
Figure 5.
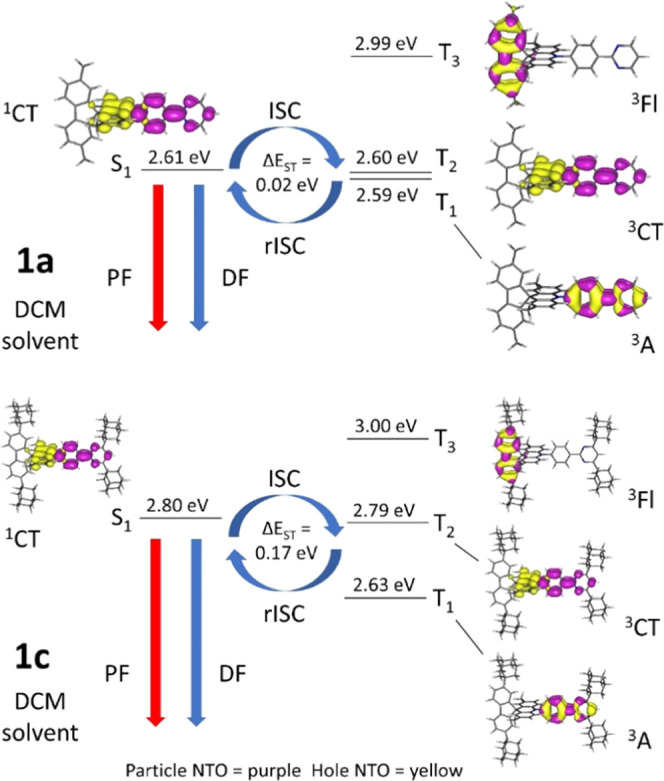
Energy diagrams illustrating TADF with natural transition orbitals (NTOs) for each state on optimized S1 excited-state geometry of 1a and 1c from TD-DFT computations with the solvation model cLR-PCM using DCM as a solvent. PF = prompt fluorescence, DF = (thermally activated) delayed fluorescence, ISC = intersystem crossing, RISC = reverse intersystem crossing, ΔEST = S1 energy – T1 energy, 1CT = singlet charge-transfer state, 3CT = triplet charge-transfer state, 3Fl = local triplet excitation state at the fluorene donor unit, and 3A = local triplet excitation state at the acceptor unit. Contours in NTOs are drawn at ± 0.04 (e/bohr3)1/2.