and the RMSD values of the amino acids can be found in the Supplementary Information (Table S8). Oleuropein had a GlideScore of −8.1 kcal/mol and hydrogen bonds were present with residues R116, N52, N209, Y217, D218, and K73. Most notably, the hydroxyl groups of oleuropein were predominantly involved in hydrogen bonding. The GlideScores of GTP, ADP, and UTP were −8.1, −7.1, and −6.9 kcal/mol, respectively. The protein–ligand interactions of oleuropein and the control compounds can be seen in Fig. 5 .
Fig. 5.

Molecular docking and blind docking results for the 6XEZ cryo-EM structure. Oleuropein and the control compounds ADP, UTP, and GTP were docked to the NiRAN domain of the 6XEZ cryo-EM structure. The GlideScores (kcal/mol) are provided for each ligand. Blind docking was also performed and the number of poses that were found to be positioned in the NiRAN domain are provided (A-D).
The molecular docking results revealed that ADP formed hydrogen bonds with N209 and K50, salt bridges with K73, K50 and R116, as well as a π-π cation with R116. The ADP that was present in the cryo-EM structure formed a π-π interaction with H75, and salt bridges with K73, R116, and K50 (Fig. 1). Blind docking revealed that oleuropein had eight poses within the NiRAN domain, while ADP had seven poses in this region (Fig. 5, Table S9). Guanosine-5′-triphosphate and UTP had 11 poses and 10 poses positioned in the NiRAN domain, respectively. The 6 M71 and 6XEZ cryo-EM structures that were obtained from the RCSB PDB, and conformation of 6 M71 that was representative of cluster 1 from the 10 μs MD simulation trajectory were also examined using the P2Rank server. In addition to the NiRAN domain being identified as a potential ligand binding site, the results revealed that there were several other pockets that may be potential allosteric sites and this included the nsp12-nsp8 interface region (Table S10).
Oleuropein is the most prominent phenolic compound in Olea europaea and belongs to the secoiridoid subclass [47]. Studies have shown that oleuropein exhibits antiviral activity in vitro against respiratory syncytial virus and para-influenza type 3 virus [48]. The pharmacokinetic profile of oleuropein in humans needs to be investigated further and its use as a potential prophylactic and therapeutic agent has been discussed in the literature [47]. Oleuropein has been screened against SARS-CoV-2 protein targets using in silico and in vitro methods, namely the spike protein and cysteine proteases [49], [50]. In general, polyphenols are being investigated for their antiviral activity against SARS-CoV-2 using a combination of molecular modelling and classical experimental methods. A number of studies have previously examined the role of the hydroxyl groups in the antioxidant activity of polyphenols and structure–activity relationships should also be performed to explore the function of hydroxyl groups in the antiviral activity of these compounds.
4. Conclusion
Overall, molecular docking was used to screen 300 ligands against the NiRAN domain of the SARS-CoV-2 RdRp. A selection of 30 compounds were then further investigated by high stringency blind docking before a final selection of nine potential lead compounds. These were docked to different conformations of the NiRAN domain identified through cluster analysis of a 10 μs MD simulation trajectory. By careful consideration of all analyses, oleuropein was identified as a lead compound. Given that this compound is relatively well-known and investigated, its potential antiviral effects can be relatively easily investigated in vitro and in vivo.
Author contributions statement
TCK and AH conceptualized the aims and methodology and were involved in supervision. EP performed data analysis, data curation, and was involved in production of the first draft of the manuscript. JL was involved in data analysis and curation and was involved in production of the first draft of the manuscript. HYMH performed formal data analysis and validation. All authors contributed to editing and reviewing the manuscript.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Epigenomic Medicine Program (TCK) is supported financially by McCord Research (Iowa, USA), which has a financial interest in dietary compounds described in this work. However, there is no conflict of interest with respect to the inhibition of the SARS-CoV-2 RNA-dependent RNA polymerase. The remaining co-authors also have no conflicts of interest.
Acknowledgements
We would like to acknowledge intellectual and financial support by McCord Research (Iowa, USA). JL is supported by an Australian Government Research Training Program Scholarship. We are indebted to Alfonso Perez Escudero and the team at Crowdfight COVID-19 for enabling access to supercomputing facilities, and to Matthew Gasperetti and the team at Hypernet Labs; Galileo, for enabling cloud computing for this project. We thank the National Computing Infrastructure (NCI), and the Pawsey Supercomputing Centre in Australia (funded by the Australian Government). Further, we thank the Spartan High Performance Computing service (University of Melbourne), and the Partnership for Advanced Computing in Europe (PRACE) for awarding the access to Piz Daint, hosted at the Swiss National Supercomputing Centre (CSCS), Switzerland.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cplett.2021.138889.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Zhu Z., et al. From SARS and MERS to COVID-19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 2020;21(1):224. doi: 10.1186/s12931-020-01479-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yuki K., Fujiogi M., Koutsogiannaki S. COVID-19 pathophysiology: A review. Clin. Immunology (Orlando Fla.) 2020;215:108427. doi: 10.1016/j.clim.2020.108427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khailany R.A., Safdar M., Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020;19 doi: 10.1016/j.genrep.2020.100682. p. 100682-100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lan J., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 5.Yan L., et al. Cryo-EM structure of an extended SARS-CoV-2 replication and transcription complex reveals an intermediate state in cap synthesis. Cell. 2021;184(1):184–193.e10. doi: 10.1016/j.cell.2020.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao Y., et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368(6492):779. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin W., et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020;368(6498):1499. doi: 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehmann K.C., et al. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015;43(17):8416–8434. doi: 10.1093/nar/gkv838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slanina H., et al. Coronavirus replication–transcription complex: Vital and selective NMPylation of a conserved site in nsp9 by the NiRAN-RdRp subunit. Proc. Natl. Acad. Sci. 2021;118(6) doi: 10.1073/pnas.2022310118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeong G.U., et al. Therapeutic strategies against COVID-19 and structural characterization of SARS-CoV-2: A review. Front. Microbiol. 2020;11(1723) doi: 10.3389/fmicb.2020.01723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahdian S., Ebrahim-Habibi A., Zarrabi M. Drug repurposing using computational methods to identify therapeutic options for COVID-19. J. Diabetes & Metabolic Disorders. 2020;19(2):691–699. doi: 10.1007/s40200-020-00546-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D.E. Shaw Research. Molecular dynamics simulations related to SARS-CoV-2. 2020; Available from: http://www.deshawresearch.com/resources_sarscov2.html..
- 13.Chen J., et al. Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex. Cell. 2020;182(6):1560–1573.e13. doi: 10.1016/j.cell.2020.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berman H.M., et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim S., et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47(D1):D1102–D1109. doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mendez D., et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47(D1):D930–D940. doi: 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.N.P. Bonvino, et al., OliveNet™: a comprehensive library of compounds from Olea europaea. Database: The J. Biol. Databases Curation, 2018. 2018: p. bay016. [DOI] [PMC free article] [PubMed]
- 18.Madhavi Sastry G., et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 19.Harder E., et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016;12(1):281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 20.A. Dwivedy, et al., Characterization of the NiRAN domain from RNA-dependent RNA polymerase provides insights into a potential therapeutic target against SARS-CoV-2. bioRxiv, 2021: p. 2021.02.03.429510. [DOI] [PMC free article] [PubMed]
- 21.Cho A.E., et al. Importance of accurate charges in molecular docking: quantum mechanical/molecular mechanical (QM/MM) approach. J. Comput. Chem. 2005;26(9):915–931. doi: 10.1002/jcc.20222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friesner R.A., et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 23.Bochevarov A.D., et al. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013;113(18):2110–2142. [Google Scholar]
- 24.Murphy R.B., Philipp D.M., Friesner R.A. A mixed quantum mechanics/molecular mechanics (QM/MM) method for large-scale modeling of chemistry in protein environments. J. Comput. Chem. 2000;21(16):1442–1457. [Google Scholar]
- 25.Krivák R., Hoksza D. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. J. Cheminformatics. 2018;10(1):39. doi: 10.1186/s13321-018-0285-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dallakyan S., Olson A. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. (Clifton N.J.) 2015;1263:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- 27.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hypernet Labs. Galileo. https://galileoapp.io/. 2020; Available from: https://galileoapp.io/.
- 29.L. Lafayette, et al., Spartan performance and flexibility; An hpc-cloud chimera, in OpenStack Summit 2016: Barcelona.
- 30.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91(1):43–56. [Google Scholar]
- 31.Abraham M.J., et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. [Google Scholar]
- 32.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1) doi: 10.1016/0263-7855(96)00018-5. pp. 33–8, 27–8. [DOI] [PubMed] [Google Scholar]
- 33.Daura X., et al. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Ed. 1999;38(1–2):236–240. [Google Scholar]
- 34.Weisberg E., et al. Repurposing of kinase inhibitors for treatment of COVID-19. Pharm. Res. 2020;37(9):167. doi: 10.1007/s11095-020-02851-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolcato G., et al. Targeting the coronavirus SARS-CoV-2: computational insights into the mechanism of action of the protease inhibitors lopinavir, ritonavir and nelfinavir. Sci. Rep. 2020;10(1):20927. doi: 10.1038/s41598-020-77700-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choy K.-T., et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Res. 2020;178:104786. doi: 10.1016/j.antiviral.2020.104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horby P.W., et al. Lopinavir-ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. The Lancet. 2020;396(10259):1345–1352. doi: 10.1016/S0140-6736(20)32013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ginsburg A.S., Klugman K.P. COVID-19 pneumonia and the appropriate use of antibiotics. The Lancet Global Health. 2020;8(12):e1453–e1454. doi: 10.1016/S2214-109X(20)30444-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khalil A., Tazeddinova D. The upshot of Polyphenolic compounds on immunity amid COVID-19 pandemic and other emerging communicable diseases: An appraisal. Natural Products and Bioprospecting. 2020;10(6):411–429. doi: 10.1007/s13659-020-00271-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobson J.M., et al. Pharmacokinetics, safety, and antiviral effects of hypericin, a derivative of St. John's wort plant, in patients with chronic hepatitis C virus infection. Antimicrob. Agents Chemother. 2001;45(2):517–524. doi: 10.1128/AAC.45.2.517-524.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patridge E., et al. An analysis of FDA-approved drugs: natural products and their derivatives. Drug Discovery Today. 2016;21(2):204–207. doi: 10.1016/j.drudis.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 42.Ogunyemi O.M., et al. Alkaloids and flavonoids from African phytochemicals as potential inhibitors of SARS-Cov-2 RNA-dependent RNA polymerase: an in silico perspective. Antiviral Chem. Chemother. 2020;28 doi: 10.1177/2040206620984076. 2040206620984076-2040206620984076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vardhan S., Sahoo S.K. In silico ADMET and molecular docking study on searching potential inhibitors from limonoids and triterpenoids for COVID-19. Comput. Biol. Med. 2020;124:103936. doi: 10.1016/j.compbiomed.2020.103936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh S., et al. Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2 via RNA-dependent RNA polymerase (RdRp) inhibition: an in-silico analysis. J. Biomol. Structure Dynamics. 2020:1–16. doi: 10.1080/07391102.2020.1796810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evangelista Falcon W., et al. Ensemble docking in drug discovery: how many protein configurations from molecular dynamics simulations are needed to reproduce known ligand binding? J. Phys. Chem. B. 2019;123(25):5189–5195. doi: 10.1021/acs.jpcb.8b11491. [DOI] [PubMed] [Google Scholar]
- 46.Kirchdoerfer R.N., Ward A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019;10(1):2342. doi: 10.1038/s41467-019-10280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.C. Nediani, et al., Oleuropein, a bioactive compound from Olea europaea L., as a potential preventive and therapeutic agent in non-communicable diseases. Antioxidants (Basel, Switzerland), 2019. 8(12): p. 578. [DOI] [PMC free article] [PubMed]
- 48.Ma S.-C., et al. In vitro evaluation of secoiridoid glucosides from the fruits of Ligustrum lucidum as antiviral agents. Chem. Pharm. Bull. 2001;49(11):1471–1473. doi: 10.1248/cpb.49.1471. [DOI] [PubMed] [Google Scholar]
- 49.Romeo A., Iacovelli F., Falconi M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020;286:198068. doi: 10.1016/j.virusres.2020.198068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.E. Pitsillou, et al., Identification of small molecule inhibitors of the deubiquitinating activity of the SARS-CoV-2 papain-like protease: in silico molecular docking studies and in vitro enzymatic activity assay 2020. 8(1171). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.