

Abstract
Although diagnosis of high-grade uterine mesenchymal tumors (high-grade UMTs) exhibiting classic morphologic features is straightforward, diagnosis is more challenging in tumors in which prototypical features are poorly developed, focal and/or co-exist with features seen in other neoplasms. Here, we sought to define the repertoire of somatic genetic alterations in diagnostically challenging UMTs with myomelanocytic differentiation, including some reported as perivascular epithelioid cell tumors (PEComas).
In 17 samples from 15 women, the tumors were histologically heterogeneous. Immunohistochemical expression of at least one melanocytic marker (HMB45, Melan-A or MiTF) was identified in all tumors, and of myogenic markers (desmin or SMA) in most tumors. Targeted massively parallel sequencing revealed several genetic alterations, most commonly in TP53 (41% mutation, 12% deletion), TSC2 (29% mutation, 6% deletion), RB1 (18% deletion), ATRX (24% mutation), MED12 (12% mutation), BRCA2 (12% deletion), CDKN2A (6% deletion) as well as FGFR3, NTRK1 and ERBB3 amplification (each 6%). Gene rearrangements (JAZF1-SUZ12; DNAJB6-PLAG1 and SFPQ-TFE3) were identified in three tumors. Integrating histopathologic, immunohistochemical and genetic findings, tumors from 4 patients were consistent with malignant PEComa (one TFE3-rearranged); 6 were classified as leiomyosarcomas; 3 showed overlapping features of PEComa and other sarcoma types (leiomyosarcoma or low-grade endometrial stromal sarcoma); and 2 were classified as sarcoma, not otherwise specified.
Our findings suggest that diagnostically challenging UMTs with myomelanocytic differentiation represent a heterogeneous group of neoplasms which harbor a diverse repertoire of somatic genetic alterations; these genetic alterations can aid classification.
Keywords: Genetics, Melanocytic differentiation, Myomelanocytic differentiation, Somatic mutations, Pathology, PEComa, Perivascular epithelioid cell tumor, Uterus
INTRODUCTION
Uterine sarcomas are uncommon mesenchymal tumors accounting for less than 5% of uterine malignancies.1 High-grade mesenchymal neoplasms arising in the uterus include leiomyosarcoma, high-grade endometrial stromal sarcoma, rhabdomyosarcoma, malignant perivascular epithelioid cell tumor (malignant PEComa) and undifferentiated uterine sarcoma, among others.2–4 Genetic studies have revealed alterations characteristic of some of these neoplasms (e.g. TSC1/TSC2 alterations and RAD51B fusions in PEComa,5–8 and DICER1 mutations and PAX3-FKHR and PAX7-FKHR fusions in rhabdomyosarcoma),9–11 and have delineated subsets of these tumors characterized by specific genetic alterations (e.g. YWHAE/NUTM2-rearranged and BCOR-altered high-grade endometrial stromal sarcoma,3,12,13 TFE3-rearranged PEComa,14 SMARCA4-deficient uterine sarcoma,15,16 and NTRK-rearranged fibrosarcoma-like uterine sarcoma).17
Most uterine sarcomas demonstrate characteristic histopathologic features. However, some tumors display ambiguous or overlapping histologic features, which may confound accurate characterization based on histopathologic features alone.2–4 As an example, uterine PEComas are rare neoplasms;6,14,18,19 and 5–10% of affected patients exhibit distant metastases at diagnosis, and 10–15% succumb to the tumor.19,20 Histologically, PEComas are characterized by spindle-shaped and/or epithelioid cells with clear-to-acidophilic cytoplasm, and may exhibit sclerosis and pericytomatous vasculature; they express markers of myogenic and melanocytic differentiation.19 PEComas exhibiting marked cytological atypia, conspicuous mitotic activity and/or tumor cell necrosis are classified as ‘malignant PEComa’.21 Although histopathologic diagnosis is straightforward in tumors homogenously or predominantly exhibiting the classic morphologic features, diagnosis is more challenging in tumors in which the prototypical features are poorly developed, seen only focally and/or present in conjunction with features seen in other neoplasms.
In this study, we sought to define the repertoire of somatic genetic alterations in a set of diagnostically challenging UMTs with ambiguous or equivocal morphologic features, and myomelanocytic differentiation. Furthermore, we sought to explore whether subsequent genomic analysis aided tumor classification.
MATERIALS AND METHODS
Case selection
This study was approved by Memorial Sloan Kettering Cancer Center’s (MSK’s) Institutional Review Board (#15–051). Seventeen mesenchymal neoplasms with myomelanocytic differentiation and ambiguous/equivocal histopathologic features which originated in the uterus (including several tumors for which the original diagnosis or differential diagnosis included PEComa), and were classified by specialist gynecologic pathologists based on review of routine histological sections and immunohistochemistry between 2010 and 2015 were identified from the archives of the Department of Pathology at MSK. Cases with available formalin-fixed, paraffin-embedded (FFPE) blocks of tumor were included in the study. Clinical data for the patients were obtained from the MSK Gynecologic Oncology clinical database. All available hematoxylin-and-eosin-stained slides were reviewed by gynecologic pathologists (RM and RAS) in an attempt to evaluate histomorphologic features suggestive of a particular subtype or line-of-differentiation. For example, fascicular arrangements of spindle cells with fusiform round-ended nuclei were suggestive of smooth muscle differentiation; tumors with tongue-like infiltration composed of small oval or rounded cells with scant cytoplasm were suggestive of endometrial stromal differentiation; and epithelioid and/or spindled cells with variably clear cytoplasm, variable nuclear pleomorphism, nested or corded architecture, pericytomatous vasculature and stromal hyalinization, allied with immunohistochemical expression of myogenic marker(s) and two or more melanocytic markers (see below) were suggestive of PEComas. Additional histopathologic data (including results of immunohistochemistry performed at the time of diagnosis) were retrieved from the MSK Pathology Laboratory Information System.
Immunohistochemical analyses
Immunohistochemical analyses were performed on representative tissue blocks using monoclonal antibodies to desmin (clone DE-R11, Dako), smooth muscle actin (SMA, clone 1A4, Dako), TFE3 (clone MRQ-37, Ventana), HMB45 (Dako), Melan-A (clone A103, LICR/in-house), pS6 (clone 91B2, CST) and pAKT(S473) (clone D9E, CST) for cases for which the original stained slides were not available for review. Immunohistochemistry was performed using a Leica Bond-3 automated platform (Leica, Buffalo Grove, IL), and a polymeric secondary kit (Refine, Leica) was used for the detection of the primary antibodies. Immunochemical results were evaluated by experienced histopathologists (RM and RAS) for the intensity of expression (weak, moderate or strong, compared to the intensity of staining of positive control tissue on the same slides) and percentage of tumor cells exhibiting expression. The results for pS6 were converted into an immunoreactive score (IRS), calculated as: intensity of staining (0 absent, 1 weak, 2 moderate, 3 strong) x percentage of cells staining (0–100), yielding IRS scores between 0 and 300. For the few cases for which tissue blocks or unstained slides for immunohistochemistry were not available, the immunohistochemical findings were obtained from the original pathology report issued by MSK gynecologic pathologists at the time of diagnosis.
TFE3 fluorescence in situ hybridization
FFPE tissue sections of 4 μm thickness with marked tumor areas were used for fluorescence in situ hybridization (FISH) as described previously,22 using TFE3 dual color break-apart probes (Zytovision, Bremerhaven, Germany), which contain a 5′ probe of 605 kb labeled in green and a 3′ probe of 505 kb labeled in orange. Signal analysis was performed in combination with morphologic correlation, and at least 100 interphase cells within the marked tumor area were evaluated. The normal result is a combination green-orange signal. The result was considered positive for TFE3 rearrangement only if >10% of cells showed a split signal pattern.
Targeted massively parallel sequencing
DNA samples extracted from FFPE tumors and matched normal tissues or blood were analyzed using the MSK Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) assay which targets all coding exons of 341–410 cancer-related genes, as previously described.23–25 Sequencing data were analyzed and somatic mutations identified, as previously described.24,25 Somatic copy number alterations (CNAs) and loss of heterozygosity (LOH) were identified using FACETS.26 The cancer cell fractions (CCFs) of all mutations were computed using ABSOLUTE (1.0.6).27 Mutation hotspots were assigned according to Chang et al.28 For comparison, reported mutation frequencies from uterine myogenic tumors exhibiting cytologic atypia, namely leiomyosarcomas29–31 and leiomyomas with bizarre nuclei (LMBN)32 were obtained.
Targeted RNA-Sequencing for fusion gene analysis
Tumor RNA was extracted from macrodissected FFPE tissue sections mounted on charged glass slides and subjected to the MSK-Solid Fusion assay, a custom targeted RNA-based panel that utilizes the Archer Anchored Multiplex PCR (AMP) technology to detect gene fusions and oncogenic isoforms in selected protein-coding exons of 62 genes.33 Archer analysis software V5.0 was used for data analysis.
RESULTS
Clinical and pathological features
We analyzed 17 tumor samples from 15 patients aged 43–73 (median 61) years. One tumor sample was analyzed in each of 13 patients, and 2 samples (primary uterine tumor and recurrence/metastasis) from 2 patients (Table 1). None of the women had stigmata of tuberous sclerosis complex or any documented germline mutations.
Table 1.
Clinical features of high-grade uterine mesenchymal tumors
| Patient | Sample ID | Age at diagnosis (years) | FIGO stage at diagnosis | Subsequent recurrences | mTORi therapy | Best response to mTORi | Follow-up duration (months) | Last follow-up status |
|---|---|---|---|---|---|---|---|---|
| 1 | UMT01 | 43 | IIIC | Soft tissue | 71.3 | NED | ||
| 2 | UMT02 | 66 | IB | Lung | 32.7 | DOD | ||
| 3 | UMT03 | 71 | IA | 43.0 | NED | |||
| 4 | UMT04 | 73 | IA | 35.5 | NED | |||
| 5 | UMT05 | 53 | IIIA | Peritoneum | Sirolimus | POD | 19.6 | DOD |
| 6 | UMT06, UMT06-R | 57 | IA | Peritoneum | LY3023414 (PI3K/mTOR inhibitor) | POD | 21.3 | DOD |
| 7 | UMT07 | 47 | IB | Soft tissue | 81.8 | NED | ||
| 8 | UMT08 | 70 | IB | Pelvis | 1.8 | NED | ||
| 9 | UMT09, UMT09-R | 65 | IIIC | Lung | LY3023414 (PI3K/mTOR inhibitor) | SD | 43.0 | AWD |
| 10 | UMT10 | 63 | IA | VS-5584 (PI3K/mTOR inhibitor) | POD | 51.6 | AWD | |
| 11 | UMT11 | 48 | IVB | Temsirolimus | PR | 9.5 | DOD | |
| 12 | UMT12 | 67 | IVB | Peritoneum | 58.2 | AWD | ||
| 13 | UMT13 | 51 | IIB | LY3023414 (PI3K/mTOR inhibitor) | POD | 8.0 | DOD | |
| 14 | UMT14 | 58 | IA | 3.6 | NED | |||
| 15 | UMT15 | 61 | IVB | Liver | Temsirolimus | PR | 7.9 | DOD |
FIGO – International Federation of Gynecology and Obstetrics; mTORi – mTOR inhibitor; POD – progression of disease; SD – stable disease; PR – partial remission; AWD – alive with disease; DOD – dead of disease; NED – no evidence of disease
The tumors showed a range of histopathologic appearances, as detailed in Table 2 (Figures 1–3). Because of the histologically ambiguous nature of the selected cohort, the morphologic clues were not sufficiently definitive to allow classification; at best, they were suggestive of a particular subtype or line-of-differentiation, as described in the Methods. The majority of tumors were composed of an admixture of spindle and epithelioid cells, with only occasional cases being composed exclusively of one cell type or the other. The tumor cells contained amphophilic or clear cytoplasm and mostly exhibited moderate to marked nuclear pleomorphism, including bizarre or giant tumor cells in several cases. Coagulative tumor necrosis was commonly seen, and the tumor mitotic rate was highly variable between tumors. Some tumors (e.g. UMT01 and UMT07) showed sclerosis (Figures 1a and 1f) and pericytomatous (staghorn) vasculature (e.g. UMT02, UMT04, UMT10).
Table 2.
Key histopathologic features of high-grade uterine mesenchymal tumors
| Sample ID | Sample location | Sample type | Original pathologic diagnosis | Size (mm) | Edge | Cytoarchitecture | Nuclear pleomorphism | Necrosis | TMR | LVI | Sclerosis | Pericytomatous vasculature | Other findings |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UMT01 | Soft tissue | Recurrence | High-grade sarcoma, consistent with malignant PEComa | 155 | NA | E (nested and corded) | ++ | CTN | 20 | Yes | Yes | Nested & corded arrangements of epithelioid cells with variable amphophilic to clear cytoplasm | |
| UMT02 | Lung | Recurrence | Malignant PEComa | 120 | I | S+E | +++ | INF | 34 | Yes | Yes | Yes | S+E cells in sheets and vague fascicles associated; marked nuclear pleomorphism incl MNGCs |
| UMT03 | Uterus | Uterus | Malignant epithelioid neoplasm with smooth muscle differentiation, most consistent with malignant PEComa; differential diagnosis includes epithelioid LMS | 50 | C | S+E | +++ | No | 5 | Yes | Vague nodules of predominantly epithelioid cells forming a nodular deposit in myometrium | ||
| UMT04 | Uterus | Uterus | Malignant myogenic neoplasm, favor LMS (on morphology); differential diagnosis of PEComa based on IHC showing rare HMB45+ | 45 | I | S + focally E | ++ | CTN | 5 | No | Yes | Yes | Spindled areas show tight fascicular arrangements; Epithelioid areas are small nodules admixed with the spindled areas |
| UMT05 | Peritoneum | Recurrence | Hybrid LMS/PEComa (primary tumor); recurrence seems to be predominantly epithelioid PEComa component | 170 | I | E+S | +++B | CTN | 30 | Yes | Sheets of highly pleomorphic S+E cells; recurrence showed more marked pleomorphism; Uterine tumor originally called LMS, but revised to hybrid LMS/PEComa after review of recurrence | ||
| UMT06 | Uterus | Uterus | Endometrial curettage: LMS with epithelioid features; Hysterectomy: malignant PEComa (based on focal HMB45+/MelanA+) | 30 | I | E+S | +++ | No | 10 | No | Biphasic tumor including fascicular spindle cell areas with moderate nuclear grade; focal areas composed of large spindle/epithelioid cells with marked nuclear pleomorphism and nodular areas composed of small epithelioid cells with moderate nuclear grade | ||
| UMT06-R | Peritoneum | Recurrence | Malignant PEComa | 60 | NA | E | +++ | CTN | 38 | No | Sheets and nodules of epithelioid cells with amphophilic or clear cytoplasm | ||
| UMT07 | Soft tissue | Recurrence | Malignant PEComa | 80 | I (focally) | E | +++ | CTN | 18 | No | Yes | Large epithelioid cells with abundant clear cytoplasm arranged in large nests/nodules separated by broad collagenous septa | |
| UMT08 | Pelvis | Recurrence | Malignant PEComa | 62 | I (focally) | S+E | +++B | No | 10 | No | Yes | Whorling fascicles of spindle and focally epithelioid cells | |
| UMT09 | Uterus | Uterus | High grade sarcoma with epithelioid features and myogenic differentiation, favor malignant PEComa | 45 | I | E (round cell) | +++ | CTN | 14 | Yes | Yes | Markedly corded appearance, with cords of epithelioid cells separated by bands of hyalinized collagen | |
| UMT09-R | Lung (core biopsy) | Recurrence | Malignant PEComa | NA | NA | E (round cell) | + | No | 12 | No | Yes | Cords and some nests of epithelioid cells separated by densely hyalinized stromal septa | |
| UMT10 | Uterus | Uterus | High grade sarcoma, favor malignant PEComa | 38 | NA | S+E | +++ | NA | 34 | No | |||
| UMT11 | Uterus | Uterus | High grade spindle and pleomorphic neoplasm, consistent with malignant PEComa | 105 | NA | S+E | +++ | CTN | High | Yes | |||
| UMT12 | Peritoneum | Recurrence | Uterine tumor: PEComa with extensive smooth muscle differentiation versus LMS (former favored after review of recurrence showing MITF+ and rare HMB45+) | 55 | C | S | ++ | CTN | 5 | No | Wide range of morphologic appearances: some areas showed bland appearing storiform fascicles of spindle cells; other areas showed much greater cellularity and nuclear atypia; still other areas composed of spindled and rare epithelioid cells with some clear cell change | ||
| UMT13 | Uterus | Uterus | Malignant PEComa | 240 | I | E+S | +++ | CTN | 10 | Yes | Yes | Spindled and epithelioid cells with eosinophilic and clear cytoplasm | |
| UMT14 | Uterus | Uterus | PEComa, with features indicative of intermediate to aggressive clinical behavior | 90 | NA | S | ++B | No | 11 | No | Scattered pleomorphic cells | ||
| UMT15 | Liver (core biopsy) | Recurrence | Malignant epithelioid neoplasm consistent with malignant PEComa | NA | NA | E | +++B | CTN | 25 | Yes | Sheets of highly pleomorphic epithelioid cells with eosinophilic cytoplasm |
NA – not assessable; TMR – tumor mitotic rate per 10 high-power fields; LVI – Lymphovascular invasion; I – irregular; C – relatively circumscribed; E – epithelioid cells; S – spindle cells; CTN – coagulative tumor necrosis; INF – infarction; Nuclear pleomorphism: + - low, ++ - moderate; +++ - marked, B - bizarre/giant cells; LMS – leiomyosarcoma; IHC – immunohistochemistry
Figure 1. Histopathologic features of selected cases of PEComa (see text and tables for further details).
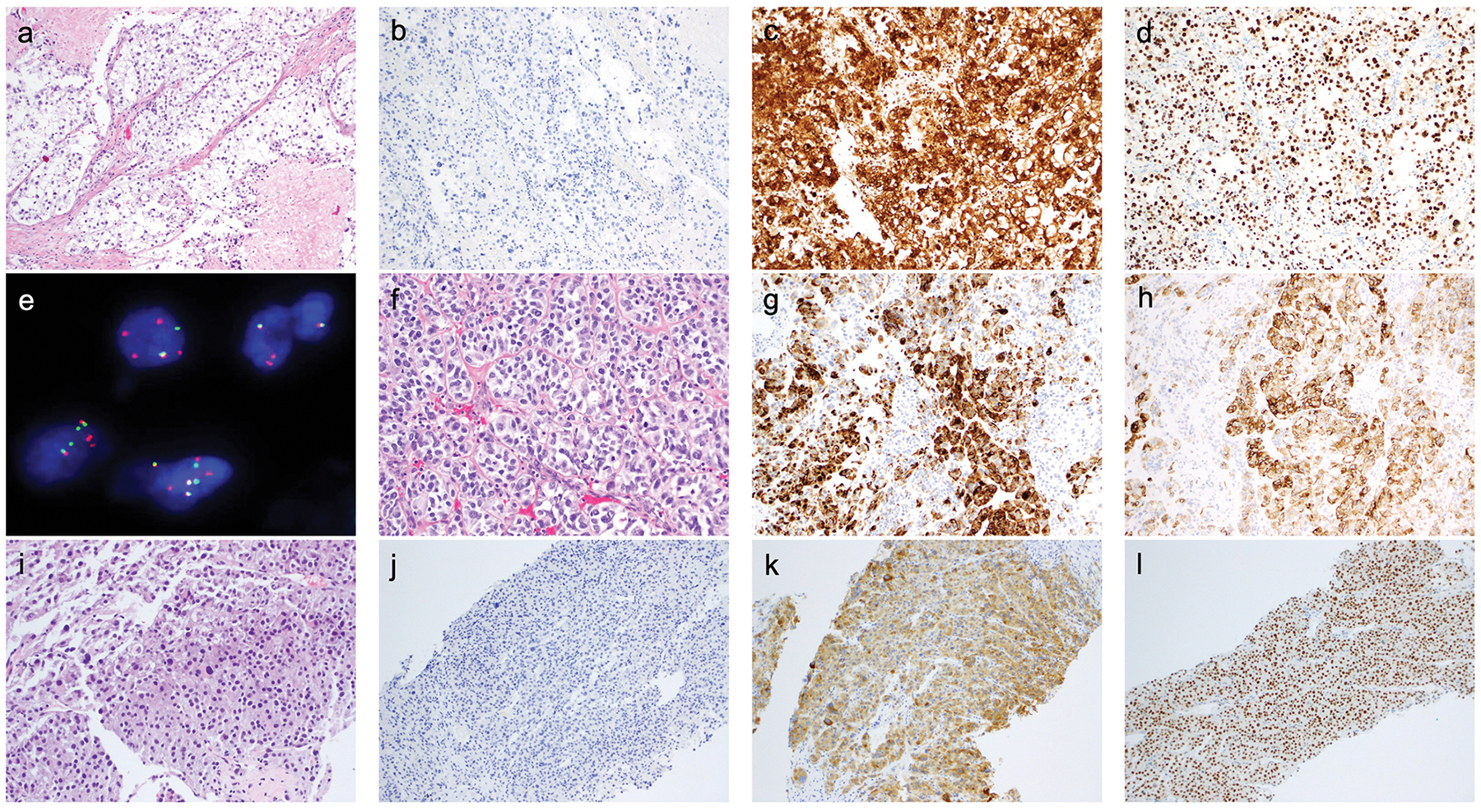
a-e) UMT07. a) H&E – nests and nodules, separated by collagenous septa, of large epithelioid cells with abundant clear cytoplasm; b) Desmin; c) HMB-45; d) TFE3; e) FISH for TFE3. The normal result is a combination green-red signal. 85% of cells show separate green (5’ TFE3) and orange (3’ TFE3) signals, indicative of TFE3 rearrangement.
f-h) UMT01. f) H&E – epithelioid cells with variably amphophilic-to-clear cytoplasm arranged in cords and nests, associated with some stromal sclerosis; g) Desmin; h) HMB-45.
i-l) UMT15. i) H&E – sheets of pleomorphic epithelioid cells with eosinophilic cytoplasm; j) Desmin; k) Melan-A; l) TFE3.
Figure 3. Histopathologic features of selected cases (see text and tables for further details).
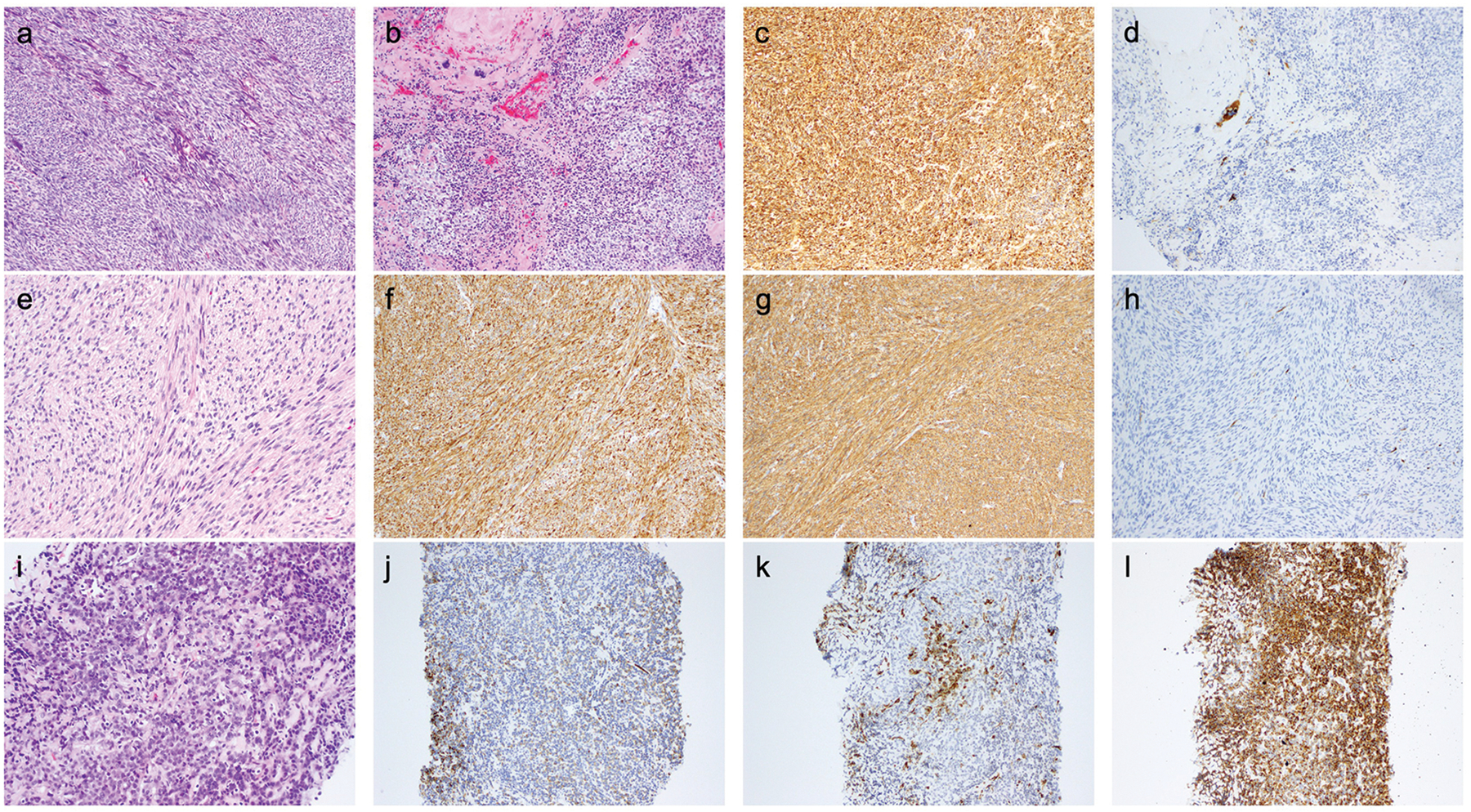
a-d) UMT04 - myogenic sarcoma, most likely leiomyosarcoma. a and b) H&E – tumor composed of tight fascicles of spindled cells with fusiform nuclei (a) and focally nodules of epithelioid cells (b) admixed with the spindled areas; c) Desmin; d) HMB-45.
e-h) UMT12 - myogenic sarcoma, most likely leiomyosarcoma. e) H&E - storiform fascicles of spindle cells with some nuclear atypia and focal clear cell change; other areas showed coagulative tumor necrosis and up to 5 mitoses per 10 high-power fields; f) Desmin; g) SMA; h) HMB-45.
i-l) UMT09-R - sarcoma associated with low-grade endometrial stromal sarcoma-like and PEComa-like features. i) H&E – cords and nests of relatively small epithelioid cells associated with stromal hyalinization; j) Desmin; k) SMA; l) HMB-45.
At least focal immunohistochemical expression of one or more melanocytic markers (HMB45, Melan-A or MiTF) was identified in all tumors and expression of 2 of these markers was seen in in 10/17 (59%); myogenic markers (desmin or SMA) were expressed in 13/17 (76%) tumors. Immunohistochemistry for pS6 and pAKT(S473) was performed to evaluate mTOR pathway activation in 12 tumor samples (10 from 10 patients, and 2 from one patient). Variable expression of pS6, a surrogate for mTOR pathway activation, was identified in all 12 tumor samples in which it could be evaluated (Table 3). pAKT(S473) was negative in all available tumor samples (n=12). Among the 11 unique patients in whose tumors pS6 immunohistochemistry was performed, the median IRS scores in tumors with an integrated diagnosis of malignant PEComa (n=3), non-PEComa (n=6) and sarcomas with features of PEComa and another entity (n=2, for which we chose the pS6 IRS in the recurrences) were 225 (mean 195, range 60–300), 70 (mean 113, range 10–300) and 68 (mean 68, range 45–90), respectively. The differences between the groups failed to reach statistical significance (one-way ANOVA: F=0.89, p=0.44).
Table 3.
Key immunohistochemical and genomic features of high-grade uterine mesenchymal tumors
| Sample ID | Desmin | SMA | HMB45 | Melan-A | TFE3 | pS6 | Other positive | TFE3 FISH | RNA sequencing | Key mutations | Key CNAs | Best diagnosis based on morphology, immunophenotype and genomics |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UMT01 | +++60 | ++20 | ++−+++70 | 0 | ++70 | ++−+++90 | MiTF | Neg | Fail | TP53, RB1, BRD4 | PRDM1 | Malignant PEComa |
| UMT02 | ++40 | +++100 | +++50 | 0 | ++40 | ++30 | MiTF | Neg | Neg | TP53, MED12 | TP53 | Leiomyosarcoma |
| UMT03 | +++100 | +++20 | +5 | 0 | +++100 | ++5 | Neg | Fail | ESR1 | Myogenic neoplasm, most likely leiomyosarcoma | ||
| UMT04 | +++100 | +++100 | ++1 | 0 | ++80 | ++40 | Neg | DNAJB6-PLAG1 | TP53, ATRX | RB1, RAD50 FGFR3 | Myogenic sarcoma, most likely leiomyosarcoma | |
| UMT05 | 0 | 0 | ++15 | ++20 | 0 | +++100 | Neg | Fail | TP53, DAXX, DICER1 | NTRK1 | Sarcoma, NOS | |
| UMT06 | +++40* | +++80 | +++5** | 0 | +70 | +30 | Neg | Neg | TP53, ATRX | Sarcoma associated with leiomyosarcoma-like and PEComa-like features | ||
| UMT06-R | 0 | +++30 | +++20 | 0 | ++100 | +++15 | Neg | Neg | TSC2, ATRX | |||
| UMT07 | 0 | 0 | +++100 | +10 | +++100 | ++30 | Positive | SFPQ-TFE3 | Malignant PEComa, TFE3-rearranged | |||
| UMT08 | +++100 | +++100 | 0 | +2 | +30 | +++70 | MiTF | Neg | Fail | KDM6A, NTRK3, JAK3 | ERCC2 | Myogenic sarcoma, most likely leiomyosarcoma |
| UMT09 | Pos | Focal | Pos | Focal | Very focal | TSC2 | ROS1 | Sarcoma associated with low-grade endometrial stromal sarcoma-like and PEComa-like features | ||||
| UMT09-R | ++50 | +++20 | +++100 | 0 | ++90 | +++30 | Neg | JAZF1-SUZ12 | TSC2 | |||
| UMT10 | Pos | Pos | Focal | Neg | Patchy weak | TP53, MED12 | RB1, BRCA2 | Leiomyosarcoma | ||||
| UMT11 | Neg | Neg | Pos | Neg | TSC2 | TP53 | Malignant PEComa | |||||
| UMT12 | +++100 | +++100 | +++5 | 0 | 0 | ++10 | MiTF | Neg | Neg | ERBB3, NCOR1 | Myogenic sarcoma, most likely leiomyosarcoma | |
| UMT13 | Neg | Patchy | Focal strong | Focal strong | ATRX | TP53, BRCA2 | Sarcoma, NOS | |||||
| UMT14 | Pos | Pos | Focal | Focal | TSC2, ASXL1 | CDKN2A | Sarcoma associated with leiomyosarcoma-like and PEComa-like features | |||||
| UMT15 | 0 | 0 | +++15 | ++100 | +++100 | +++100 | Neg | Neg | TP53 | TSC2, RB1 | Malignant PEComa |
Immunohistochemistry: Results described as a combination of staining intensity (+ weak, ++ moderate, +++ strong) and percentage of tumor cells exhibiting expression; for UMT09, UMT10, UMT11, UMT13 and UMT14, unstained slides for immunohistochemistry were not available; immunohistochemical findings are as described in original report.
Desmin: expression in spindle and part of round cell areas, negative in other round cell areas; HMB45: *mainly in cells in septa separating nests of epithelioid cells;
positive in bizarre large cells, negative everywhere else.
Key immunohistochemical and genetic findings that (in combination with the histopathologic features) contributed to the diagnosis
RNA sequencing: Fail – sequencing failure due to poor RNA quality; Neg – negative for gene rearrangements
Seven patients in our cohort received systemic therapy including an mTOR inhibitor; the best responses were partial remission (PR, n=2), stable disease (SD, n=1) and progression of disease (POD, n=4) (Table 1).
Genetic alterations
FISH for TFE3 was performed in 12 cases with available tissue and showed a clear split signal indicative of TFE3 rearrangement in 1 of 12 (8%) tumors (Figure 1e). In parallel, 8 tumors from which RNA was obtainable were subjected to targeted RNA-Sequencing using ArcherFusion Plex, and gene rearrangements were identified in three tumors: SFPQ-TFE3 in UMT07, DNAJB6-PLAG1 in UMT04 and JAZF1-SUZ12 (exon3-exon2) in UMT09-R (Figure 4).
Figure 4. Somatic genetic alterations.
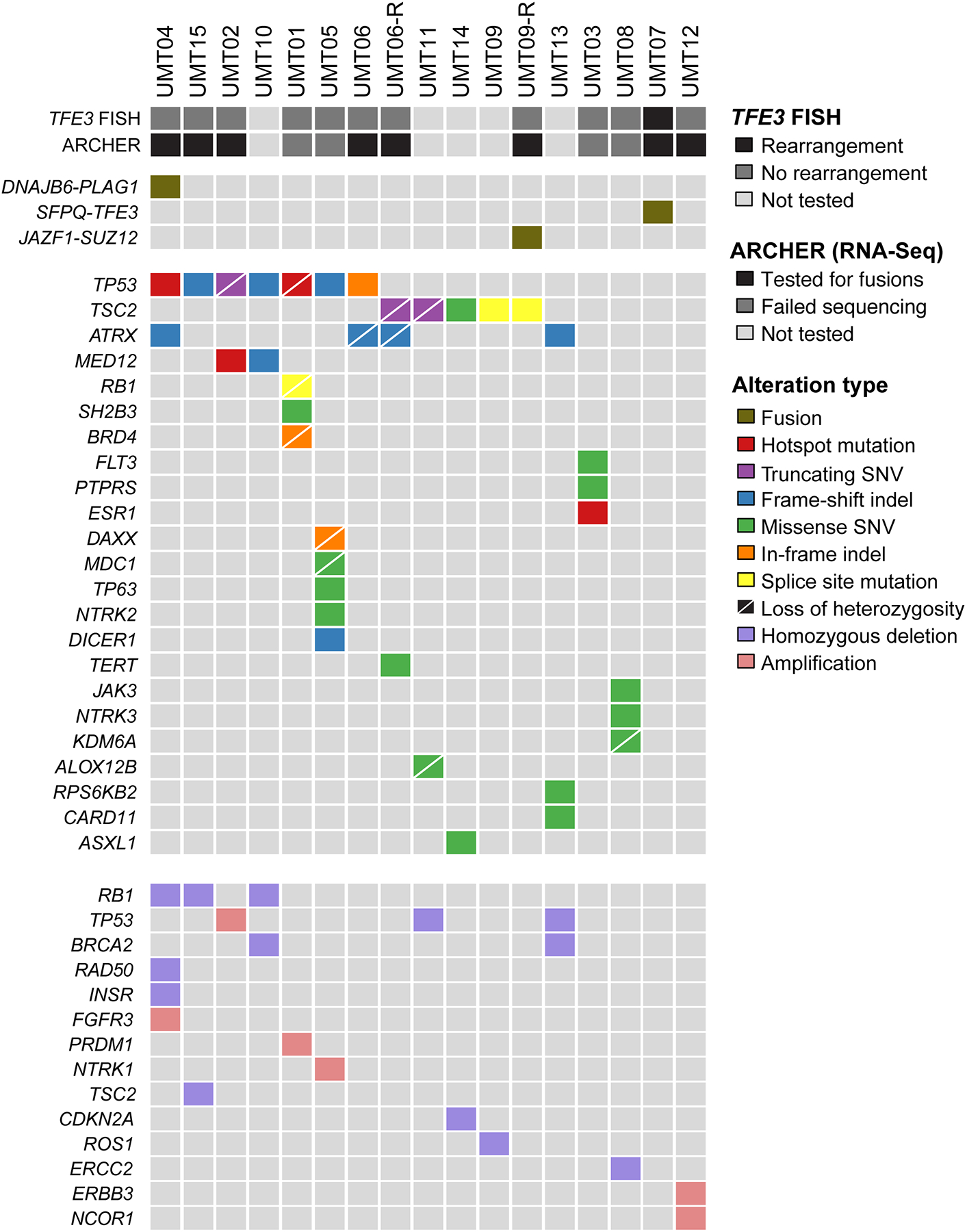
The most common mutations involved TP53 (41%) and TSC2 (29%). Other likely oncogenic mutations were found in: ATRX (24%), MED12 (12%), and ESR1 (Y537S hotspot), DAXX (in-frame indel with LOH) and DICER1 (frameshift indel) and RB1 (splice site; each 6%. At the gene copy number level, homozygous deletions of RB1 (18%), BRCA2 (12%), TP53 (12%), TSC2 (6%) and CDKN2A (6%) were the most common. Amplifications were present in FGFR3, NTRK1 and ERBB3 (each 6%). Gene rearrangements identified by fluorescence in situ hybridization (FISH) or Archer targeted RNA-sequencing (top), non-synonymous somatic mutations (middle) and amplifications and homozygous deletions (bottom) identified in 17 tumors from 15 patients using targeted massively parallel sequencing. Mutation types are color-coded according to the legend. Loss of heterozygosity is depicted by a diagonal bar. Indel, small insertion and deletion; SNV, single nucleotide variant.
Tumor and normal tissues of all cases were subjected to high-depth massively parallel sequencing (median coverage of 747x (325x-1289x) and 392x (189x-586x), respectively), which revealed that the tumors were heterogeneous at the genetic level. We observed recurrent somatic mutations and CNAs affecting cell cycle-related genes, including TP53, CDKN2A and RB1 in 59% (10/17) of tumors. The most common genetic alterations were TP53 mutations (7/17; 41%) and TSC2 mutations, which were identified in 29% (5/17). No TSC2 mutations were detected in the TFE3-rearranged tumor. Furthermore, there were likely oncogenic mutations affecting ATRX (n=4/17, 24%), MED12 (2/17, 12%), and ESR1 (Y537S hotspot), DAXX (in-frame indel with LOH) and DICER1 (frameshift indel) and RB1 (splice site; each 1/17, 6%; Figure 4; Supplementary Figure S1). At the gene copy number level, homozygous deletions of RB1 (3/17, 18%), BRCA2 (2/17, 12%), TP53 (2/17, 12%), TSC2 (1/17, 6%) and CDKN2A (1/17, 6%) were the most common. Amplifications were present in FGFR3, NTRK1 and ERBB3 (each 1/17, 6%; Figures 4 and 5).
Figure 5. DNA copy number alterations.
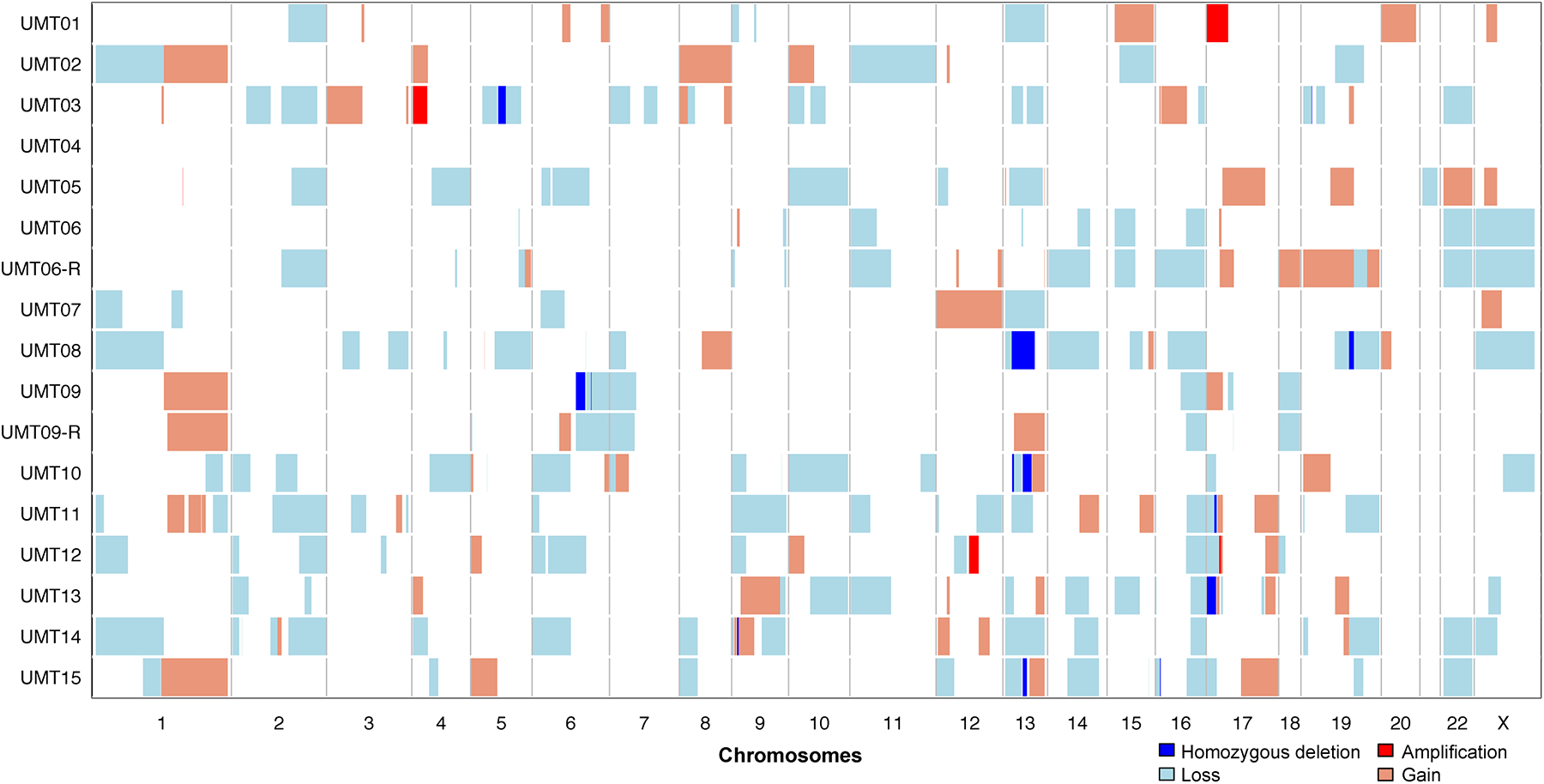
DNA copy number alterations of 17 tumors from 15 patients subjected to targeted massively parallel sequencing showed homozygous deletions at the loci encoding RB1 (18%), BRCA2 (12%), TP53 (12%), TSC2 (6%) and CDKN2A (6%) were the most common. Amplifications were present in FGFR3, NTRK1 and ERBB3 (each 6%). Chromosomes are shown along the x-axis, and cases along the y-axis. Homozygous deletions, dark blue; losses, light blue; gains, orange; amplifications, red.
Two tumors (UMT07 and UMT12) did not harbor any non-synonymous mutations in the 341–410 cancer-related genes analyzed. Instead, UMT07 harbored a SFPQ-TFE3 rearrangement; and UMT12 showed ERBB3 and NCOR1 amplification, along with diffuse myogenic marker expression and minimal melanocytic marker expression, most consistent with a myogenic sarcoma.
DISCUSSION
Although most uterine sarcomas demonstrate characteristic histopathologic features, diagnostic challenges may be posed by high-grade uterine mesenchymal tumors which display ambiguous or overlapping histologic features.2–4
The initial diagnosis or differential diagnosis in many of the tumors in this study included malignant PEComa, based on morphological and immunohistochemical evaluation, but without supportive molecular genetic evidence, which was not available for most of these cases at the time of diagnosis. The histopathologic diagnosis of uterine PEComa has traditionally relied on the finding of one or more suggestive histologic features (epithelioid +/− spindle cell shape, nested architecture, clear cytoplasm, variable nuclear atypia and mitotic activity, multinucleate giant cells and spider-like cells, pericytomatous vasculature and stromal hyalinization), allied with immunohistochemical evidence of myomelanocytic differentiation.6,19,34–36
Due to the overlap in morphologic and immunophenotypic features between epithelioid smooth muscle tumors and PEComas, some authors have proposed that uterine PEComa is not a distinct entity but rather a variant of epithelioid smooth muscle tumor expressing melanocytic markers.37–41 However, others argue that the morphologic features and genetic alterations seen in PEComa (e.g. in TSC2) together with the rarity and focality of melanocytic marker expression in muscle tumors support the existence of PEComa as a distinct entity.18
In hepatic and renal PEComas (angiomyolipomas), expression of melanocytic markers can be variable in intensity and extent,42 and uniform criteria or cut-offs for melanocytic marker expression are lacking; in these tumors, the immunohistochemical panel for diagnostic confirmation generally includes 2 melanocytic markers. Based on the finding that the extent of melanocytic marker expression in uterine PEComas was variable (weak, heterogenous or strong in different tumors), the authors of one study proposed that expression of two melanocytic markers with co-expression of at least one muscle marker in a tumor exhibiting typical morphologic features is necessary for a diagnosis of uterine PEComa.18 However, uterine smooth muscle tumors can also display at least focal positivity for melanocytic markers such as HMB-45 (at least focally in 31–36% of conventional leiomyosarcomas),43,44 Melan-A (32%) and MiTF (57%).38,39,41,43–45 Epithelioid smooth muscle tumors have been reported to exhibit HMB-45 in 56–80%,38,44 Melan-A in 56%,44 and MiTF in 22%.44 In one study of five leiomyosarcomas with spindled and epithelioid morphology, HMB-45 expression was identified in clear-cell epithelioid areas and also in spindled areas in four tumors; expression was seen in 1–25% of cells in 2 tumors, and in 26–50% of cells in 2 tumors.38 Challenges in the distinction of PEComa from smooth muscle tumors are compounded by cases such as UMT06/UMT06-R in this study (see below) and the reported development of a clear cell, diffusely HMB-45-positive, predominantly SMA-negative metastasis from a HMB-45-negative primary epithelioid leiomyosarcoma,39 which highlight the genotypic and phenotypic overlap between PEComa and leiomyosarcoma. Furthermore, it has been reported in one study of 35 uterine leiomyosarcomas that 11% of tumors expressed HMB-45, and none expressed Melan-A.46 Many of the tumors in the present study that were classified as ‘not PEComa’ following review of the histologic, immunohistochemical and genomic features exhibited varying degrees (usually focal) of expression of HMB-45 and Melan-A (Table 3). These data argue against placing too much weight on melanocytic marker expression (especially if weak or focal in a tumor with equivocal morphologic features) when making a pathologic diagnosis of uterine PEComa.
Genomic alterations in PEComas and smooth muscle neoplasms
PEComas harbor TSC1 and TSC2 mutations5,47 and rearrangements in TFE37,8 and RAD51B.6,8 However, these alterations are not universally found in tumors that are considered to be diagnostic of PEComas. Two recent studies explored genetic alterations in PEComas. One study included 11 uterine PEComas (of which 4 underwent targeted massively parallel sequencing and 5 underwent RNA sequencing),8 and found TSC2 mutations in 8/13 (62%) PEComas of all sites, which included 2/4 (50%) uterine PEComas.8 The other study used FISH to investigate TFE3 and RAD51B rearrangements in 32 uterine PEComas.6 These studies revealed the presence of rearrangements involving TFE3 and RAD51B in a subset of uterine PEComas.6,8 TFE3 rearrangements and TSC2 mutations were found to be mutually exclusive.8
We found TSC2 mutations in 29% of our cases, all of which were classified as malignant PEComas or myogenic sarcomas with PEComa-like features. TSC2 mutations were not identified in leiomyosarcomas in 2 studies (n=76),29,31 but missense mutations of unknown significance in TSC2 were reported in 4% (2/49 cases) in one series.30 The histopathologic features of these TSC2-mutant cases were not described; one of them also harbored a TP53 mutation, and the other showed alterations in several other genes, including ATRX.30 The latter 2 abnormalities are among the most common mutations reported in uterine leiomyosarcoma.29–31 Along with RB1 and, to a lesser extent, MED12, the frequencies of these “leiomyosarcoma-associated” mutations are generally similar to those in our cohort. Furthermore, TP53 and MED12 mutations and FH mutations and deletions were the most common alterations in a study of LMBN.32 In addition, one case harbored a truncating single nucleotide variant in TSC2 (as well as an in-frame indel in TP53).32
Tumors classified as PEComas: TSC2, TFE3 and immunohistochemical findings
Based on our findings, we propose a set of immunohistochemical and genetic criteria of varying utility for the diagnosis of uterine PEComa (Table 4).
Table 4.
Criteria for the diagnosis of uterine PEComas: comments based on our findings in 17 high-grade uterine mesenchymal tumors
| Criterion | Comments based on the findings in the present study | Verdict on utility for diagnosis of uterine PEComa |
|---|---|---|
| Melanocytic marker expression | Most of the tumors that were interpreted as PEComa in light of the combined evidence from morphology and genetic analysis showed non-focal expression (>5% of tumor) of at least 2 melanocytic markers (HMB-45, Melan-A and/or MiTF). Given that smooth muscle tumors are known to express melanocytic markers (usually one marker, and usually focally), the identification of expression of 2 or more melanocytic markers seems to be supportive of a diagnosis of PEComa. However, in the presence of other supportive evidence (e.g. TSC2 mutation in PEC14), the identification of one melanocytic marker, along with absent myogenic marker expression, may be sufficient for a diagnosis of PEComa. |
Useful. Readily available and therefore recommended for evaluation of tumors considered to be uterine PEComa on morphological grounds. Expression of 2 markers ((HMB45, Melan-A) would be supportive of a PEComa diagnosis (except in cases with TFE3 rearrangements, which are generally only positive for HMB-45). |
| TFE3 alterations | Unequivocal identification of TFE3 rearrangement (seen in one case in our cohort) was seen in a tumor that showed strong HMB45 expression and absent myogenic marker expression. This immunoprofile is characteristic of PEComas harbouring TFE3 rearrangement. | Useful. Identification of bona fide TFE3 rearrangement (via FISH or sequencing) is sufficient for a diagnosis of TFE3-rearranged PEComa. Strong immunoexpression of HMB45 and absent expression of myogenic markers is characteristic of TFE3-rearranged PEComas. However, immunohistochemistry for TFE3 is not specific for a diagnosis of PEComa. |
| TSC2 alterations | Although these might be considered definitional of PEComa, given their association with PEComas at other anatomic site and mTOR pathway activation, TSC2 alterations have also been identified in rare smooth muscle tumors in humans and rodent models (see discussion). Therefore, they cannot be used in isolation, but may be useful in conjunction with other criteria, e.g. melanocytic marker expression | Somewhat useful. While their identification cannot unequivocally make a diagnosis of PEComa, the finding of TSC2 alterations (in the absence of other features indicating alternative diagnoses) along with supportive immunohistochemical evidence, may be helpful in the diagnosis of PEComa |
| Myogenic marker expression | While myogenic marker expression may be seen in conjunction with melanocytic marker expression and/or TSC2 alterations, cases that meet criteria for PEComa may lack myogenic marker expression, particularly when they harbor TFE3 rearrangements | Potentially useful. Not all PEComas show myogenic marker expression (especially those harboring TFE3 rearrangements), but these markers may be useful in exploring differential diagnostic possibilities in cases in which other criteria for PEComa are not met |
One tumor (UMT07) showed evidence of TFE3 translocation, along with additional complex genomic aberrations of the region, by FISH (Figure 1e). The presence of an SPFQ-TFE3 translocation, reported previously in PEComas,8 was confirmed by targeted RNA-Sequencing. Typical of reported examples of TFE3-translocated PEComas,7,14,18 UMT07 was also characterized by large alveolar nests of pleomorphic epithelioid cells with abundant clear cytoplasm, absent myogenic marker expression and strong immunoexpression of TFE3 (Figures 1a–d). We did not identify RAD51B rearrangements in our cohort.
UMT01, UMT11 and UMT15 were malignant PEComas. UMT01 was composed of alveolar groups of epithelioid cells with clear cytoplasm associated with some sclerosis. It exhibits convincing evidence of myomelanocytic differentiation in a substantial proportion of tumor cells (Figures 1f–h). UMT11 was a pleomorphic spindle and epithelioid cell tumor and UMT15 was composed of highly pleomorphic epithelioid cells; both exhibited melanocytic marker expression and coagulative necrosis (Figures 1i–k), as well as TSC2 alterations (truncating mutation with LOH in UMT11 and TSC2 homozygous deletion in UMT15). The phenotype is in keeping with malignant PEComa. UMT15 showed strong diffuse expression of TFE3 (Figure 1l), but no evidence of TFE3 rearrangement was identified.
Tumors classified as sarcomas with PEComa-like features
Based on the combination of histopathologic, immunohistochemical and genetic findings, a few tumors showed overlapping features of PEComa and other sarcoma types, as detailed below. The nature and biological significance of these difficult-to-classify sarcomas, specifically whether they represent divergent differentiation or collision tumors, is unclear.
UMT06-R (lung recurrence) showed a truncating mutation in TSC2 with LOH, as well as a frameshift mutation in ATRX with LOH. The matching primary uterine tumor (UMT06) shared the ATRX alteration and also had a TP53 in-frame indel, but lacked detectable alterations in TSC2. Especially in the primary tumor, HMB45 expression was confined to the bizarre epithelioid cells (Figure 2a–d). Loss of TSC2 function (via germline TSC2 mutation coupled with LOH of TSC2) has been demonstrated to be associated with uterine smooth muscle tumors in rodent models48–51 and studies in human tumors have shown that approximately 4% of LMBN32 and leiomyosarcoma (including one case showing a missense TSC2 mutation and a frameshift indel in ATRX)30 harbor somatic TSC2 alterations. These data call into question the specificity of TSC2 alterations for a diagnosis of PEComa, and suggest an alternative diagnostic possibility (sarcoma associated with leiomyosarcoma-like and PEComa-like features) for UMT06-R/UMT06. In a similar vein, UMT14 harbored missense mutations in TSC2 and ASXL1, and a homozygous deletion of CDKN2A. It was composed of spindle cells with moderate nuclear pleomorphism (including bizarre cells) and up to 11 mitoses per 10 high-power fields but lacking coagulative tumor necrosis or lymphovascular invasion. The tumor expressed SMA and desmin, but showed only focal HMB45 and Melan-A expression. Based on the available evidence, this tumor also likely represents a sarcoma associated with leiomyosarcoma-like and PEComa-like features.
Figure 2. Histopathologic features of selected cases (see text and tables for further details).
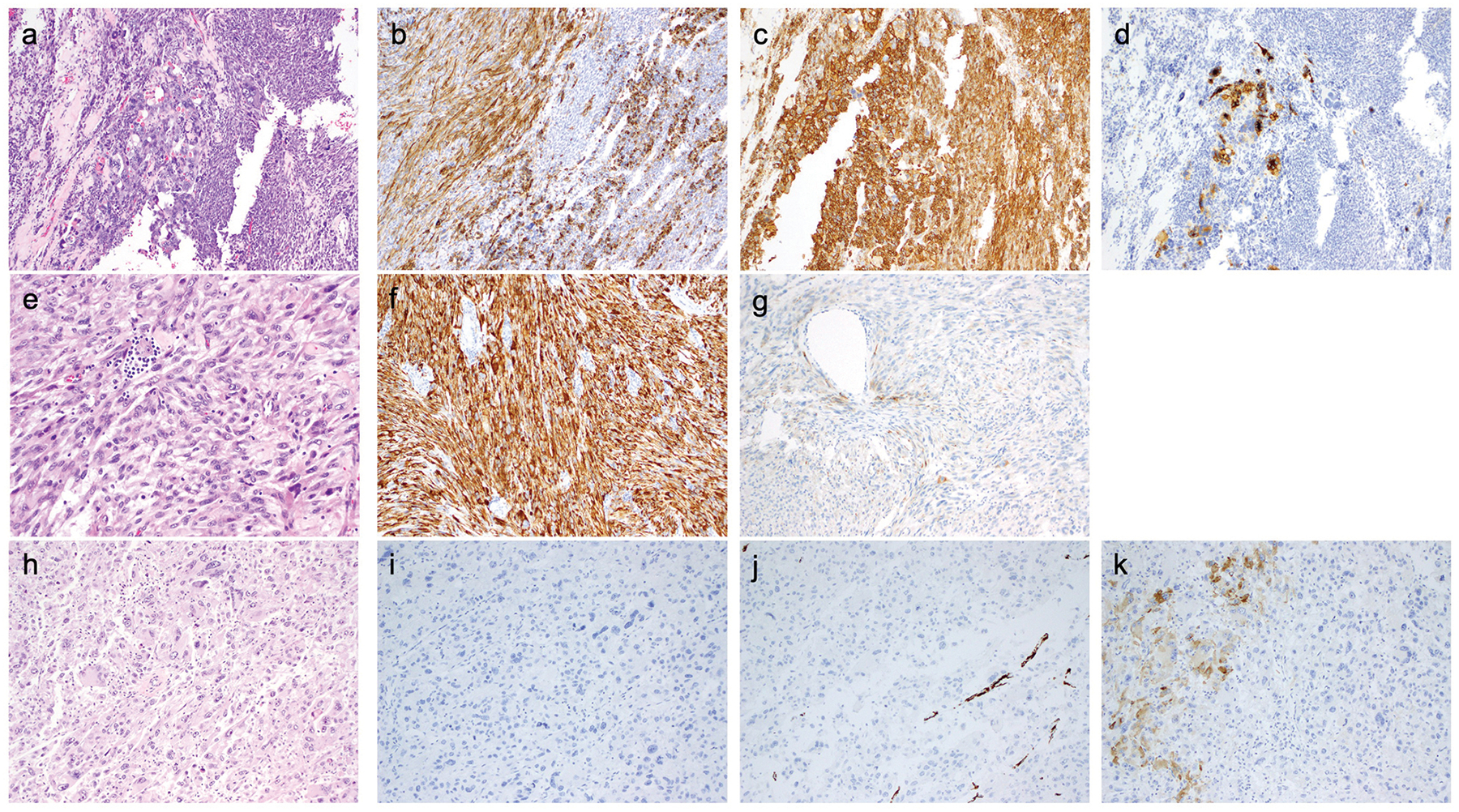
a-d) UMT06 - sarcoma associated with leiomyosarcoma-like and PEComa-like features. a) H&E – biphasic tumor composed of a moderately atypical spindle cell component and an epithelioid cell component showing moderate to marked nuclear pleomorphism; b) Desmin; c) SMA; d) HMB-45.
e-g) UMT08 - myogenic sarcoma, most likely leiomyosarcoma. e) H&E – predominantly atypical spindle cells in a whirling fascicular arrangement; f) Desmin; g) Melan-A.
h-k) UMT05 - sarcoma, not otherwise specified. h) H&E – sheets of highly pleomorphic epithelioid cells (the primary tumor was reportedly composed predominantly of spindle cells); i) Desmin; j) SMA; k) HMB-45.
UMT09 and the lung recurrence (UMT09-R) were composed of relatively small round epithelioid cells with sclerotic collagenous septa separating cords of tumor cells (Figure 3i), which expressed myogenic and melanocytic markers (Figure 3j–l). Both tumors showed a splice site mutation in TSC2, and in the recurrence, a JAZF1-SUZ12 fusion (characteristic of low-grade endometrial stromal sarcoma, LGESS52) was identified. Prototypical morphologic features of LGESS were not identified in the primary tumor or in the recurrence. There was diffuse and strong expression of HMB45, a phenomenon that has been described in ~20% of tumors reported as LGESS.53 Overall, however, given the morphological appearance and the identification of the JAZF1-SUZ12 fusion, in conjunction with TSC2 mutation and diffuse HMB45 expression, the tumor in this patient most likely represents a sarcoma associated with LGESS-like and PEComa-like features.
The finding of these tumors with hybrid/mixed features raises several tantalizing hypotheses: for example, rather than arising de novo, some uterine PEComas may derive from progression and divergent differentiation of an antecedent smooth muscle or stromal neoplasm (or vice versa). Alternatively, these entities may represent either a mesenchymal tumor with intratumoral heterogeneity or predominantly spindled PEComas, only one part of which demonstrates melanocytic differentiation. Further studies (including comparative genomic and transcriptomic analyses of different components of hybrid/mixed tumors) are required to test these hypotheses.
Tumors classified as myogenic neoplasms
The mutational frequencies in TP53, ATRX, RB1 and MED12 in our series are 41%, 24%, 6% and 12%, respectively, which compares with figures reported in uterine leiomyosarcoma29–31 of 32–49%, 25–30%, 16–22% and 2–21%, respectively (Figure 6). TSC2 mutations were not identified in leiomyosarcomas in two independent studies (n=76),29,31 but missense mutations of unknown significance in TSC2 were reported in 4% (2/49) in one series.30 The most common mutations in leiomyomas with bizarre nuclei (LMBN) are reported to be in TP53 (29%), FH (21–54%) and MED12 (17%)(Figure 6).32,54,55
Figure 6. Comparison of the mutational profiles of tumors included in this study with those reported in uterine smooth muscle tumors.
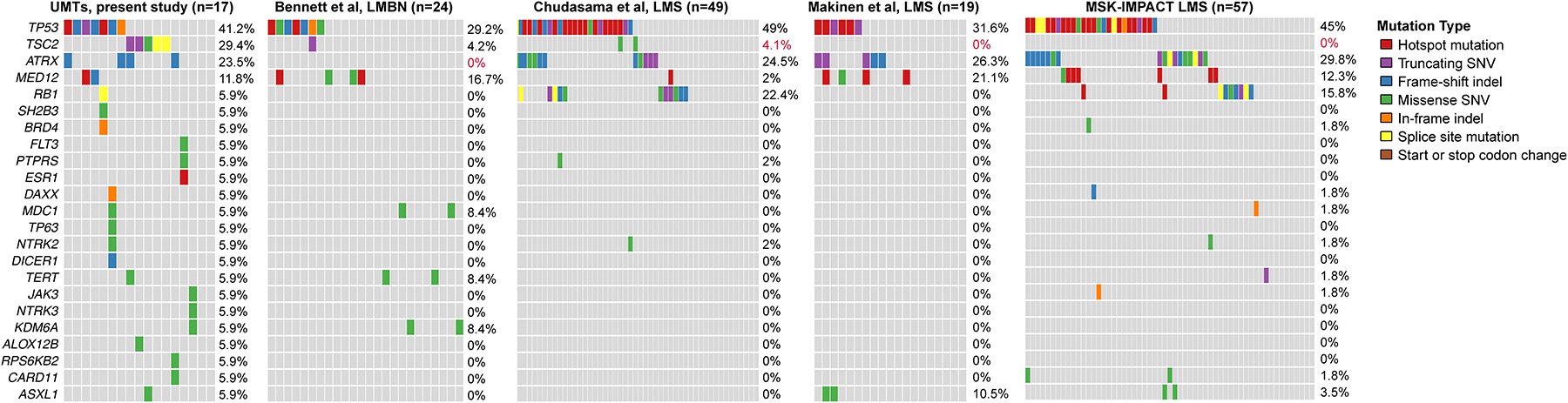
Comparison of somatic non-synonymous mutations identified in tumors in the present study (n=17 from 15 women, this study) with those of uterine leiomyosarcomas29–31 and leiomyomas with bizarre nuclei.32 Red font, statistically significantly different, Fisher’s exact test, p<0.05. Indel, small insertion and deletion; SNV, single nucleotide variant.
UMT03 and UMT08 were composed of pleomorphic spindle and epithelioid cells exhibiting strong expression of SMA and desmin with weak/focal or absent HMB45 expression (Figure 2e–g). UMT03 harbored an ESR1 hotspot mutation; ESR1 mutation has been rarely found (<1%) in uterine leiomyosarcomas.56 Based on the pathologic features and ESR1 mutation, the most likely diagnosis for this tumor is leiomyosarcoma. UMT08 harbored missense mutations in KDM6A (with LOH), JAK3 and NTRK3 and an ERCC2 homozygous deletion. KDM6A missense mutations are rarely found in LMBN32 and an in-frame indel in JAK3 was described in one leiomyosarcoma in the MSK-IMPACT cohort29 (Figure 6).
ATRX and/or DAXX alterations have been described in uterine smooth muscle tumors (leiomyomas and leiomyosarcomas),31,57–59 and loss of their expression in these tumors has been reported to be associated with poor prognosis.57,58 ATRX and DAXX mutations are identified in 20–30%30,31,60,61 and ~2%60,61 of leiomyosarcomas, respectively. We identified mutations in ATRX in 4 tumors (UMT04, UMT13, UMT06/06-R) from 3 women, and DAXX mutations in one tumor (UMT05). Based on this genotype and the pathologic phenotype, UMT04 most likely represents a leiomyosarcoma. UMT05 and UMT13 were highly pleomorphic malignancies lacking significant evidence of myogenic differentiation and showing only focal melanocytic marker expression (Figure 2h–k) – based on the available evidence, these tumors are probably best classified as sarcoma, not otherwise specified. UMT05 showed gain of TFE3 copies by FISH: 30% of tumor cells had 3–5 copies, and 40% of tumor cells had 6 or more copies; the significance of this finding is uncertain. UMT06/06-R was discussed above.
A DNAJB6-PLAG1 fusion was identified in UMT04. This tumor was composed of a predominantly fascicular spindle cell component along with a focal component of small epithelioid cells (Figure 3a–b). Myogenic markers were diffusely positive (Figure 3c), while HMB45 expression was very focal (Figure 3d). The tumor also harbored mutations in TP53 and ATRX. The morphology and genomic findings are most consistent with leiomyosarcoma. Rearrangements involving PLAG1 (with fusion partners TRPS1 and RAD51B) have recently been described in myxoid leiomyosarcoma.62,63 No myxoid areas were identified in UMT04. DNAJB6 is a RanGTP-regulated protein required for microtubule organization during mitosis, and mutations have been described in patients with myofibrillar myopathy, a group of rare inherited muscular disorders.64,65 DNAJB6 has also been shown to regulate tumor growth and metastasis through the canonical Wnt pathway and AKT signaling.66
UMT02 and UMT10 were spindle and epithelioid cell malignancies exhibiting marked nuclear pleomorphism and immunohistochemical evidence of extensive myogenic differentiation, with variable (50% of tumor cells and focally, respectively) melanocytic marker expression. However, they both harbored mutations in MED12 and TP53. MED12 mutations have been well described in uterine leiomyomas and leiomyosarcomas31,67–72, and the co-occurrence of MED12 and TP53 alterations has been reported in leiomyosarcoma.31,73 Taken together, the pathological and genomic features are most in keeping with a diagnosis of leiomyosarcoma for UMT02 and UMT10.
UMT12 was a spindle cell malignancy with a fascicular growth pattern (Figure 3e), exhibiting diffuse muscle marker expression and very focal HMB45 expression (Figures 3f–h). The only genetic alterations identified were amplifications of ERBB3 (mutation of which has been reported in a retroperitoneal leiomyosarcoma74) and NCOR1 (known to interact with the ERBB2 promoter75), which could conceivably modulate the PI3K-AKT pathway. This tumor is most likely a leiomyosarcoma with focal aberrant HMB45 expression.
mTOR pathway and inhibition
Immunohistochemical markers that have been used as surrogate markers of PI3K/Akt/mTOR pathway activation include pS6, pS6K1, pAKT, p21 and p27.76 In our cohort, we performed immunohistochemistry for pS6 and pAKT as surrogate markers of mTOR pathway activation, since mTOR inhibitors are often considered for treatment of patients with malignant PEComa. pAKT was not expressed in any of the tumors. The median IRS for pS6 showed a trend with the highest levels seen in tumors with an integrated diagnosis of malignant PEComa, with lower figures in tumors with an integrated diagnosis of non-PEComa and sarcomas with features of PEComa and another entity; however, there was some overlap, and these trends did not reach statistical significance, which may be attributable, at least partially, to the small numbers in each of these groups. Of the seven patients who received therapy including an mTOR inhibitor, pS6 results were available in 4 of these patients, and showed IRS of 90, 210, 30 and 300 in patients whose best response to mTOR inhibitors was PR, SD, POD and POD, respectively. The numbers are too small to allow definitive conclusions to be drawn about correlations between pS6 expression and response to mTOR inhibitors. In the literature, some studies have shown associations of expression of mTOR pathway markers with prognosis,76 but others have found no associations between expression of these markers and response to agents targeting these pathways.77,78 Further studies employing a larger panel of markers of PI3K/Akt/mTOR pathway activation, and ideally also incorporating functional assays, will be needed to elucidate the status of these pathways in these tumors, and to explore associations between pathway activation status and genotype, pathologic phenotype and response to mTOR inhibition.
High-grade uterine mesenchymal tumors exhibiting histopathologic and immunophenotypic features that are equivocal and do not permit ready classification represent a heterogeneous group of neoplasms which harbor a diverse repertoire of somatic genetic alterations; the results of our study suggest that these genetic alterations can aid classification of such tumors, although given their diversity, they do not permit clear-cut separation of these tumors into distinct genetic or phenotypic entities unless features thought to be characteristic of a certain entity are found (i.e. TSC mutations in PEComa).
The goal of our study was to determine, in diagnostically challenging high-grade uterine mesenchymal tumors with ambiguous morphological features, whether integrated classification based on the addition of molecular genetic analysis to histopathologic and immunophenotypic evaluation (a simplified schema for which is presented in Table 5) allowed more robust classification of the tumors. As many of the tumors included in the study were initially diagnosed as ‘malignant PEComa’, our hypothesis here was that some tumors diagnosed as ‘malignant PEComa’ based on morphology and immunophenotype may represent other entities exhibiting ‘PEComa’-like phenotypes. The results, which appear to support our hypothesis, indicate that some tumors that had been classified on phenotypic grounds as ‘malignant PEComa’ may represent (or at least contain a component of) other entities based on their well-described genetic alterations e.g. JAZF1 or YWHAE fusions in endometrial stromal sarcoma).
Table 5.
Suggested integrated phenotypic-genotypic diagnostic approach to high-grade uterine mesenchymal tumors with ambiguous histologic appearances
| 1. Histopathologic evaluation | ||||
|---|---|---|---|---|
| Favored diagnosis | Leiomyosarcoma | PEComa | ESS | Sarcoma, NOS |
| Confirm morphologic impression with: | ||||
| 2. Immunohistochemistry | ||||
| Favored diagnosis | Suggestive of leiomyosarcoma | Suggestive of PEComa | Suggestive of ESS | Consider hybrid tumor or sarcoma NOS with a descriptive diagnosis |
| If immunophenotype is inconsistent with morphologic impression or the diagnosis remains in question, proceed to: | ||||
| 3. Molecular profiling | ||||
| Favored diagnosis | Leiomyosarcoma | PEComa | ESS | Consider hybrid tumor or sarcoma NOS with a descriptive diagnosis |
Note: Clearly the spectrum of tumors in the differential diagnosis, the number of immunohistochemical markers that are used to aid diagnosis of uterine mesenchymal tumors and the number of genetic alterations that may be found is much larger in reality, but this highly simplified approach is presented to illustrate one possible scheme for integrated phenotypic-genotypic classification. ESS – endometrial stromal sarcoma
Our findings also suggest that we should cautious about making a definitive diagnosis of uterine PEComa based solely on pathologic phenotype. Tumors which exhibit classic morphological features (which from our cases with an integrated diagnosis of PEComa include: epithelioid and/or spindled cells with variably clear cytoplasm, variable nuclear pleomorphism, nested or corded architecture, stromal hyalinization) allied with supportive immunohistochemical evidence, i.e. expression of myogenic marker(s) and two or more melanocytic markers, are likely to represent PEComas. In the absence of molecular genetic evidence, tumors exhibiting underdeveloped or focal prototypical PEComa-like features may be diagnosed as “epithelioid and spindle cell neoplasm, with morphologic/immunohistochemical features suggestive of PEComa; molecular profiling is recommended for definitive classification”. In tumors exhibiting PEComa-like morphologic features along with appearances typical of other tumor types, the possibility of non-PEComa diagnosis or a diagnosis of sarcoma associated with features of PEComa and these entities should be considered. To specifically elucidate the spectrum of genetic and phenotypic characteristics of PEComas and diagnostic criteria that will allow their clear-cut diagnosis and distinction from other UMTs, further studies of larger numbers of low-grade and high-grade PEComa-like neoplasms will be required.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the staff of the Integrated Genomics Operation of the Center for Molecular Oncology, Memorial Sloan Kettering Cancer Center for their expert assistance with sequencing.
Funding disclosures:
This work was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Britta Weigelt is funded in part by Cycle for Survival, Stand Up To Cancer and Breast Cancer Research Foundation grants.
David M. Hyman is currently a salaried employee of Loxo Oncology Inc.
Footnotes
DISCLOSURE / CONFLICT OF INTEREST
This work was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748. Britta Weigelt is funded in part by Cycle for Survival, Stand Up To Cancer and Breast Cancer Research Foundation grants. David M. Hyman is currently a salaried employee of Loxo Oncology Inc. The other authors have no relevant conflicts of interest.
REFERENCES
- 1.D’Angelo E, Prat J. Uterine sarcomas: A review. Gynecol. Oncol 2010;116:131–139. doi: 10.1016/j.ygyno.2009.09.023. [DOI] [PubMed] [Google Scholar]
- 2.Oliva E Practical issues in uterine pathology from banal to bewildering: The remarkable spectrum of smooth muscle neoplasia. Mod. Pathol 2016;29:S104–S120. doi: 10.1038/modpathol.2015.139. [DOI] [PubMed] [Google Scholar]
- 3.Lee CH, Nucci MR. Endometrial stromal sarcoma - the new genetic paradigm. Histopathology 2015;67:1–19. doi: 10.1111/his.12594. [DOI] [PubMed] [Google Scholar]
- 4.Nucci MR. Practical issues related to uterine pathology: Endometrial stromal tumors. Mod. Pathol 2016;29:S92–S103. doi: 10.1038/modpathol.2015.140. [DOI] [PubMed] [Google Scholar]
- 5.Pan CC, Chung MY, Ng KF, et al. Constant allelic alteration on chromosome 16p (TSC2 gene) in perivascular epithelioid cell tumour (PEComa): Genetic evidence for the relationship of PEComa with angiomyolipoma. J. Pathol 2008;214:387–393. doi: 10.1002/path.2289. [DOI] [PubMed] [Google Scholar]
- 6.Bennett JA, Braga AC, Pinto A, et al. Uterine PEComas. Am. J. Surg. Pathol 2018;42:1370–1383. doi: 10.1097/PAS.0000000000001119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Argani P, Aulmann S, Illei PB, et al. A distinctive subset of PEComas harbors TFE3 gene fusions. Am. J. Surg. Pathol 2010;34:1395–1406. doi: 10.1097/PAS.0b013e3181f17ac0. [DOI] [PubMed] [Google Scholar]
- 8.Agaram NP, Sung YS, Zhang L, et al. Dichotomy of genetic abnormalities in PEComas with therapeutic implications. Am. J. Surg. Pathol 2015;39:813–825. doi: 10.1097/PAS.0000000000000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doros L, Yang J, Dehner L, et al. DICER1 Mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr. Blood Cancer 2012. doi: 10.1002/pbc.24020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dehner LP, Jarzembowski JA, Hill DA. Embryonal rhabdomyosarcoma of the uterine cervix: A report of 14 cases and a discussion of its unusual clinicopathological associations. Mod. Pathol 2012. doi: 10.1038/modpathol.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis RJ, Barr FG. Fusion genes resulting from alternative chromosomal translocations are overexpressed by gene-specific mechanisms in alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. U. S. A 1997. doi: 10.1073/pnas.94.15.8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis N, Soslow RA, Delair DF, et al. ZC3H7B-BCOR high-grade endometrial stromal sarcomas: A report of 17 cases of a newly defined entity. Mod. Pathol 2018. doi: 10.1038/modpathol.2017.162. [DOI] [PubMed] [Google Scholar]
- 13.Marinõ-Enriquez A, Lauria A, Przybyl J, et al. BCOR Internal Tandem Duplication in High-grade Uterine Sarcomas. Am. J. Surg. Pathol 2018. doi: 10.1097/PAS.0000000000000993. [DOI] [PubMed] [Google Scholar]
- 14.Schoolmeester JK, Dao LN, Sukov WR, et al. TFE3 translocation-associated perivascular epithelioid cell neoplasm (PEComa) of the gynecologic tract: Morphology, immunophenotype, differential diagnosis. Am. J. Surg. Pathol 2015;39:394–404. doi: 10.1097/PAS.0000000000000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolin DL, Dong F, Baltay M, et al. SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus): a clinicopathologic entity distinct from undifferentiated carcinoma. Mod. Pathol 2018. doi: 10.1038/s41379-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 16.Kolin DL, Quick CM, Dong F, et al. SMARCA4-deficient Uterine Sarcoma and Undifferentiated Endometrial Carcinoma Are Distinct Clinicopathologic Entities. Am. J. Surg. Pathol 2020. doi: 10.1097/PAS.0000000000001375. [DOI] [PubMed] [Google Scholar]
- 17.Chiang S, Cotzia P, Hyman DM, et al. NTRK Fusions Define a Novel Uterine Sarcoma Subtype with Features of Fibrosarcoma. Am. J. Surg. Pathol 2018. doi: 10.1097/PAS.0000000000001055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schoolmeester JK, Howitt BE, Hirsch MS, et al. Perivascular epithelioid cell neoplasm (PEComa) of the gynecologic tract: Clinicopathologic and immunohistochemical characterization of 16 cases. Am. J. Surg. Pathol 2014;38:176–188. doi: 10.1097/PAS.0000000000000133. [DOI] [PubMed] [Google Scholar]
- 19.Folpe AL, Mentzel T, Lehr HA, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: A clinicopathologic study of 26 cases and review of the literature. Am. J. Surg. Pathol 2005;29:1558–1575. doi: 10.1097/01.pas.0000173232.22117.37. [DOI] [PubMed] [Google Scholar]
- 20.Fadare O Perivascular epithelioid cell tumor (PEComa) of the uterus: An outcome-based clinicopathologic analysis of 41 reported cases. Adv. Anat. Pathol 2008;15:63–75. doi: 10.1097/PAP.0b013e31816613b0. [DOI] [PubMed] [Google Scholar]
- 21.Bleeker JS, Quevedo JF, Folpe AL. Malignant Perivascular epithelioid cell neoplasm: Risk stratification and treatment strategies. Sarcoma 2012;2012:541626. doi: 10.1155/2012/541626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Llamas-Velasco M, Mentzel T, Requena L, et al. Cutaneous PEComa does not harbour TFE3 gene fusions: Immunohistochemical and molecular study of 17 cases. Histopathology 2013;63:122–129. doi: 10.1111/his.12145. [DOI] [PubMed] [Google Scholar]
- 23.Cheng DT, Mitchell TN, Zehir A, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagnostics 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith ES, Da Cruz Paula A, Cadoo KA, et al. Endometrial Cancers in BRCA1 or BRCA2 Germline Mutation Carriers: Assessment of Homologous Recombination DNA Repair Defects. JCO Precis. Oncol 2019:1–11. doi: 10.1200/po.19.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weigelt B, Bi R, Kumar R, et al. The landscape of somatic genetic alterations in breast cancers from atm germline mutation carriers. J. Natl. Cancer Inst 2018;110:1030–1034. doi: 10.1093/jnci/djy028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen R, Seshan VE. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang MT, Bhattarai TS, Schram AM, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov 2018;8:174–183. doi: 10.1158/2159-8290.CD-17-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chudasama P, Mughal SS, Sanders MA, et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun 2018;9:144. doi: 10.1038/s41467-017-02602-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mäkinen N, Aavikko M, Heikkinen T, et al. Exome Sequencing of Uterine Leiomyosarcomas Identifies Frequent Mutations in TP53, ATRX, and MED12. PLoS Genet 2016;12:e1005850. doi: 10.1371/journal.pgen.1005850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett JA, Weigelt B, Chiang S, et al. Leiomyoma with bizarre nuclei: A morphological, immunohistochemical and molecular analysis of 31 cases. Mod. Pathol 2017;30:1476–1488. doi: 10.1038/modpathol.2017.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med 2014;20:1479–1484. doi: 10.1038/nm.3729. [DOI] [PubMed] [Google Scholar]
- 34.Folpe AL, Kwiatkowski DJ. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum. Pathol 2010;41:1–15. doi: 10.1016/j.humpath.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 35.Fadare O Perivascular epithelioid cell tumors (PEComas) and smooth muscle tumors of the uterus [3]. Am. J. Surg. Pathol 2007;31:1454–1455. doi: 10.1097/PAS.0b013e318039b218. [DOI] [PubMed] [Google Scholar]
- 36.Valencia-Guerrero A, Pinto A, Anderson WJ, et al. PNL2: A Useful Adjunct Biomarker to HMB45 in the Diagnosis of Uterine Perivascular Epithelioid Cell Tumor (PEComa). Int. J. Gynecol. Pathol 2019:doi: 10.1097/PGP.0000000000000653. doi:10.1097/pgp.0000000000000653. [DOI] [PubMed] [Google Scholar]
- 37.Toledo G, Oliva E. Smooth muscle tumors of the uterus: A practical approach. Arch. Pathol. Lab. Med 2008;132:595–605. doi: 10.1043/1543-2165(2008)132[595:SMTOTU]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 38.Silva EG, Deavers MT, Bodurka DC, et al. Uterine Epithelioid Leiomyosarcomas with Clear Cells: Reactivity with HMB-45 and the Concept of PEComa. Am. J. Surg. Pathol 2004;28:244–249. doi: 10.1097/00000478-200402000-00013. [DOI] [PubMed] [Google Scholar]
- 39.Silva EG, Bodurka DC, Scouros MA, et al. A uterine leiomyosarcoma that became positive for HMB45 in the metastasis. Ann. Diagn. Pathol 2005;9:43–45. doi: 10.1053/j.anndiagpath.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 40.Fadare O Uterine PEComa: Appraisal of a controversial and increasingly reported mesenchymal neoplasm. Int. Semin. Surg. Oncol 2008;5:7. doi: 10.1186/1477-7800-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vang R, Kempson RL. Perivascular epithelioid cell tumor ('PEcoma’) of the uterus: A subset of HMB-45-positive epithelioid mesenchymal neoplasms with an uncertain relationship to pure smooth muscle tumors. Am. J. Surg. Pathol 2002;26:1–13. doi: 10.1097/00000478-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 42.Makhlouf HR, Ishak KG, Shekar R, et al. Melanoma markers in angiomyolipoma of the liver and kidney: A comparative study. Arch. Pathol. Lab. Med 2002;126:49–55. doi:. [DOI] [PubMed] [Google Scholar]
- 43.Simpson KW, Albores-Saavedra J. HMB-45 reactivity in conventional uterine leiomyosarcomas. Am. J. Surg. Pathol 2007;31:95–98. doi: 10.1097/01.pas.0000213346.57391.70. [DOI] [PubMed] [Google Scholar]
- 44.Oliva E, Wang W, Branton P, et al. Expression of melanocytic (“PEComa”) markers in smooth muscle tumors of the uterus: an immunohistochemical analysis of 86 cases. Mod Pathol 2006;19:191A. [Google Scholar]
- 45.Zamecnik M, Voltr L, Chlumska A. HMB45+ cells in mixed stromal-smooth muscle tumour of the uterus [1]. Histopathology 2006;48:463–464. doi: 10.1111/j.1365-2559.2005.02251.x. [DOI] [PubMed] [Google Scholar]
- 46.Howitt BE, Schoolmeester JK, Quade BJ, et al. Immunohistochemical analysis of HMB-45, MelanA and CathepsinK in a series of 35 uterine leiomyosarcoma. Lab Invest 2013;93:279A.23318885 [Google Scholar]
- 47.Qin W, Bajaj V, Malinowska I, et al. Angiomyolipoma have common mutations in TSC2 but no other common genetic events. PLoS One 2011;6:e24919. doi: 10.1371/journal.pone.0024919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hunter DS, Klotzbücher M, Kugoh H, et al. Aberrant expression of HMGA2 in uterine leiomyoma associated with loss of TSC2 tumor suppressor gene function. Cancer Res 2002;62:3766–3772. [PubMed] [Google Scholar]
- 49.Everitt JI, Wolf DC, Howe SR, et al. Rodent model of reproductive tract leiomyomata: Clinical and pathological features. Am. J. Pathol 1995;146:1556–1567. [PMC free article] [PubMed] [Google Scholar]
- 50.Kubo Y, Kikuchi Y, Mitani H, et al. Allelic Loss at the Tuberous Sclerosis (Tsc2) Gene Locus in Spontaneous Uterine Leiomyosarcomas and Pituitary Adenomas in the Eker Rat Model. Japanese J. Cancer Res 1995;86:828–832. doi: 10.1111/j.1349-7006.1995.tb03092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yeung RS, Xiao G‐H, Everitt JI, et al. Allelic loss at the tuberous sclerosis 2 locus in spontaneous tumors in the Eker rat. Mol. Carcinog 1995;14:28–36. doi: 10.1002/mc.2940140107. [DOI] [PubMed] [Google Scholar]
- 52.Koontz JI, Soreng AL, Nucci M, et al. Frequent fusion of the JAZF1 and JJAZ1 genes in endometrial stromal tumors. Proc. Natl. Acad. Sci. U. S. A 2001;98:6348–6353. doi: 10.1073/pnas.101132598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Albores-Saavedra J, Dorantes-Heredia R, Chablé-Montero F, et al. Endometrial stromal sarcomas: Immunoprofile with emphasis on HMB45 reactivity. Am. J. Clin. Pathol 2014;141:850–855. doi: 10.1309/AJCPS88CMJRXZBWA. [DOI] [PubMed] [Google Scholar]
- 54.Gregová M, Hojný J, Němejcová K, et al. Leiomyoma with Bizarre Nuclei: a Study of 108 Cases Focusing on Clinicopathological Features, Morphology, and Fumarate Hydratase Alterations. Pathol. Oncol. Res 2019:doi: 10.1007/s12253-019-00739-5. doi:10.1007/s12253-019-00739-5. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Q, Poropatich K, Ubago J, et al. Fumarate Hydratase Mutations and Alterations in Leiomyoma with Bizarre Nuclei. Int. J. Gynecol. Pathol 2018;37:421–430. doi: 10.1097/PGP.0000000000000447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gaillard SL, Andreano KJ, Gay LM, et al. Constitutively active ESR1 mutations in gynecologic malignancies and clinical response to estrogen-receptor directed therapies. Gynecol. Oncol 2019;154:199–206. doi: 10.1016/j.ygyno.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 57.Slatter TL, Hsia H, Samaranayaka A, et al. Loss of ATRX and DAXX expression identifies poor prognosis for smooth muscle tumours of uncertain malignant potential and early stage uterine leiomyosarcoma. J. Pathol. Clin. Res 2015;1:95–105. doi: 10.1002/cjp2.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liau JY, Tsai JH, Jeng YM, et al. Leiomyosarcoma with alternative lengthening of telomeres is associated with aggressive histologic features, loss of ATRX expression, and poor clinical outcome. Am. J. Surg. Pathol 2015;39:236–244. doi: 10.1097/PAS.0000000000000324. [DOI] [PubMed] [Google Scholar]
- 59.Ahvenainen TV., Mäkinen NM, von Nandelstadh P, et al. Loss of ATRX/DAXX expression and alternative lengthening of telomeres in uterine leiomyomas. Cancer 2018;124:4650–4656. doi: 10.1002/cncr.31754. [DOI] [PubMed] [Google Scholar]
- 60.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bailey MH, Tokheim C, Porta-Pardo E, et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018;173:371–385.e18. doi: 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arias-Stella JA, Benayed R, Oliva E, et al. Novel PLAG1 Gene Rearrangement Distinguishes a Subset of Uterine Myxoid Leiomyosarcoma from Other Uterine Myxoid Mesenchymal Tumors. Am. J. Surg. Pathol 2019;43:382–388. doi: 10.1097/PAS.0000000000001196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoon JY, Mariño-Enriquez A, Stickle N, et al. Myxoid smooth muscle neoplasia of the uterus: comprehensive analysis by next-generation sequencing and nucleic acid hybridization. Mod. Pathol 2019;32:1688–1697. doi: 10.1038/s41379-019-0299-4. [DOI] [PubMed] [Google Scholar]
- 64.Fichna JP, Maruszak A, Żekanowski C. Myofibrillar myopathy in the genomic context. J. Appl. Genet 2018;59:431–439. doi: 10.1007/s13353-018-0463-4. [DOI] [PubMed] [Google Scholar]
- 65.Ruggieri A, Saredi S, Zanotti S, et al. DNAJB6 myopathies: Focused review on an emerging and expanding group of myopathies. Front. Mol. Biosci 2016;3. doi: 10.3389/fmolb.2016.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meng E, Shevde LA, Samant RS. Emerging roles and underlying molecular mechanisms of DNAJB6 in cancer. Oncotarget 2016;7:53984–53996. doi: 10.18632/oncotarget.9803. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Mäkinen N, Kämpjärvi K, Frizzell N, et al. Characterization of MED12, HMGA2, and FH alterations reveals molecular variability in uterine smooth muscle tumors. Mol. Cancer 2017;16:101. doi: 10.1186/s12943-017-0672-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kämpjärvi K, Mäkinen N, Kilpivaara O, et al. Somatic MED12 mutations in uterine leiomyosarcoma and colorectal cancer. Br. J. Cancer 2012;107:1761–1765. doi: 10.1038/bjc.2012.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ravegnini G, Mariño-Enriquez A, Slater J, et al. MED12 mutations in leiomyosarcoma and extrauterine leiomyoma. Mod. Pathol 2013;26:743–749. doi: 10.1038/modpathol.2012.203. [DOI] [PubMed] [Google Scholar]
- 70.Bertsch E, Qiang W, Zhang Q, et al. MED12 and HMGA2 mutations: Two independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod. Pathol 2014;27:1144–1153. doi: 10.1038/modpathol.2013.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwetye KE, Pfeifer JD, Duncavage EJ. MED12 exon 2 mutations in uterine and extrauterine smooth muscle tumors. Hum. Pathol 2014;45:65–70. doi: 10.1016/j.humpath.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 72.Pérot G, Croce S, Ribeiro A, et al. MED12 alterations in both human benign and malignant uterine soft tissue tumors. PLoS One 2012;7. doi: 10.1371/journal.pone.0040015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cuppens T, Moisse M, Depreeuw J, et al. Integrated genome analysis of uterine leiomyosarcoma to identify novel driver genes and targetable pathways. Int. J. Cancer 2018;142:1230–1243. doi: 10.1002/ijc.31129. [DOI] [PubMed] [Google Scholar]
- 74.González-Alonso P, Chamizo C, Moreno V, et al. Pyrosequencing-based assays for rapid detection of HER2 and HER3 mutations in clinical samples uncover an E332E mutation affecting HER3 in retroperitoneal leiomyosarcoma. Int. J. Mol. Sci 2015;16:19447–19457. doi: 10.3390/ijms160819447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Myers E, Hill ADK, Kelly G, et al. Associations and interactions between Ets-1 and Ets-2 and coregulatory proteins, SRC-1, AIB1, and NCoR in breast cancer. Clin. Cancer Res 2005;11:2111–2122. doi: 10.1158/1078-0432.CCR-04-1192. [DOI] [PubMed] [Google Scholar]
- 76.Pantuck AJ, Seligson DB, Klatte T, et al. Prognostic relevance of the mTOR pathway in renal cell carcinoma: Implications for molecular patient selection for targeted therapy. Cancer 2007;109:2257–2267. doi: 10.1002/cncr.22677. [DOI] [PubMed] [Google Scholar]
- 77.Yarchoan M, Ma C, Troxel AB, et al. pAKT Expression and Response to Sorafenib in Differentiated Thyroid Cancer. Horm. Cancer 2016;7:188–195. doi: 10.1007/s12672-016-0253-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roldan-Romero JM, Beuselinck B, Santos M, et al. PTEN expression and mutations in TSC1, TSC2 and MTOR are associated with response to rapalogs in patients with renal cell carcinoma. Int. J. Cancer 2020;146:1435–1444. doi: 10.1002/ijc.32579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.