

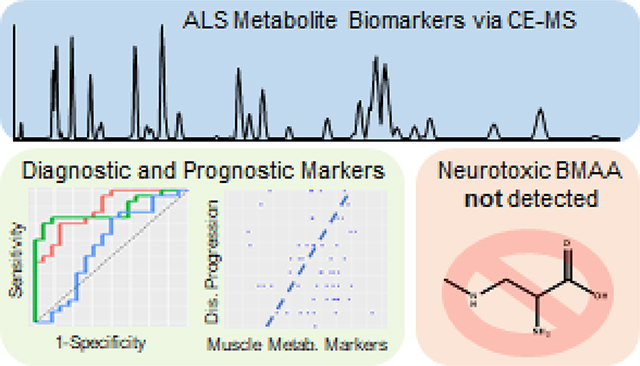
Abstract
By employing chip-based capillary zone electrophoresis coupled to high resolution mass spectrometry, we profiled the plasma metabolome of 134 patients diagnosed with sporadic amyotrophic lateral sclerosis (81 males and 53 females) and 118 individuals deemed healthy (49 males and 69 females). The most significant markers (p<0.01) were creatine, which was 49% elevated, and creatinine and methylhistidine which were decreased by 20% and 24%, respectively, in ALS patients. The ratio of creatine versus creatinine increased 370% and 200% for male and female ALS patients, respectively. In addition, male ALS patients on average had 5–13% lower amounts of 7 essential amino acids while females did not significantly differ from healthy controls. We developed two models using the metabolite abundances: 1) A classification model for the separation of ALS and healthy samples; and 2) A classification model for the prediction of disease progression based on the ALS functional rating score. Utilizing a Monte Carlo cross-validation approach, a linear discriminant analysis model achieved a mean area under the receiver operating characteristic curve (AUC) of 0.85 (0.06) with a mean sensitivity of 80% (9%) and specificity of 78% (10%), for the separation of ALS and controls, respectively. A support vector machine classifier predicted progression categories with an AUC of 0.90 (0.06) with a mean sensitivity 73% (10%) and specificity 86% (5%). Lastly, using a previously reported assay with a stable isotope labeled (13C315N2) spike-in standard, we were unable to detect the exogenous neurotoxic metabolite, β-methylamino-L-alanine (BMAA), in the free or protein bound fraction of any of the 252 plasma samples.
Keywords: Amyotrophic Lateral Sclerosis, Biomarkers, BMAA, CZE-MS/MS, Metabolites, Plasma
Graphical Abstract
INTRODUCTION
Amyotrophic lateral sclerosis (ALS)1 is a devastatingly progressive neurodegenerative disease that often strikes seemingly healthy middle-age individuals at random. It is characterized by the deterioration of both upper and lower motor neurons in the brain, brain stem, and spinal cord leading to loss of voluntary muscle action and eventual total body paralysis. Patients typically succumb to respiratory failure, a direct result of the deterioration of the muscles that control breathing, 3–5 years following the onset of symptoms.2 While 10% of patients survive five or more years, these individuals endure significant medical interventions, associated costs, and decreased quality of life. Currently only two drugs have been approved for treatment of ALS; however, neither Riluzole nor Radicava have proven to be that effective and offer meager improvements in survival (i.e., 2–3 months)3 and function in a subset of patients.4,5 Consequently, the ALS community is in dire need of effective therapies to reverse, halt, or even slow disease progression.
The journey to treat ALS has been hampered for several reasons over the years.6 A significant reason is the lack of an objective biomarker that can be used to aid diagnosis, assess progression, confirm target engagement, and serve as endpoints for trial evaluation. ALS is diagnosed by exclusion of other confounding diseases and the presentation of clinical symptoms indicative of both upper and lower motor neuron loss.7 This highly subjective approach is lengthy with a median of 11.5 months from symptom onset to diagnosis.8 This delay often results in unnecessary surgeries, expenses, and notably prevents early intervention where future therapies will be most likely be effective.
To date, no biomarker exists to assess disease progression or the efficacy of therapeutic intervention. Progression is monitored in the clinic by using a 12-question survey where each question addresses a specific area of physical disability and is scored on a five-point scale (0–4). The sum is referred to as the patient’s ALS functional rating score (ALS-FRS).9 Although the advantages of the survey are clear (e.g., cheap and fast), the limitations include but are not limited to: 1) several questions are subjective; 2) the scale is not linear with the degree of physical impairment; 3) (un)conscious bias; and 4) it is an indirect measure of the presentation of the underlying disease processes and thus a poor indicator of disease progression and response to therapy. Disturbingly, it is quite possible that previous clinical trials have identified a successful therapy; however, due to the length of any given trial and other parameters6,10 the effect was not yet observed at the phenotypic level (i.e., ALS-FRS) and these trials were deemed a failure. Thus, a sorely needed game changer in ALS research that would impact everything from basic disease biology to drug discovery and clinical care, is the identification of a quantitative marker(s) indicative of disease and progression.
Numerous studies have been undertaken over the past couple of decades to identify a fluid based quantitative biomarker for ALS.11,12 These efforts have focused on proteins, small molecules, and microRNA.11–13 The most studied and promising biomarker identified thus far are neurofilaments14. Neurofilaments provide structure to the motor neurons and are released into the CSF upon cellular injury. Research has shown elevated levels of phosphorylated neurofilament heavy protein (pNFL) in ALS patients compared to other neurodegenerative diseases and healthy controls.15–17 In addition, higher levels of pNFH in the CSF were associated with decreased survival but notably did not correlate with ALS-FRS.17 Although promising, limitations for the use of neurofilaments as biomarkers for ALS include susceptibility to protease degradation (i.e., instability)18,19 and conflicting studies on their use for prognosis.20,21 Other notable protein biomarkers for ALS include various ones related to immune response and neuroinflammation.12,22–24
While small molecules have received considerably less attention than proteins as biomarkers for ALS, disruptions in metabolism are hallmark features of disease pathogenesis.25 Thus, the assessment of metabolite markers is a promising avenue for biomarker discovery. Due to its direct role in a major pathway of neuronal death associated with ALS (i.e., excitotoxicity), several studies have measured glutamate in CSF or plasma; however, with conflicted results on its difference in ALS patients.26–28 With the emergence of mass spectrometry based metabolomics, investigations have focused on the discovery of metabolite signatures for both clinical purposes and insight into disease mechanisms.29,30 For example, Berry and coworkers identified a plasma derived 32-metabolite panel with high specificity (90%) yet moderate sensitivity (58%) in the separation of ALS patients from disease mimics and healthy controls.31
In this contribution, capillary zone electrophoresis (CZE) provided a rapid and highly efficient separation followed by detection by high resolution mass spectrometry of metabolites derived from plasma taken from 134 and 118 sporadic ALS (sALS) and control patients, respectively. A combination of univariate and multivariate methods was used to assess significance and develop signatures of disease status separation and progression. Muscle metabolism markers were most significantly different between healthy and diseased patients, but several essential amino acids were found lower in males with ALS. Results presented herein demonstrate the potential for a panel of plasma metabolites to aid in the diagnosis of sALS and serve as an objective marker to monitor disease progression. A secondary goal of this work was to quantify β-methylamino-L-alanine (BMAA), a nonconical neurotoxic amino acid produced by cyanobacteria which has been previously associated with ALS etiology32, in plasma from ALS and control patients. Importantly, we were unable to detect BMAA at a low limit of detection (LOD) in the free (LOD=2.3 ng/mL) or protein bound (LOD=1 ng/mL) fractions in any of the 252 plasma samples.
MATERIALS AND METHODS
Materials
Ammonium acetate, hydrochloric acid (HCl), methylamine hydrochloride, barium hydroxide, α-casein and 0.22 μm nylon centrifuge tube filters were obtained from Sigma Aldrich (St. Louis, MO). LC-MS grade water, methanol and formic acid were purchased from Fisher Scientific (Hampton, NH). BMAA (catalog No. ULM-10493–1.2) and 13C3,15N2-BMAA (SIL-BMAA, catalog No. CNLM-10424–1.2) were obtained from Cambridge Isotope Laboratories (Tewksbury, MA).
Sample Collection and Preparation
Samples in the study were provided by The Neurodegenerative Disease biobank at Macquarie University. The biobank recruited, obtained informed consent and collected biological samples from all participants in accordance with relevant guidelines and regulations. The majority of participants were of European descent. Whole blood was collected from participants in 10 ml EDTA tubes according to standard protocols. Blood was centrifuged at 2,100g for 10 minutes, following which plasma was removed and stored at −80°C, on the day of collection. Ethics approval for the study was obtained from Macquarie University’s human research ethics committee (Approval no. 5201600387).
The cohort consists of 134 patients and 118 controls. The majority of patients (n=129) were diagnosed with classic ALS i.e. both upper and lower motor neurons affected. The remainder (n=6) were just affected in either the upper or lower motor neurons. At the time of cohort design, all patients were classified as sporadic i.e. no family history of ALS. Controls (n=118) were deemed to be healthy and unrelated to an ALS patient. Neither patients nor controls were directed to fast prior to sample collection.
Samples were randomly assigned to one of six batches for detection of protein bound BMAA and one of three batches for detection of free BMAA and metabolite profiling. Three quality control samples of pooled plasma spiked with BMAA and SIL-BMAA were prepared with each batch.
Detection of Protein Bound BMAA
250 μg of protein (BCA assay) was spiked with SIL-BMAA to give a final concentration of 6.25 ng/mL. The mixture was acid hydrolyzed in 6N HCl overnight at 95°C. Samples were lyophilized and reconstituted in the 908 Devices metabolites diluent, resulting in a 1:100 overall dilution. Three quality control samples (i.e., positive controls) were prepared and analyzed with each batch to evaluate intra- and inter-batch precision. QC samples were created by performing a combination of β-elimination followed by Michael Addition of methylamine to chemically convert phosphoserines in α-casein to BMAA33. This protein was then added to pooled plasma and underwent sample preparation as described. A 10-point calibration curve was created by spiking in increasing amounts of unlabeled BMAA along with a constant amount of SIL-BMAA into pooled plasma. Final concentrations of the calibrators following dilution ranged from 5,000, 1,000, 500, 100, 50, 10, 5, 2.5, 1 and 0 ng/mL. The ratio of the response (unlabeled/labeled) was plotted as a function of BMAA concentration. The linear fit was excellent (R2>0.999) and limits of detection and quantitation, calculated as described34, were 1.0 and 3.3 ng/mL, respectively. The intra- and inter-batch precision of BMAA quantitation was 9.0% and 14.4% CV, respectively. These data were only used for detection of protein bound BMAA and not metabolomic analysis.
Detection of Free BMAA and Relative Quantitation of Metabolites
For metabolite profiling and detection of free BMAA, analytes were extracted using the following method. 90 μL of a cold extraction solution consisting of an 80:20 mixture of methanol and water with 100 mM ammonium acetate and 123 ng/mL (1 μM) SIL-BMAA internal standard was added to 10 μL of each plasma sample. Each sample was subsequently vortexed and kept on ice for 10 minutes, then filtered using a 0.22 μm nylon centrifugal filter. A 9-point calibration curve was created by spiking increasing amounts of unlabeled BMAA along with a constant amount of SIL-BMAA into pooled plasma. Final concentrations of the calibrators ranged from 1,000, 500, 100, 50, 10, 1, 0.5, 0.01 and 0 ng/mL. Three quality control samples (i.e., positive controls) were prepared and analyzed with each batch to evaluate intra- and inter-batch precision. QC samples were created by spiking 10 ng/mL unlabeled BMAA into 10 μL of pooled plasma and were prepared using the extraction method described. The ratio of the response (unlabeled/labeled) was plotted as a function of BMAA concentration. The linear fit was excellent (R2>0.999) and limits of detection and quantitation, calculated as described34, were 2.3 and 7.6 ng/mL, respectively. Representative calibration curves for both sample preparations (e.g., protein bound and free BMAA) are shown in Figure S1. The intra- and inter-batch precision of BMAA quantitation was 14.9% and 15.6% CV, respectively. These data were used for both detection of free BMAA and relative quantitation of metabolites.
CZE-MS Analysis
Hydrolyzed samples were analyzed using ZipChip™ HS chips and extracted metabolite samples were analyzed using ZipChip™ HSX chips from 908 Devices (Boston, MA) coupled to a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). 20 μL of analyte solution was placed into the sample well and 5.0 nL was subsequently injected into the chip. A field strength of 1000 V/cm and an ESI voltage of 3 kV were applied for separation over 3 minutes using a background electrolyte solution consisting of 2% formic acid in 50% methanol in water. The capillary temperature was set to 200°C. For MS1 analysis, mass resolving power was 15,000 (FWHM at m/z = 200) with an AGC target of 1 × 106, maximum injection time of 20 ms, and scan range of m/z 70–210. Separate methods were applied for MS1 and MS2 analyses due to limited peak width (FWHM was ~0.5 s). For MS2 analysis, resolving power was 15,000 (FWHM at m/z = 200) with an AGC target of 2 × 105, maximum injection time of 200 ms, isolation window of 2.0 m/z, and NCE of 35. Three QC standards were analyzed at the beginning, middle and end of each day followed by a blank injection. Further details on the method used for CZE-MS detection of BMAA can be found here.34
Data Analysis and Availability
BMAA analysis, PRM raw data files were imported into Skyline where peak areas were extracted for unlabeled BMAA (precursor 119.0815, products 102.0550, 88.0393, 76.0393, 73.0760, and 56.0495 m/z) and SIL-BMAA (precursor 124.0856, products 106.0621, 92.0464, 79.0430, 77.0768, and 59.0532 m/z) and exported to a .csv file.
Metabolites were identified based on MS1 MMA ± 10 ppm. The migration times were compared to data provided by the manufacturer from a pooled plasma sample to confirm identification and to distinguish isomers (e. g., Leu vs Ile). RAW files were imported into Skyline35 where MS1 extracted ion based quantitation was performed.36 Peak areas were then exported to a .csv file and imported into R Studio. The precision of relative quantitation was estimated for individual chips, sample preparation for each day, and total experimental variability using the median coefficient of variation (CV) of the pooled replicates. The within chip precision was 14%, 6%, and 19% (CV) for chips 1–3, respectively. The precision in sample preparation for batches 1–3 was 6%, 10%, and 11% CV, respectively. Total experimental precision was 35% CV (Figure S2).
All RAW files from this study are available on the Chorus Project mass spectrometry data repository (Project ID=1664). All Skyline files are located on the PanoramaWeb37 repository in the ‘ALS Metabolite Biomarker” project folder under Bereman Lab. Data for the BMAA calibration, quality control samples, metabolite abundances used in this study are available in Supplemental File 1.
Univariates Statistics
Data were log2 transformed and the significance of the difference in the mean analyte abundance between ALS and control were compared using the Welch two tailed t-test. The Benjamini Hochberg method38 was used to correct for multiple hypothesis testing. Percent change was calculated in non-log space as the difference in the average analyte abundance (ALS-control) divided by control times 100. For assessing the relationship between ALS-FRS and abundance, a simple linear regression was performed and the probability that the slope was different from zero was used for significance.
Multivariate Statistics
RStudio v2.1, along with several packages including caret, applied predictive modeling, ggplot2, and pROC, were used for quality control and multivariate modeling. K-means clustering was performed following principal component transformation. We were particularly interested in two quality checks: 1) An outlier data point(s) that clearly was separated from the majority of the data; and 2) A group of data that clustered and associated with sex or disease status. Neither were qualitatively observed (Figure S3).
For the disease classification model, data were log2 transformed, centered, and scaled. Then a test sample set was randomly chosen (n=42) and withheld from the model building process. A technique called recursive feature elimination39 was then used to choose features to maximize model performance. Three models were evaluated including linear discriminant analysis (LDA), random forest (RF), and support vector machine (SVM). Feature selection was performed within the model building process such as to encompass the variability of feature selection in the final results. Using a repeated (n=5) 5-fold cross-validation approach, the model was first constructed using all 40 metabolites with 80% of the data. Features were then ranked based on performance and subset sizes ranging from 1:10, 15, 20, 25, 30, 35, and 40 metabolites were evaluated on the hold out sample set (20%).22 This inner-loop was repeated 4 additional times (i.e. 5-fold) and then the whole process was repeated 5 times. Both the number of randomly selected variables at each split (mtry=2:5) and cost parameter (2−2 2−1 20… 212) were tuned for the RF and SVM, respectively. The sigma parameter for the radial Bessel function in SVM was held constant at its predicted value (σ=0.034).40 The average area under the receiver operating characteristic curve, calculated from the resampled data sets (n=25), were plotted as a function of subset size for each model type to identify the optimal size. Since models were cross validated on identical subsets of data, a paired t-test was used to statistically determine the highest performing model based on AUC.41 The best model, based on area under the ROC curve, was then used to predict the classes of the hold out sample set in efforts to validate performance. To ensure the performance of the classifier was not due to random effect, this whole feature selection and training procedure was repeated with the data set after randomization of the class labels (i.e., negative control). ROC analysis was performed on the predictions from the hold out data set using both LDA models.
For the second classifier, we calculated the rate of progression for each patient by taking the change in FRS at time of sampling divided by the length of time since diagnosis (Δ points per month). Patients were then designated as fast (rate ≥3 pts/mo), average (3>rate ≥1 pts/mo), and slow (rate<1 pt/mo) categories of progression. This process led to 14, 42, and 42 fast, average and slow progressors in the data set (36 patients were removed due to missing clinical assessment data). Due to the severe class imbalance, which can bias the sensitivity of classifiers towards the majority class, we performed an imputation method called Synthetic Minority Oversampling Technique (SMOTE).42 After performing this combination of up- and down-sampling of the minority and the majority classes, we had 28, 33, and 30 fast, average, and slow progressors, respectively. The SVM was tuned and trained with a 5-fold repeated (n=50) cross-validation approach. The model was then compared to an analogous classifier constructed on the same data with the class labels randomized (i.e., negative control). The average of the AUC, sensitivity, and specificity (1 vs. all classes) was used to evaluate performance of each hold out sample data set. These data were then compared using boxplots.
Results
Figure 1 describes the overall experimental design. Plasma was sampled from individuals with ALS and individuals considered healthy. Materials were prepared as described for capillary zone electrophoresis. The median age of the healthy individuals was 61.7 with an interquartile range of 17.5 years. The median age of the ALS patients was 65.3 with an interquartile range of 15.1 years. The median ALS-FRS was 32 points, though scores ranged from 12 to 47 points out of the 48 possible in the scale. Figure 2 displays volcano plots from pairwise comparison of ALS and controls as a function of sex. The -log10 (p-value) is plotted as a function of the log2 (fold-change). The two most significant markers were the elevation of creatine and the decrease of creatinine in ALS patients. Supplemental File 1 (Table 1) summarizes the findings for the metabolites quantified in the study. The distribution of abundances in all metabolites as a function of disease and sex can be viewed in Supplemental File 2.
Figure 1:

Summary of sample preparation and experimental design and clinical characteristics. The median ages of the ALS patients and the healthy individuals were 65.3 and 61.7 years (IQRs of 15.3 and 17.5 years), respectively.
Figure 2:

Volcano plots of A) ALS vs. Control and separated by sex B) Males and C) Females. Shaded region represents significance (p<0.05). Creatinine and creatine are annotated.
Figure 3 shows the correlation of muscle associated metabolites methylhistidine, creatine and creatinine as a function of disease progression as estimated by the ALS-FRS. A simple linear regression is fitted to male and female ALS patients separately. The gray distribution denotes the error in the fit. A significant slope was measured in all cases except for creatine in females. Methylhistidine explained 17% of the variation in FRS followed by creatinine (15%) and creatine (3%). The covariance between methylhistidine and creatinine was high (r=0.49) suggesting a relationship with underlying disease processes (i.e. skeletal muscle breakdown). As shown by the graphs creatine and creatinine had an inverse relationship with disease progression. Figure 3D displays the distribution in the ratio (creatine/creatinine) between ALS and healthy controls as a function of sex. A 370% and 200% increase (p<0.001) were observed for men and women ALS patients compared to control. Other notable metabolites with moderate significance in both females and males (p<0.1) were stachydrine and acetyl-L-carnitine which both decreased with increased disease progression. The relationship of all metabolites with the FRS can be viewed in Supplemental File 3.
Figure 3:

Correlation of muscle metabolism markers with ALS-FRS for A) methylhistidine B) creatine and C) creatinine. Circle=female, Triangle=male. Slopes of regression lines were statistically significant (p<0.05) except creatine in females. D) Ratio (creatine/creatinine) between ALS patients and controls as a function of sex. Blue=healthy red=SALS
The results for the development of a diagnostic metabolite signature are shown in Figure 4. Figure 4A displays the mean area under the curve as a function of the number of metabolites during the feature selection process for the LDA, RF and SVM classifiers. All three models showed a similar increase followed by incremental improvements in performance with more metabolite features. The highest performance amongst the individual models was achieved with the 35 feature LDA model (p<0.001) with an AUC of 0.85 (0.06) and a mean sensitivity and specificity of 80% (0.09) and 78% (0.10), respectively. Figure S4 compares the performance of the models on the resampled data sets using traditional binary classification metrics.
Figure 4:

A) The mean area under the curve is plotted as a function of the number of metabolites in the model. The performance of a support vector machine, linear discriminant analysis, and random forest models were compared. B) Training the same models on the data set in which the class labels were randomized. C) Normalized variable importance, based on area under the curve for individual metabolites, of the metabolites in the optimized LDA model. Green highlights the top 6 most important features. D) ROC analysis of the predicted classes from the test set using the full feature (n=35) LDA (red) and reduced feature (n=6) LDA model (green). The blue curve was created by using the LDA model constructed from the randomized data set. Dashed line represents the performance of a theoretical random classifier.
Figure 4B shows the results of the same feature selection and model building process only with the class labels from the original data randomized (i.e., negative control). As expected for a random classifier, all three models trend near an area under the curve of 0.5. These results further support the significance of the models described. The importance of each feature, based on normalized AUC, from the LDA model is shown in Figure 4C. Based on these data, we took the top six metabolites (highlighted in green box) and trained them on the same sets of data. Performance of the full (n=35) and reduced feature (n=6) LDA models were statistically the same on the resampled data sets (Figure S4 and Figure S5). Furthermore, albeit different ROC curves, Figure 4D shows the performance of both the full (red) and reduced feature (green) LDA model were the same (AUC=0.85) on the hold out data set (n=42). The blue line denotes the performance (AUC=0.58) of the LDA model trained with the randomized data on the hold data set.
A support vector machine classifier was built to classify patients as fast, average, or slow progression based on the ALS-FRS. Figure 5 displays boxplots of the performance as measured by AUC, sensitivity and specificity from a repeated (n=50) 5-fold cross validation. The model achieved a median area under the curve of 0.91 (IQR=0.86–0.94) and a median sensitivity and specificity of 74% (IQR=66–80%) and 87% (IQR=83–90%). To further assess the significance of this finding, we trained an analogous classifier on the same data after permutation of the class labels. These results indicate the performance of the classifier trained on the permuted data is random with an AUC of 0.53 (IQR=0.47–0.61). Figure S6 shows the average variable importance of each metabolite in prediction of rate. Abundances of individual metabolites as a function of disease progression rate can be viewed in Supplemental File 4.
Figure 5:

The performance of the SVM in classifying disease progression as measured by AUC, sensitivity and specificity. A second SVM was trained after permutation of the class labels (svmR).
Lastly, representative electropherograms showing the failure to detect BMAA from the extracted metabolite fraction (i.e. free BMAA) and hydrolyzed fraction (i.e., protein bound BMAA) of plasma samples in Figure S7 and Figure S8. Extracted ion electropherograms clearly show the migration at approximately 1 minute of SIL-BMAA (m/z 124.0856) and the absence of endogenous BMAA (m/z 119.0815). In contrast Figure S9 shows a representative positive control from which BMAA was released from a synthetically made BMAA incorporated protein standard via acid hydrolysis.
DISCUSSION
In this study, chip-based capillary zone electrophoresis coupled to high resolution mass spectrometry enabled confident identification and quantification of polar metabolites with cycle times similar to clinical assays of approximately 4 minutes. This technology was then applied to a large plasma sample cohort for detection of small molecule biomarkers and the cyanotoxin BMAA.
The two most significant markers identified were elevation of creatine and lower amounts of creatinine in ALS patients which confirm previous findings in both the CSF and plasma of ALS patients.29,31 This result is likely due to changes in muscle mass and catabolism characteristic of ALS. Notably the elevation of creatine kinase43, the enzyme responsible for the creation of the high energy compound creatine phosphate, has been shown to be beneficial in both ALS patients44 and animal models of ALS.45 These results are suggestive of the enormous energy requirements of the disease and/or possibly the lower activity of creatine kinase45 which is partly compensated by its elevated levels in disease patients.43 This defect in energy production is further supported by the 2–3.7 fold increase in the (creatine/creatinine) in ALS patients. Regardless, the perturbation of the creatine kinase system has repeatedly been reported in ALS; yet modulation has shown only to be therapeutic in animal models of the disease.46,47 Methylhistidine, a marker of skeletal muscle breakdown, was found to be lower in ALS patients. To our knowledge this is the first report of changes in this marker in the plasma of ALS patients. A previous literature report in a small set of ALS patients (n=7) identified higher levels of methylhistidine in urine48. Notably aside from creatine in females, all three muscle markers correlated with disease progression as estimated by the FRS. Thus, these markers show potential for diagnosis, prognosis and monitoring therapeutic intervention.
Another finding worth noting is the small decrease (5–13%) of seven essential amino acids in the plasma of male ALS patients compared to their healthy counterparts. While we note four of the seven became insignificant or mildly significant (FDR <0.1) after multiple hypothesis correction, it is reasonable to believe that the finding is genuine. Basic probability theory shows the chances of identifying 7 of 9 essential amino acids at random from a total of 20 amino acids is low (9C7/20C7 = 0.0004). Also it is worth noting the other two essential amino acids (i.e. Trp and Val) that did not reach the significance threshold (p>0.05) trended lower in male ALS patients. The reason for this finding and its consequences are unknown; however, it could be related to the dietary changes and often malnutrition that are associated with disease progression. Although, the majority of essential amino acids were poorly correlated with FRS (Supplemental File 3). While essential amino acids are required for protein synthesis, an underrecognized role is their involvement in various ALS associated cellular signaling pathways including the mechanistic target of rapamycin, oxidative stress (Nrf2), and calcium sensing pathways49. We do note three male reversals of ALS, which are extremely rare50, using a supplement containing high levels of amino acids amongst many other constituents (Richard Bedlack MD PhD, Personal Communication, January 26, 2020).
Machine learning was used to develop two models for the separation of ALS patients and healthy controls and for predicting disease progression based on FRS. A linear discriminant model achieved a sensitivity and specificity of 80% and 78% in the classification of disease status. Several muscle metabolism markers were the most important features in the model which supports the univariate findings. Notably the majority of features from the optimized RFE model were uninformative in the classification process. Similar performance was achieved with a LDA model using six metabolites. Three of the six metabolites (creatine, creatinine, and proline) were previously included in the 32-metabolite model as described by Berry et. al.; however, histidine, methylhistidine, and dihydrothymine were neither significant nor used in their model. Although encouraging, several of these markers (creatine, methylhistidine, creatinine) are associated with muscle metabolism and not specific to ALS. For example, there are several diseases and conditions that result in muscle wasting and some are considered in the differential diagnosis of ALS (e.g., Kennedy’s Disease, Human Immunodeficiency Virus, Multifocal Motor Neuropathy).51 This would inhibit the algorithm for use as a screening test and limit it as a sole diagnostic procedure but could be a useful aid for diagnosis in conjunction with clinical findings.
An often underappreciated theme in basic ALS research is the enormous heterogeneity of clinical ALS.52 Although the average survival is 3–5 years after the onset of symptoms, some forms of ALS are extremely lethal with patients succumbing within months of diagnosis. In contrast, with other forms of the disease, patients survive well past the five-year mark. Thus, it would be incredibly useful to be able to predict disease progression not only for the benefit of the patient but importantly for disease categorization, which then would inform downstream clinical trials and eventual therapies. Based on the metabolite profile, we were able to classify patients as slow, average or fast progressors with a sensitivity and specificity of 74% and 87%. The most important metabolite in prediction of rate of disease progression was an unidentified feature (m/z=130.0869); however, its significance is questionable since it was also highly predictive in the randomized data set. Acetyl-L-Carnitine (ALC) was found to be important (AUC=0.73) relative to its value in the randomized data set (AUC=0.54). ALC is believed to be helpful in a wide range of neurological diseases as it decreases oxidative stress and increases energy through the metabolism of long chain fatty acids in the mitochondria.53 Most notable, supplementation with high quantities of ALC in ALS patients was shown beneficial in a double blind placebo controlled clinical trial.54 Essential amino acids Phe, Leu, Val and Met were shown to be important relative to the model trained on the randomized data set; however, other essential amino acids were not. Interestingly, muscle metabolism markers were not useful in the prediction of rate of disease progression. Limitations of these analyses include lack of a separate validation set and the noted shortcomings of the FRS to provide an accurate measure of disease state. Further research is needed to validate the model using both additional cross-sectional studies and longitudinal sampling but the potential of such a model is enormous for the clinical care of ALS patients.
We have spent significant efforts developing new analytical technologies for confident and accurate quantification of BMAA.34 The impetus behind these efforts has been driven by past studies that have quantified BMAA in high amounts yet often with nonspecific analytical techniques.55 BMAA has been previously detected in the protein bound and free fractions of biological samples. In these studies, we applied these tools in efforts to detect BMAA in human plasma samples. We report the failure to detect BMAA in any of the 134 ALS or 118 control samples. Thus, these findings lead to one of three possible conclusions: 1) BMAA is not involved in the etiology of sporadic ALS26,55–59 in this sample cohort; 2) BMAA may be involved yet below the detection limits of the analytical technologies used in this study; or 3) transient acute or sub chronic exposures to BMAA may be the trigger for neurodegeneration60 in susceptible individuals61 yet difficult to detect at any single time point. Regardless, these findings do not support the accumulation of BMAA to significant levels within the body.
CONCLUSIONS
High throughput metabolite profiling identified several muscle metabolism markers that were differentially abundant and correlated with disease progression in plasma of ALS patients. Models were developed using machine learning to distinguish health status and categorize patients based on progression rates with high sensitivity and specificity. Ongoing studies are examining the markers in plasma longitudinally collected from ALS patients in efforts to refine and validate these models. Longitudinal samples are essential in biomarker discovery as they account for the intraindividual variability, which will be critical for assessing the suitability of our models for clinical purposes.
Supplementary Material
ACKNOWLEDGEMENTS
All measurements were performed in the Molecular Education, Technology and Research Innovation Center (METRIC) at NC State University. M.S.B. is grateful for support (#19-SI-458) from the ALS Biomarker Consortium: ALS Association; ALS Finding a Cure; CreATe; NEALS; MDA; Packard Association for ALS. D.C.M. is thankful for support from the Chancellor’s Innovation Fund at NC State University. GJG is supported by FightMND, CreATe; MNDRIA and National Health and Medical Research Council (NHMRC). Biospecimens and related clinical data used in this research were obtained from the Neurodegenerative Disease Biobank, Macquarie University, New South Wales, Australia. Thank you to Prof Mark Baker for being the spark of this collaborative project.
Footnotes
COMPETING INTERESTS
The Authors declare no competing interests.
DATA AVAILABILITY
All raw data raw data are publicly available as discussed in Methods Section
REFERENCES
- (1).Zarei S; Carr K; Reiley L; Diaz K; Guerra O; Altamirano PF; Pagani W; Lodin D; Orozco G; Chinea A A Comprehensive Review of Amyotrophic Lateral Sclerosis. Surg. Neurol. Int 2015, 6, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Niedermeyer S; Murn M; Choi PJ Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155 (2), 401–408. [DOI] [PubMed] [Google Scholar]
- (3).Amyotrophic Lateral Sclerosis/Riluzole Study Group II; Lacomblez L; Bensimon G; Meininger V; Leigh PN; Guillet P Dose-Ranging Study of Riluzole in Amyotrophic Lateral Sclerosis. Lancet 1996, 347 (9013), 1425–1431. [DOI] [PubMed] [Google Scholar]
- (4).Jackson C; Heiman-Patterson T; Kittrell P; Baranovsky T; McAnanama G; Bower L; Agnese W; Martin M Radicava (edaravone) for Amyotrophic Lateral Sclerosis: US Experience at 1 Year after Launch. Amyotroph. Lateral Scler. Frontotemporal Degener 2019, 20 (7–8), 605–610. [DOI] [PubMed] [Google Scholar]
- (5).Cruz MP Edaravone (Radicava): A Novel Neuroprotective Agent for the Treatment of Amyotrophic Lateral Sclerosis. P T 2018, 43 (1), 25–28. [PMC free article] [PubMed] [Google Scholar]
- (6).Petrov D; Mansfield C; Moussy A; Hermine O ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci 2017, 9, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Brooks BR; Miller RG; Swash M; Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord 2000, 1 (5), 293–299. [DOI] [PubMed] [Google Scholar]
- (8).Paganoni S; Macklin EA; Lee A; Murphy A; Chang J; Zipf A; Cudkowicz M; Atassi N Diagnostic Timelines and Delays in Diagnosing Amyotrophic Lateral Sclerosis (ALS). Amyotroph. Lateral Scler. Frontotemporal Degener 2014, 15 (5–6), 453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).The Amyotrophic Lateral Sclerosis Functional Rating Scale. Assessment of Activities of Daily Living in Patients with Amyotrophic Lateral Sclerosis. The ALS CNTF Treatment Study (ACTS) Phase I-II Study Group. Arch. Neurol 1996, 53 (2), 141–147. [PubMed] [Google Scholar]
- (10).Fogel DB Factors Associated with Clinical Trials That Fail and Opportunities for Improving the Likelihood of Success: A Review. Contemp Clin Trials Commun 2018, 11, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Taga A; Maragakis NJ Current and Emerging ALS Biomarkers: Utility and Potential in Clinical Trials. Expert Rev. Neurother 2018, 18 (11), 871–886. [DOI] [PubMed] [Google Scholar]
- (12).Vu LT; Bowser R Fluid-Based Biomarkers for Amyotrophic Lateral Sclerosis. Neurotherapeutics 2017, 14 (1), 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Joilin G; Leigh PN; Newbury SF; Hafezparast M An Overview of MicroRNAs as Biomarkers of ALS. Front. Neurol 2019, 10, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Poesen K; Van Damme P Diagnostic and Prognostic Performance of Neurofilaments in ALS. Front. Neurol 2018, 9, 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Brettschneider J; Petzold A; Süssmuth SD; Ludolph AC; Tumani H Axonal Damage Markers in Cerebrospinal Fluid Are Increased in ALS. Neurology 2006, 66 (6), 852–856. [DOI] [PubMed] [Google Scholar]
- (16).Reijn TS; Abdo WF; Schelhaas HJ; Verbeek MM CSF Neurofilament Protein Analysis in the Differential Diagnosis of ALS. J. Neurol 2009, 256 (4), 615–619. [DOI] [PubMed] [Google Scholar]
- (17).Ganesalingam J; An J; Bowser R; Andersen PM; Shaw CE pNfH Is a Promising Biomarker for ALS. Amyotroph. Lateral Scler. Frontotemporal Degener 2013, 14 (2), 146–149. [DOI] [PubMed] [Google Scholar]
- (18).Pant HC Dephosphorylation of Neurofilament Proteins Enhances Their Susceptibility to Degradation by Calpain. Biochem. J 1988, 256 (2), 665–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Goldstein ME; Sternberger NH; Sternberger LA Phosphorylation Protects Neurofilaments against Proteolysis. J. Neuroimmunol 1987, 14 (2), 149–160. [DOI] [PubMed] [Google Scholar]
- (20).Steinacker P; Feneberg E; Weishaupt J; Brettschneider J; Tumani H; Andersen PM; von Arnim CAF; Böhm S; Kassubek J; Kubisch C; Lulé D; Müller H-P; Muche R; Pinkhardt E; Oeckl P; Rosenbohm A; Anderl-Straub S; Volk AE; Weydt P; Ludolph AC; Otto M Neurofilaments in the Diagnosis of Motoneuron Diseases: A Prospective Study on 455 Patients. J. Neurol. Neurosurg. Psychiatry 2016, 87 (1), 12–20. [DOI] [PubMed] [Google Scholar]
- (21).Lu C-H; Macdonald-Wallis C; Gray E; Pearce N; Petzold A; Norgren N; Giovannoni G; Fratta P; Sidle K; Fish M; Orrell R; Howard R; Talbot K; Greensmith L; Kuhle J; Turner MR; Malaspina A Neurofilament Light Chain: A Prognostic Biomarker in Amyotrophic Lateral Sclerosis. Neurology 2015, 84 (22), 2247–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bereman MS; Beri J; Enders JR; Nash T Machine Learning Reveals Protein Signatures in CSF and Plasma Fluids of Clinical Value for ALS. Sci. Rep 2018, 8 (1), 16334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Baldacci F; Lista S; Palermo G; Giorgi FS; Vergallo A; Hampel H The Neuroinflammatory Biomarker YKL-40 for Neurodegenerative Diseases: Advances in Development. Expert Rev. Proteomics 2019, 16 (7), 593–600. [DOI] [PubMed] [Google Scholar]
- (24).Thompson AG; Gray E; Bampton A; Raciborska D; Talbot K; Turner MR CSF Chitinase Proteins in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90 (11), 1215–1220. [DOI] [PubMed] [Google Scholar]
- (25).Tefera TW; Borges K Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front. Neurosci 2016, 10, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Perry TL; Krieger C; Hansen S; Eisen A Amyotrophic Lateral Sclerosis: Amino Acid Levels in Plasma and Cerebrospinal Fluid. Ann. Neurol 1990, 28 (1), 12–17. [DOI] [PubMed] [Google Scholar]
- (27).Rothstein JD; Tsai G; Kuncl RW; Clawson L; Cornblath DR; Drachman DB; Pestronk A; Stauch BL; Coyle JT Abnormal Excitatory Amino Acid Metabolism in Amyotrophic Lateral Sclerosis. Ann. Neurol 1990, 28 (1), 18–25. [DOI] [PubMed] [Google Scholar]
- (28).Spreux-Varoquaux O; Bensimon G; Lacomblez L; Salachas F; Pradat PF; Le Forestier N; Marouan A; Dib M; Meininger V Glutamate Levels in Cerebrospinal Fluid in Amyotrophic Lateral Sclerosis: A Reappraisal Using a New HPLC Method with Coulometric Detection in a Large Cohort of Patients. J. Neurol. Sci 2002, 193 (2), 73–78. [DOI] [PubMed] [Google Scholar]
- (29).Wuolikainen A; Moritz T; Marklund SL; Antti H; Andersen PM Disease-Related Changes in the Cerebrospinal Fluid Metabolome in Amyotrophic Lateral Sclerosis Detected by GC/TOFMS. PLoS One 2011, 6 (4), e17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wuolikainen A; Jonsson P; Ahnlund M; Antti H; Marklund SL; Moritz T; Forsgren L; Andersen PM; Trupp M Multi-Platform Mass Spectrometry Analysis of the CSF and Plasma Metabolomes of Rigorously Matched Amyotrophic Lateral Sclerosis, Parkinson’s Disease and Control Subjects. Mol. Biosyst 2016, 12 (4), 1287–1298. [DOI] [PubMed] [Google Scholar]
- (31).Lawton KA; Brown MV; Alexander D; Li Z; Wulff JE; Lawson R; Jaffa M; Milburn MV; Ryals JA; Bowser R; Cudkowicz ME; Berry JD; Northeast ALS Consortium. Plasma Metabolomic Biomarker Panel to Distinguish Patients with Amyotrophic Lateral Sclerosis from Disease Mimics. Amyotroph. Lateral Scler. Frontotemporal Degener 2014, 15 (5–6), 362–370. [DOI] [PubMed] [Google Scholar]
- (32).Bradley WG; Mash DC Beyond Guam: The cyanobacteria/BMAA Hypothesis of the Cause of ALS and Other Neurodegenerative Diseases. Amyotroph. Lateral Scler 2009, 10 Suppl 2, 7–20. [DOI] [PubMed] [Google Scholar]
- (33).Adamczyk M; Gebler JC; Wu J Identification of Phosphopeptides by Chemical Modification with an Isotopic Tag and Ion Trap Mass Spectrometry. Rapid Commun. Mass Spectrom 2002, 16 (10), 999–1001. [DOI] [PubMed] [Google Scholar]
- (34).Beri J; Kirkwood KI; Muddiman DC; Bereman MS A Novel Integrated Strategy for the Detection and Quantification of the Neurotoxin β-N-Methylamino-L-Alanine in Environmental Samples. Anal. Bioanal. Chem 2018, 410 (10), 2597–2605. [DOI] [PubMed] [Google Scholar]
- (35).Adams KJ; Pratt B; Bose N; Dubois LG; St John-Williams L; Perrott KM; Ky K; Kapahi P; Sharma V; MacCoss MJ; Moseley MA; Colton CA; MacLean BX; Schilling B; Thompson JW; Alzheimer’s Disease Metabolomics Consortium. Skyline for Small Molecules: A Unifying Software Package for Quantitative Metabolomics. J. Proteome Res 2020, 19 (4), 1447–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Schilling B; Rardin MJ; MacLean BX; Zawadzka AM; Frewen BE; Cusack MP; Sorensen DJ; Bereman MS; Jing E; Wu CC; Verdin E; Kahn CR; Maccoss MJ; Gibson BW Platform-Independent and Label-Free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline: Application to Protein Acetylation and Phosphorylation. Mol. Cell. Proteomics 2012, 11 (5), 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sharma V; Eckels J; Taylor GK; Shulman NJ; Stergachis AB; Joyner SA; Yan P; Whiteaker JR; Halusa GN; Schilling B; Gibson BW; Colangelo CM; Paulovich AG; Carr SA; Jaffe JD; MacCoss MJ; MacLean B Panorama: A Targeted Proteomics Knowledge Base. J. Proteome Res 2014, 13 (9), 4205–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Benjamini Y; Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B Stat. Methodol 1995, 57 (1), 289–300. [Google Scholar]
- (39).Guyon I; Weston J; Barnhill S; Vapnik V Gene Selection for Cancer Classification Using Support Vector Machines. Mach. Learn 2002, 46 (1), 389–422. [Google Scholar]
- (40).Caputo B; Sim K; Furesjo F; Smola A Appearance-Based Object Recognition Using SVMs: Which Kernel Should I Use? 2001.
- (41).Hothorn T; Leisch F; Zeileis A; Hornik K The Design and Analysis of Benchmark Experiments. J. Comput. Graph. Stat 2005, 14 (3), 675–699. [Google Scholar]
- (42).Chawla NV; Bowyer KW; Hall LO; Kegelmeyer WP SMOTE: Synthetic Minority Over-Sampling Technique. 1 2002, 16, 321–357. [Google Scholar]
- (43).Harrington TM; Cohen MD; Bartleson JD; Ginsburg WW Elevation of Creatine Kinase in Amyotrophic Lateral Sclerosis. Arthritis & Rheumatism 1983, 26 (2), 201–205. [DOI] [PubMed] [Google Scholar]
- (44).Rafiq M; Lee E; Bradburn M; McDermott C; Shaw P ELEVATED CREATINE KINASE SUGGESTS BETTER PROGNOSIS IN PATIENTS WITH AMYOTROPHIC LATERAL SCLEROSIS. J. Neurol. Neurosurg. Psychiatry 2013, 84 (11), e2–e2.25346970 [Google Scholar]
- (45).Tai H; Cui L; Guan Y; Liu M; Li X; Shen D; Li D; Cui B; Fang J; Ding Q; Zhang K; Liu S Correlation of Creatine Kinase Levels with Clinical Features and Survival in Amyotrophic Lateral Sclerosis. Front. Neurol 2017, 8, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Rosenfeld J; King RM; Jackson CE; Bedlack RS; Barohn RJ; Dick A; Phillips LH; Chapin J; Gelinas DF; Lou J-S Creatine Monohydrate in ALS: Effects on Strength, Fatigue, Respiratory Status and ALSFRS. Amyotroph. Lateral Scler 2008, 9 (5), 266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Klivenyi P; Ferrante RJ; Matthews RT; Bogdanov MB; Klein AM; Andreassen OA; Mueller G; Wermer M; Kaddurah-Daouk R; Beal MF Neuroprotective Effects of Creatine in a Transgenic Animal Model of Amyotrophic Lateral Sclerosis. Nat. Med 1999, 5 (3), 347–350. [DOI] [PubMed] [Google Scholar]
- (48).Wang J; Thornton JC; Pierson RN Urinary Excretion of Creatinine and 3-Methylhistidine for Estimation of Skeletal Muscle Mass in Humans: An Overview. In Quality of the Body Cell Mass; Springer; New York, 2000; pp 89–94. [Google Scholar]
- (49).He F; Wu C; Li P; Li N; Zhang D; Zhu Q; Ren W; Peng Y Functions and Signaling Pathways of Amino Acids in Intestinal Inflammation. Biomed Res. Int 2018, 2018, 9171905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Harrison D; Mehta P; van Es MA; Stommel E; Drory VE; Nefussy B; van den Berg LH; Crayle J; Bedlack R; Pooled Resource Open-Access ALS Clinical Trials Consortium. “ALS Reversals”: Demographics, Disease Characteristics, Treatments, and Co-Morbidities. Amyotroph. Lateral Scler. Frontotemporal Degener 2018, 19 (7–8), 495–499. [DOI] [PubMed] [Google Scholar]
- (51).Ghasemi M Amyotrophic Lateral Sclerosis Mimic Syndromes. Iran J Neurol 2016, 15 (2), 85–91. [PMC free article] [PubMed] [Google Scholar]
- (52).Mezzapesa DM; D’Errico E; Tortelli R; Distaso E; Cortese R; Tursi M; Federico F; Zoccolella S; Logroscino G; Dicuonzo F; Simone IL Cortical Thinning and Clinical Heterogeneity in Amyotrophic Lateral Sclerosis. PLoS One 2013, 8 (11), e80748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Ferreira GC; McKenna MC L-Carnitine and Acetyl-L-Carnitine Roles and Neuroprotection in Developing Brain. Neurochem. Res 2017, 42 (6), 1661–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Beghi E; Pupillo E; Bonito V; Buzzi P; Caponnetto C; Chiò A; Corbo M; Giannini F; Inghilleri M; Bella VL; Logroscino G; Lorusso L; Lunetta C; Mazzini L; Messina P; Mora G; Perini M; Quadrelli ML; Silani V; Simone IL; Tremolizzo L; Italian ALS Study Group. Randomized Double-Blind Placebo-Controlled Trial of Acetyl-L-Carnitine for ALS. Amyotroph. Lateral Scler. Frontotemporal Degener 2013, 14 (5–6), 397–405. [DOI] [PubMed] [Google Scholar]
- (55).Chernoff N; Hill DJ; Diggs DL; Faison BD; Francis BM; Lang JR; Larue MM; Le T-T; Loftin KA; Lugo JN; Schmid JE; Winnik WM A Critical Review of the Postulated Role of the Non-Essential Amino Acid, β-N-Methylamino-L-Alanine, in Neurodegenerative Disease in Humans. J. Toxicol. Environ. Health B Crit. Rev 2017, 20 (4), 1–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Duncan MW Beta-Methylamino-L-Alanine (BMAA) and Amyotrophic Lateral Sclerosis-Parkinsonism Dementia of the Western Pacific. Ann. N. Y. Acad. Sci 1992, 648, 161–168. [DOI] [PubMed] [Google Scholar]
- (57).Duncan MW; Marini AM Debating the Cause of a Neurological Disorder. Science 2006, 313 (5794), 1737. [DOI] [PubMed] [Google Scholar]
- (58).Meneely JP; Chevallier OP; Graham S; Greer B; Green BD; Elliott CT β-Methylamino-L-Alanine (BMAA) Is Not Found in the Brains of Patients with Confirmed Alzheimer’s Disease. Sci. Rep 2016, 6, 36363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Montine TJ; Li K; Perl DP; Galasko D Lack of Beta-Methylamino-L-Alanine in Brain from Controls, AD, or Chamorros with PDC. Neurology 2005, 65 (5), 768–769. [DOI] [PubMed] [Google Scholar]
- (60).Cox PA; Davis DA; Mash DC; Metcalf JS; Banack SA Dietary Exposure to an Environmental Toxin Triggers Neurofibrillary Tangles and Amyloid Deposits in the Brain. Proc. Biol. Sci 2016, 283 (1823). 10.1098/rspb.2015.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Scott LL; Downing TG A Single Neonatal Exposure to BMAA in a Rat Model Produces Neuropathology Consistent with Neurodegenerative Diseases. Toxins 2017, 10 (1). 10.3390/toxins10010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.