

Abstract
At present, cardiovascular disease is one of the important factors of human death, and there are many kinds of proteins involved. Sirtuins family proteins are involved in various physiological and pathological activities of the human body. Among them, there are more and more studies on the relationship between sirtuin2 (SIRT2) protein and cardiovascular diseases. SIRT2 can effectively inhibit pathological cardiac hypertrophy. The effect of SIRT2 on ischaemia‐reperfusion injury has different effects under different conditions. SIRT2 can reduce the level of reactive oxygen species (ROS), which may help to reduce the severity of diabetic cardiomyopathy. SIRT2 can affect a variety of cardiovascular diseases, energy metabolism and the ageing of cardiomyocytes, thereby affecting heart failure. SIRT2 also plays an important role in vascular disease. For endothelial cell damage used by oxidative stress, the role of SIRT2 is bidirectional, which is related to the degree of oxidative stress stimulation. When the degree of stimulation is small, SIRT2 plays a protective role, and when the degree of stimulation increases to a certain level, SIRT2 plays a negative role. In addition, SIRT2 is also involved in the remodelling of blood vessels and the repair of skin damage.
Keywords: diabetic cardiomyopathy, heart failure, ischaemia‐reperfusion injury, oxidative stress, pathological cardiac hypertrophy, SIRT2
1. INTRODUCTION
Cardiovascular diseases have become one of the important factors that endanger human health, and post‐translational modifications of related proteins play an important role in this. 1 , 2 , 3 , 4 , 5 Among them, acetylation and deacetylation have increasingly become the focus of research. The seven‐member sirtuins family (SIRT1‐7) belongs to the third deacetylase. In order to catalyse the deacetylation reaction, this protein requires the presence of nicotinamide adenine dinucleotide (NAD+) to have deacetylation activity. Sirtuins are involved in a variety of physiological and pathological activities in the human body, including ageing, energy responses to low calorie availability and stress resistance, apoptosis, inflammation, regulate mitochondrial biogenesis and biological rhythm. We currently know the substrates of the sirtuins family proteins including p53, 6 nuclear factor‐kappa B (NF‐κB), 7 forkhead box protein O1 (FOXO1), 8 forkhead box protein O3 (FOXO3), 9 peroxisome proliferator‐activated receptor‐γ (PPAR‐γ), 10 peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC1‐α), 11 acetyl‐CoA synthetases (AceCSs), 12 α‐tubulin, 13 cell division cycle 20 (CDC20), 14 CDC20‐homologue 1 (CDH1), 14 phosphoenolpyruvate carboxykinase (PEPCK1), 15 histone H3 lysine 56 (H3K56) 16 and histone H4 lysine 16 (H4K16). 17 The positioning of sirtuins in the cells is also different. At the same time, the substrates and binding partners used are also different. 18 For SIRT2, it is mainly located in the cytoplasm, but under certain conditions, it will also enter the nucleus and play the corresponding function. 13 At present, sirtuin 1 (SIRT1) is the most widely studied in the cardiovascular system, and SIRT2 is relatively little studied. In this review, we summarized the different functions and simple mechanisms of SIRT2 in the cardiovascular system, which is expected to contribute to finding therapeutic targets for cardiovascular diseases.
2. SIRT2 AND HEART‐RELATED DISEASES
2.1. SIRT2 and pathological myocardial hypertrophy
Myocardial hypertrophy is a powerful form of compensation, which is divided into physiological myocardial hypertrophy and pathological myocardial hypertrophy. The significance of physiological myocardial hypertrophy is positive. It is made by the body to some changes in the body or some external stimuli. The adaptive changes often occur in pregnant women and athletes. 19 This is a reversible change. After the stimulation disappears, the myocardium returns to normal levels. However, under the action of some pathological stimulation, myocardial cells will undergo pathological myocardial hypertrophy. 20 This change is within a certain range. If the cause is persistent and cannot be eliminated, the hypertrophic cardiac muscle cannot maintain normal function for a long time and eventually turn to heart failure. 21 Chronic heart failure generally develops gradually on the basis of compensatory hypertrophy of the myocardium.
Post‐translational modification is very important in the development of pathological myocardial hypertrophy, and acetylation is one of them. 22 The family of histone deacetylases (HDAC) plays a key role in regulating pathological cardiac hypertrophy. HDACs are divided into four categories. Class I HDAC can promote the occurrence and development of pathological cardiac hypertrophy, which in turn aggravates the condition. 23 Class II HDAC is contrary to it and plays a negative role in regulating cardiac hypertrophy. 24 The sirtuins family belong to class III HDAC. 25 And so far, the role of class IV HDAC is unclear.
There was evidence that SIRT2 is related to pathological myocardial hypertrophy and plays a negative role in regulating the occurrence and development of pathological myocardial hypertrophy, thereby reducing the severity of illness and heart failure. SIRT2 can deacetylate the liver kinase B1 (LKB1), thereby promoting the activation of the AMP‐activated protein kinase (AMPK) pathway. 26 The loss of SIRT2 inhibits the activation of AMPK, promotes ageing‐related and/or angiotensin II (Ang II)‐induced pathological cardiac hypertrophy and weakens the metformin‐mediated cardiac protection. SIRT2 can affect the stability of microtubules (MTs) by deacetylating tubulin 27 and has a certain effect on the FOXO3a pathway related to oxidative stress. 9 In addition, the deacetylation of FOXO1 and PGC1‐α under the influence of SIRT2 is also related to pathological myocardial hypertrophy. 28 At the same time, we noticed that the above four proteins, namely tubulin, FOXO3a, FOXO1, PGC1‐α are all downstream proteins of the LKB1‐AMPK signalling pathway. Therefore, it is reasonable to believe that in the protective effect of SIRT2 on pathological cardiac hypertrophy, LKB1‐AMPK signalling pathway plays a vital role. In addition to the LKB1‐AMPK pathway, the calcineurin/nuclear factor of activated T‐cells (NFAT) pathway may also be involved. NFAT is a transcription factor and NFATc2 is one of its isomers. SIRT2 can bind to NFATc2 and deacetylate it, inhibit NFATc2’s activity and then inhibit this signal transduction pathway and inhibit the occurrence of pathological myocardial hypertrophy. 29 The latest research showed that constitutive photomorphogenic 9 signalosome complex subunit 6 (CSN6) and plant homeodomain finger protein 19 (PHF19) exacerbate the pathological myocardial hypertrophy induced by Ang II by inhibiting the expression of SIRT2, and the overexpression of SIRT2 can reduce the effect of CSN6 in promoting myocardial hypertrophy. 30 , 31
In short, for pathological myocardial hypertrophy, the role of SIRT2 is positive. Therefore, finding a highly effective SIRT2 agonist can effectively suppress the occurrence and development of pathological myocardial hypertrophy and then avoid the development of heart failure.
2.2. SIRT2 and myocardial ischaemia‐reperfusion injury
The meaning of myocardial ischaemia‐reperfusion is that the coronary artery is partially or completely acutely blocked due to some reasons, and then recanalized after a period of time. Although the perfusion of the ischaemic myocardium can be restored to normal at this time, myocardial tissue damage is instead progressive aggravation. 32 After vascular recanalization, a series of traumatic changes in myocardial ultrastructure, energy metabolism, cardiac function and electrophysiology caused by myocardial ischaemia become more serious. At this time, the patient may experience severe arrhythmia or even sudden death.
Cardiac ischaemia or hypoxia caused by acute myocardial infarction (AMI) can cause the ATP content in cardiomyocytes to drop rapidly. In order to adapt to the hypoxic environment, cardiomyocytes obtain energy through anaerobic respiration, that is, glycolysis, which leads to accumulation of lactic acid, an increase in intracellular H+ content and activation of Na+‐H+ ion exchange channel. The energy reduction caused by hypoxia will inhibit the function of 3Na+‐2K+ ATPase and increase the accumulation of Na+in the cell. In order to expel a large amount of Na+in the cell, the 2Na+‐Ca2+ ion exchange channel is activated. Eventually, it causes Ca2+ overload in myocardial cells, triggering a series of adverse reactions. 33 Prolonged ischaemia also induces mitochondria to release cytochrome c, which is an important protein in the classical apoptosis pathway. A prospective observational study found that the higher the level of SIRT2 in the plasma of patients with AMI, the higher the Killip class of the patient, and the lower the left ventricular ejection fraction (LVEF). This indicates that the level of SIRT2 is closely related to AMI, and AMI can be used to predict major adverse cardiovascular events (MACE) after AMI. 34 Another study conducted on AMI patients also found that mutations in the SIRT2 gene promoter are closely related to AMI. 35
Reperfusion can provide oxygen and nutrition, meanwhile remove toxic substances produced by necrotic cells to restore the ischaemic and hypoxic state of myocardial cells. However, the rapid recovery of oxygen content and extracellular pH 36 will cause the accumulation of ROS 37 and aggravate Ca2+ overload situation. This will lead to apoptosis and necrosis and furthermore aggravate pathological injury than before reperfusion. Among the present studies of sirtuins family, only SITR1 is involved in ischaemia‐reperfusion injury and few studies focussed on SIRT2. Some studies observed SIRT2 could aggravate ischaemia‐reperfusion injury by inhibiting the binding of 14‐3‐3 zeta and B‐cell lymphoma‐2 associated death promoter (BAD), furthermore releasing BAD into mitochondria from the cytoplasm. In mitochondria, BAD binds to B‐cell lymphoma‐2 (BCL‐2) and/or B‐cell lymphoma‐extra large (BCL‐XL) and inhibits the anti‐apoptotic functions of them to promote cell apoptosis. In experiments, the levels of 14‐3‐3 zeta and BAD are elevated in SIRT2 depleted H9C2 cells, and the interaction between these two proteins was enhanced. 38 However, other studies have shown that toll‐like receptor 4 (TLR4) can reduce the expression and activity of SIRT2, increase the acetylation of p53, increase the level of ROS in cardiomyocytes and promote cell apoptosis. The overexpression of SIRT2 can inhibit the acetylation of p53 and thus inhibit the damage of TLR4 to cardiomyocytes. 39 The reason why SIRT2 has these two opposite functions needs further research to discover.
2.3. SIRT2 and diabetic cardiomyopathy
Diabetes is a complex chronic disease, and it appears in multiple states worldwide. 40 The causes of diabetes are diverse and eventually lead to absolute or relative lack of insulin, which leads to diabetes. Cardiovascular complication is one of the main complications of diabetes and one of the main causes of death of diabetic patients. 41 The disease causes extensive focal necrosis of the myocardium on the basis of metabolic disorders 42 and microvascular lesions, followed by abnormal heart function. The disease is characterized by atherosclerosis and left ventricular (LV) dysfunction not related to coronary artery disease, and this dysfunction changes from diastolic dysfunction to systolic dysfunction as the disease progresses. 43 Eventually, the patients develop heart failure, arrhythmia, cardiogenic shock and even sudden death.
Microtubules are present in various cells. Their most important roles are to function as cytoskeletal filaments. MTs are composed of heterodimers containing α‐tubulin and β‐tubulin. The latter two can undergo various post‐translational modifications. After acetylation modification, this heterodimer will make MTs more stable. 44 However, previous studies have found that in rats with type 1 diabetes mellitus (T1MD), when the content of MTs in cardiomyocytes is too high, that is, the state of MTs is stable, ventricular contractile function will be impaired, 45 which in turn causes diabetic cardiomyopathy.
As we mentioned earlier, one of the substrates of SIRT2 is α‐tubulin. SIRT2 makes it deacetylated; therefore, the above processes will be inhibited, and eventually, the stability of MTs will be reduced. This can well relieve ventricular systolic dysfunction and delay the development of cardiomyopathy caused by diabetes. 46 However, only T1DM is mentioned here. The relationship between type 2 diabetes mellitus (T2DM)‐induced cardiomyopathy and SIRT2 will be studied in the future.
At present, there are very few direct studies on the relationship between SIRT2 and diabetic cardiomyopathy. It is interesting that other literatures indicate the use of HDAC inhibitors, such as sodium butyrate, 47 MPT0E014, 48 RGFP966 (a kind of selective HDAC3 inhibitor), 49 suberoylanilide hydroxamic acid (SAHA), 50 can well control the occurrence and development of diabetic cardiomyopathy. However, because the sirtuins family belongs to the third category of HDACs, the effects of these two conditions for diabetic cardiomyopathy are completely opposite. This phenomenon may be caused by the interaction between different types and even different subspecies of HDACs. The specific reason needs to be studied in the future.
In addition, there are reports in the literature that the generation of high glucose‐induced ROS can cause diabetic cardiomyopathy. And experiments have shown that the use of diallyl trisulfide (DATS) can inhibit the generation of ROS, which in turn inhibits c‐Jun N‐terminal kinase (JNK)/NF‐κB signalling, thereby inhibiting high glucose‐induced myocardium apoptosis. 51 SIRT2 can reduce the content of ROS, which provides another idea for the treatment of diabetic cardiomyopathy.
2.4. SIRT2 and heart failure
Heart failure is caused by impaired systolic and/or diastolic function of the heart. The amount of venous circulatory blood cannot be fully discharged from the heart, resulting in blood stasis in the venous system and insufficient blood can be perfused in the arterial system, resulting in impaired cardiac circulation. This disorder manifests as pulmonary congestion and vena cava congestion. Heart failure is not an independent disease, but the end stage of the development of heart disease. The consequences of almost all cardiovascular diseases without active treatment are heart failure, such as the aforementioned myocardial hypertrophy, myocardial ischaemia‐reperfusion injury and myocardial infarction, 52 hypertension, 53 valvular heart disease, 54 cardiomyopathy, 55 arrhythmia. 56 The vast majority of heart failures begin with left heart failure, which first manifests as pulmonary congestion.
In addition to the aforementioned causes of heart failure, the energy metabolism in cardiomyocytes is also very important for the development of heart failure. Because the function of the heart is to transport blood to the whole body, it is working all the time, and the demand for energy is very high. So the energy metabolism in myocardial cells, especially the state of mitochondria, is closely related to heart failure. PGC1‐α is a cofactor for PPAR‐γ in adipocytes and can generate heat when the body temperature is low. 57 In the heart, PGC1‐α is the main regulator of mitochondrial biogenesis and metabolism. 58 For decades, the relationship between PGC1‐α and cardiac energy regulation has attracted more and more attention, and its mechanism has also been explored more and more clearly. The expression and function of PGC1‐α are key determinants of cardiac energy status. At present, studies have shown that the reduction of PGC1‐α expression is the main cause of heart failure due to mitochondrial damage and metabolic defects in the heart. 59 At the same time, some studies have proved the relationship between SIRT1 and the expression level of PGC1‐α. It has been found in the myocardial tissue of heart failure that SIRT1 can deacetylate the histone H3 lysine 9 (H3K9) in the promoter of PGC1‐α, resulting in genes Inactivation, thereby interfering with the expression of PGC1‐α. 60 Although the relationship between SIRT2 and PGC1‐α in cardiomyocytes and the role of SIRT2 in cardiac mitochondrial function and energy metabolism are currently unclear, related experiments in adipocytes have shown that reduced SIRT2 function directly inhibits the deacetylation of PGC1‐α and β‐oxidation and the expression of mitochondrial gene. 61 Therefore, we speculate that SIRT2 will promote the deacetylation of PGC1‐α in cardiomyocytes, which will reduce the expression of PGC1‐α and eventually lead to heart failure. This result is hoped to be confirmed by experiments in the future.
In addition, the ageing of cardiomyocytes and heart failure is also closely related, and experiments have shown that the balance between the survival and death of cardiomyocytes plays a key role in heart failure. 62 For SIRT2, the relationship with the nervous system was mainly studied before. 63 Recently, it has been reported that budding uninhibited by benzimidazole‐related 1 (BUBR1) can delay the senescence of mouse cells and prolong the lifespan of mice, while SIRT2 can deacetylate BUBR1 and inhibit its degradation. This function of SIRT2 reverses the reduction in LV chamber dimension and ensures the normal heart function. 64 SIRT2 can also regulate the activity of NFATc2 to protect the heart from diseases related to ageing of cardiomyocytes and inhibit heart failure. 29
3. SIRT2 AND VASCULAR‐RELATED DISEASES
3.1. SIRT2 and atherosclerosis
Atherosclerosis is the main cause of coronary heart disease, cerebral infarction 65 and peripheral vascular disease. 66 If patients with atherosclerosis are not actively treated in time, the consequences are very serious and even life‐threatening. Risk factors for atherosclerosis include hypercholesterolemia, 67 metabolic syndrome, 68 hypertension, 69 obesity 70 and diabetes. 71 The basis of atherosclerosis is lipid metabolism disorder. 72 It is characterized by the lesion of the affected artery starting from the intima, generally the accumulation of lipids and complex carbohydrates, followed by bleeding and thrombosis, and then fibrous tissue hyperplasia and calcification, and the gradual transformation and calcification of the middle layer of the artery, and finally thickening and stiffening of the arterial wall, and narrowing of the vessel lumen. 73 Lesions often involve large and medium muscular arteries. Once they develop enough to block the arterial lumen, the tissue or organ supplied by the artery will be ischaemic or necrotic. 74 When the thrombus falls off for some reason, the thrombus will move to various organs of the body with the flow of blood flow, causing secondary damage to the remaining organs.
Macrophages play a very important role in the development of atherosclerosis. 75 Macrophages have two phenotypes, namely "inflammatory" M1 phenotype and "regulatory" M2 phenotype, and these two phenotypes coexist in the lesion 76 and switch back and forth according to the different conditions of the lesion, 77 and exercise different functions. The roles of these two macrophage phenotypes in the occurrence and development of atherosclerosis is very complex. In short, the M1 phenotype inhibits the stability of atherosclerotic plaques and leads to more serious results, the M2 phenotype will make the plaque more stable and relieve the damage caused by atherosclerosis. 78 Therefore, adjusting the ratio of M1 and M2 phenotypes in the plaque can regulate the development of atherosclerosis. Experiments have shown that overexpression of SIRT2 protein in HUVEC can reduce the expression level of inducible nitric oxide synthase (iNOS) (macrophage M1 type marker) and increase the expression level of arginase‐1 (ARG‐1) (macrophage M2 type marker). This proved that SIRT2 can transform macrophages in HUVEC from M1 type to M2 type, thereby stabilizing plaque in atherosclerosis. SIRT2 plays a protective role in this. 79
Under normal circumstances, vascular endothelial cells will produce physiological doses of ROS to participate in the transmission of cell signals, but excessive ROS will lead to the generation of oxidative stress, which in turn causes atherosclerosis. 80 In atherosclerotic HUVEC, if MicroRNA‐140‐5p is overexpressed, the content of ROS and malondialdehyde (MDA) in the cell increases, while the expression of SIRT2 is inhibited. Moreover, SIRT2 agonists can also inhibit the positive effect of MicroRNA‐140‐5p on oxidative stress. Therefore, it is reasonable to believe that SIRT2 mediates the effect of MicroRNA‐140‐5p on oxidative stress, and the function of SIRT2 is to suppress negative effects of oxidative stress. 81 In addition, in the experiment of tumour necrosis factor‐alpha (TNF‐α) inducing HUVEC to produce ROS, resveratrol can activate the SIRT2 pathway to increase the expression of SIRT2, reduce the production of ROS under the above conditions and inhibit the negative effects of oxidative stress. 82
3.2. SIRT2 and oxidative stress‐induced endothelial cell damage
Oxidative stress‐induced endothelial cell damage is inseparable from many diseases. In addition to the aforementioned atherosclerosis, there are also hypertension, peripheral vascular disease, 83 and diabetes. 80 The result of oxidative stress occurring in endothelial cells is mainly to promote their own apoptosis and cause the death of endothelial cells. 84 In the experiment of vascular endothelial injury induced by high glucose, SIRT2 can prevent the injury by inhibiting the p53 and NF‐κB pathways. 85 But what is interesting is that depending on the degree of oxidative stress, SIRT2 may have two completely opposite effects. Because SIRT2 can bind FOXO3a and make it deacetylated, and FOXO3a will increase the expression of the target protein manganese superoxide dismutase and BCL‐2 interacting mediator of cell death (Bim). 86 Among them, the former has an antioxidant effect, 87 which can reduce the level of ROS in the cell, showing a protective effect, while the latter is a pro‐apoptotic protein, 88 which causes apoptosis and shows a harmful effect. When endothelial cells are stimulated by a lesser degree of oxidative stress, the effect of manganese superoxide dismutase prevails. When the oxidative stress is more intense, Bim's effect will be greater.
3.3. SIRT2 and vascular remodelling
In the case of changes in the internal and external environment, in order to ensure the normal supply of blood flow, the blood vessel wall can change its structure to maintain an appropriate lumen size and allow blood to flow smoothly throughout the body. This process is called vascular remodelling. 89 Vascular remodelling is associated with many diseases or conditions, such as pregnancy, 90 pulmonary artery blood pressure, 91 ageing. As mentioned earlier, MTs in cardiomyocytes are related to the occurrence of diabetic cardiomyopathy. There are also MTs in HUVCE, the structure of which is the same as that in cardiac muscle cells. Similarly, when α‐tubulin is deacetylated by SIRT2, the stability of MTs will be greatly reduced, and the migration ability of endothelial cells will be enhanced, which suggests that SIRT2 is a key regulator of vascular remodelling. 92 In the presence of small trauma in vitro, the enhanced endothelial cell migration ability helps repair skin damage, so SIRT2 plays an active role in this situation. 93
3.4. SIRT2 and energy metabolism
Normal energy metabolism is a prerequisite for maintaining cell physiological functions. Among them, glucose metabolism and lipid metabolism are the most important. 94 Disorders of energy metabolism of vascular endothelial cells can damage the functions of endothelial cells and cause various vascular diseases. 95 If this metabolic disorder occurs in the coronary arteries, then some physiological functions of the heart will also be affected. 96 SIRT2 can inhibit the degradation of phosphoenolpyruvate carboxykinase (PEPCK‐C) to regulate gluconeogenesis. 15 SIRT2 can also inhibit the acetylation of FOXO1 and promote its combination with PPAR‐γ to inhibit fat formation. 97 After energy restriction (ER) on Sprague‐Dawley male rats fed a high‐fat diet for 12 months, it was found that the expression of SIRT2 was suppressed. This indicated that there is a link between SIRT2 and energy metabolism. 98 Experiments have found that the use of SIRT2 inhibitors will cause a significant decrease in the survival rate of porcine vascular endothelial cell line (PIEC). Treatment with SIRT2 inhibitors will increase the apoptosis and necrosis of PIEC, and the content of ATP in the cells will also decrease, indicating that under physiological conditions, SIRT2 is essential to maintain the normal energy metabolism of endothelial cells and maintain their survival of. 99
4. CONCLUSION
In fact, SIRT2 plays a key and important role in cardiovascular diseases, and there are many aspects of the protein available for research, which is very useful for clarifying the future research direction and finding effective clinical drugs for treating cardiovascular diseases. As mentioned above, Table 1 lists the cardiovascular diseases related to SIRT2 and the corresponding molecules that interact with SIRT2. We can see that SIRT2 is involved in many diseases of the cardiovascular system. Figures 1 and 2 vividly introduce the mechanism of SIRT2's participation in cardiovascular diseases. At the same time, it can be seen that SIRT2 has a big difference in the impact of cardiovascular diseases, and sometimes the impact on the same disease is not even the same. This situation has a great relationship with the degree and type of stimuli suffered by the cardiovascular system, and the specific situation requires researchers to do further research. In short, SIRT2 provides a very promising idea for researchers to find drugs for treating cardiovascular diseases.
TABLE 1.
This table summarizes the effects of SIRT2 on different cardiovascular states and shows the corresponding proteins that are currently known to interact with SIRT2
| Disease | Known effects | Related molecule and Ref. |
|---|---|---|
| Pathological myocardial hypertrophy | Inhibition | LKB1, 26 tubulin, 27 FOXO3a, 9 FOXO1 and PGC1‐α, 28 NFATc2, 29 CSN6, 30 PHF19 31 |
| Myocardial ischaemia‐reperfusion injury | Inhibition/Promotion | 14‐3‐3 zeta, 38 BAD, 38 p53 39 |
| Diabetic cardiomyopathy | Inhibition | α‐tubulin 45 |
| Heart failure | Inhibition | BUBR1, 64 NFATc2 29 |
| Atherosclerosis | Inhibition | MicroRNA‐140‐5p 81 |
| Oxidative stress‐induced endothelial cell damage | Inhibition/Promotion (according to the degree of oxidative stress) | p53, 85 NF‐κB, 85 FOXO3a 86 |
| Vascular remodelling | Regulation | α‐tubulin |
| Energy metabolism in endothelial cells | Maintain normal energy metabolism | PEPCK‐C, 15 FOXO1 97 |
FIGURE 1.

The figure shows that SIRT2 as a deacetylase has a role on a variety of proteins, which ultimately affects the heart in different ways
FIGURE 2.
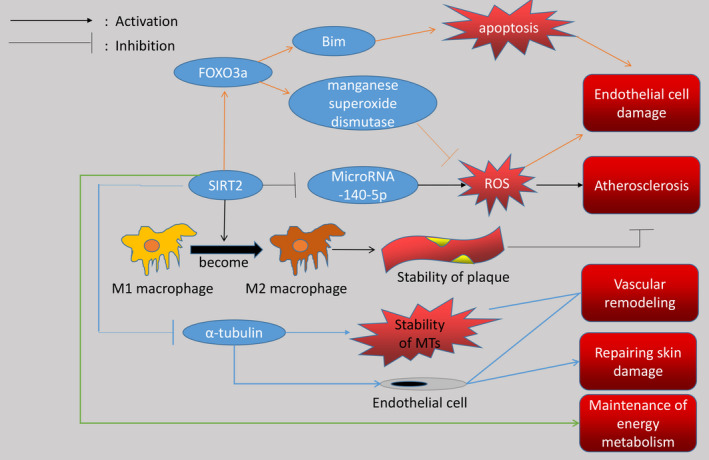
The figure shows that SIRT2 as a deacetylase not only affects a variety of proteins, but also changes the type of macrophages. Interestingly, SIRT2 has two effects after deacetylation of FOXO3a, which is related to the degree of stimulation given to blood vessels. When blood vessels are stimulated by mild oxidative stress, the manganese superoxide dismutase pathway plays a major role, and when blood vessels are stimulated by severe oxidative stress, the Bim pathway plays a major role
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this paper.
AUTHOR CONTRIBUTION
Boquan Wu: Writing‐original draft (lead); Writing‐review & editing (lead). Shilong You: Formal analysis (supporting). Hao Qian: Formal analysis (supporting). Shaojun Wu: Methodology (supporting). Saien Lu: Formal analysis (supporting). Ying Zhang: Resources (equal). Yingxian Sun: Resources (equal). Naijin Zhang: Resources (equal).
ACKNOWLEDGEMENTS
This work was supported by National Natural Science Foundation of China (Grant No. 81670231, 81900355, 81900372, 81970211).
Wu B, You S, Qian H, et al. The role of SIRT2 in vascular‐related and heart‐related diseases: A review. J Cell Mol Med. 2021;25:6470–6478. 10.1111/jcmm.16618
Contributor Information
Ying Zhang, Email: yzhang02@cmu.edu.cn.
Yingxian Sun, Email: yxsun@cmu.edu.cn.
Naijin Zhang, Email: njzhang@cmu.edu.cn.
REFERENCES
- 1. Zhang N, Zhang Y, Wu B, You S, Sun Y. Role of WW domain E3 ubiquitin protein ligase 2 in modulating ubiquitination and degradation of septin4 in oxidative stress endothelial injury. Redox Biol. 2020;30:101419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang N, Zhang Y, Qian H, Wu S, Cao L, Sun Y. Selective targeting of ubiquitination and degradation of PARP1 by E3 ubiquitin ligase WWP2 regulates isoproterenol‐induced cardiac remodeling. Cell Death Differ. 2020;27:2605‐2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Y, You S, Tian Y, et al. WWP2 regulates SIRT1‐STAT3 acetylation and phosphorylation involved in hypertensive angiopathy. J Cell Mol Med. 2020;24:9041‐9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang Y, Qian H, Wu B, et al. E3 Ubiquitin ligase NEDD4 family‐regulatory network in cardiovascular disease. Int J Biol Sci. 2020;16:2727‐2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Qian H, Zhang Y, Wu B, et al. Structure and function of HECT E3 ubiquitin ligases and their role in oxidative stress. J Transl Int Med. 2020;8:71‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Winnik S, Auwerx J, Sinclair DA, Matter CM. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J. 2015;36:3404‐3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang BL. Sirt1 and the mitochondria. Mol Cells. 2016;39:87‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yamamoto H, Schoonjans K, Auwerx J. Sirtuin functions in health and disease. Mol Endocrinol. 2007;21:1745‐1755. [DOI] [PubMed] [Google Scholar]
- 9. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD‐dependent histone deacetylase. Nature. 2000;403:795‐800. [DOI] [PubMed] [Google Scholar]
- 10. Masri S, Sassone‐Corsi P. Sirtuins and the circadian clock: bridging chromatin and metabolism. Sci Signal. 2014;7(342):re6. [DOI] [PubMed] [Google Scholar]
- 11. Jin YH, Kim YJ, Kim DW, et al. Sirt2 interacts with 14‐3‐3 beta/gamma and down‐regulates the activity of p53. Biochem Biophys Res Commun. 2008;368:690‐695. [DOI] [PubMed] [Google Scholar]
- 12. Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl‐CoA synthetases. Proc Natl Acad Sci USA. 2006;103:10230‐10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+‐dependent tubulin deacetylase. Mol Cell. 2003;11:437‐444. [DOI] [PubMed] [Google Scholar]
- 14. Kim HS, Vassilopoulos A, Wang RH, et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20:487‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang W, Wang S, Xiao M, et al. Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol Cell. 2011;43:33‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300‐mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Serrano L, Martínez‐Redondo P, Marazuela‐Duque A, et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013;27:639‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417‐435. [DOI] [PubMed] [Google Scholar]
- 19. Li L, Xu J, He L, et al. The role of autophagy in cardiac hypertrophy. Acta Biochim Biophys Sin (Shanghai). 2016;48:491‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frieler RA, Mortensen RM. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation. 2015;131:1019‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387‐407. [DOI] [PubMed] [Google Scholar]
- 22. Greco CM, Condorelli G. Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat Rev Cardiol. 2015;12:488‐497. [DOI] [PubMed] [Google Scholar]
- 23. Montgomery RL, Davis CA, Potthoff MJ, et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790‐1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal‐responsive repressors of cardiac hypertrophy. Cell. 2002;110:479‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoon S, Eom GH. HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med J. 2016;52:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang X, Chen XF, Wang NY, et al. SIRT2 acts as a cardioprotective deacetylase in pathological cardiac hypertrophy. Circulation. 2017;136:2051‐2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fassett JT, Hu X, Xu X, et al. AMPK attenuates microtubule proliferation in cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2013;304:H749‐H758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim TT, Dyck JR. Is AMPK the savior of the failing heart? Trends Endocrinol Metab. 2015;26:40‐48. [DOI] [PubMed] [Google Scholar]
- 29. Sarikhani M, Maity S, Mishra S, et al. SIRT2 deacetylase represses NFAT transcription factor to maintain cardiac homeostasis. J Biol Chem. 2018;293:5281‐5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mei ZL, Wang HB, Hu YH, Xiong L. CSN6 aggravates Ang II‐induced cardiomyocyte hypertrophy via inhibiting SIRT2. Exp Cell Res. 2020;396:112245. [DOI] [PubMed] [Google Scholar]
- 31. Gu W, Cheng Y, Wang S, Sun T, Li Z. PHD finger protein 19 promotes cardiac hypertrophy via epigenetically regulating SIRT2. Cardiovasc Toxicol. 2021;21(6):451‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Braunwald E, Kloner RA. Myocardial reperfusion: a double‐edged sword? J Clin Invest. 1985;76:1713‐1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hausenloy DJ, Yellon DM. Myocardial ischemia‐reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng M, Du X, Zhao L, Sun H, Chen M, Yang X. Elevated plasma sirtuin2 level predicts heart failure after acute myocardial infarction. J Thorac Dis. 2021;13:50‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang W, Gao F, Zhang P, et al. Functional genetic variants within the SIRT2 gene promoter in acute myocardial infarction. PLoS One. 2017;12:e0176245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lemasters JJ, Bond JM, Chacon E, et al. The pH paradox in ischemia‐reperfusion injury to cardiac myocytes. EXS. 1996;76:99‐114. [DOI] [PubMed] [Google Scholar]
- 37. Hearse DJ, Humphrey SM, Chain EB. Abrupt reoxygenation of the anoxic potassium‐arrested perfused rat heart: a study of myocardial enzyme release. J Mol Cell Cardiol. 1973;5:395‐407. [DOI] [PubMed] [Google Scholar]
- 38. Lynn EG, McLeod CJ, Gordon JP, Bao J, Sack MN. SIRT2 is a negative regulator of anoxia‐reoxygenation tolerance via regulation of 14‐3‐3 zeta and BAD in H9c2 cells. FEBS Lett. 2008;582:2857‐2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Katare PB, Nizami HL, Paramesha B, Dinda AK, Banerjee SK. Activation of toll like receptor 4 (TLR4) promotes cardiomyocyte apoptosis through SIRT2 dependent p53 deacetylation. Sci Rep. 2020;10:19232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:4‐14. [DOI] [PubMed] [Google Scholar]
- 41. Garcia MJ, McNamara PM, Gordon T, Kannel WB. Morbidity and mortality in diabetics in the Framingham population. Sixteen year follow‐up study. Diabetes. 1974;23:105‐111. [DOI] [PubMed] [Google Scholar]
- 42. Fuentes‐Antrás J, Picatoste B, Ramírez E, Egido J, Tuñón J, Lorenzo Ó. Targeting metabolic disturbance in the diabetic heart. Cardiovasc Diabetol. 2015;14:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pepe A, Meloni A, Rossi G, et al. Cardiac complications and diabetes in thalassaemia major: a large historical multicentre study. Br J Haematol. 2013;163:520‐527. [DOI] [PubMed] [Google Scholar]
- 44. Conacci‐Sorrell M, Ngouenet C, Eisenman RN. Myc‐nick: a cytoplasmic cleavage product of Myc that promotes alpha‐tubulin acetylation and cell differentiation. Cell. 2010;142:480‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koide M, Hamawaki M, Narishige T, et al. Microtubule depolymerization normalizes in vivo myocardial contractile function in dogs with pressure‐overload left ventricular hypertrophy. Circulation. 2000;102:1045‐1052. [DOI] [PubMed] [Google Scholar]
- 46. Yuan Q, Zhan L, Zhou QY, et al. SIRT2 regulates microtubule stabilization in diabetic cardiomyopathy. Eur J Pharmacol. 2015;764:554‐561. [DOI] [PubMed] [Google Scholar]
- 47. Chen Y, Du J, Zhao YT, et al. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc Diabetol. 2015;14:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee TI, Kao YH, Tsai WC, Chung CC, Chen YC, Chen YJ. HDAC inhibition modulates cardiac PPARs and fatty acid metabolism in diabetic cardiomyopathy. PPAR Res. 2016;2016:5938740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu Z, Tong Q, Zhang Z, et al. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5‐ERK1/2 pathway. Clin Sci (Lond). 2017;131:1841‐1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bocchi L, Motta BM, Savi M, et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) restores cardiomyocyte contractility in a rat model of early diabetes. Int J Mol Sci. 2019;20:1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuo WW, Wang WJ, Tsai CY, Way CL, Hsu HH, Chen LM. Diallyl trisufide (DATS) suppresses high glucose‐induced cardiomyocyte apoptosis by inhibiting JNK/NFκB signaling via attenuating ROS generation. Int J Cardiol. 2013;168:270‐280. [DOI] [PubMed] [Google Scholar]
- 52. Cruz MS, da Silva AMG, de Souza KSC, Luchessi AD, Silbiger VN. miRNAs emerge as circulating biomarkers of post‐myocardial infarction heart failure. Heart Fail Rev. 2020;25:321‐329. [DOI] [PubMed] [Google Scholar]
- 53. Di Palo KE, Barone NJ. Hypertension and heart failure: prevention, targets, and treatment. Heart Fail Clin. 2020;16:99‐106. [DOI] [PubMed] [Google Scholar]
- 54. Nakamura M, Yoshida H, Arakawa N, Mizunuma Y, Makita S, Hiramori K. Endothelium‐dependent vasodilatation is not selectively impaired in patients with chronic heart failure secondary to valvular heart disease and congenital heart disease. Eur Heart J. 1996;17:1875‐1881. [DOI] [PubMed] [Google Scholar]
- 55. Cecchi F, Sgalambro A, Baldi M, et al. Microvascular dysfunction, myocardial ischemia, and progression to heart failure in patients with hypertrophic cardiomyopathy. J Cardiovasc Transl Res. 2009;2:452‐461. [DOI] [PubMed] [Google Scholar]
- 56. Aistrup GL, Balke CW, Wasserstrom JA. Arrhythmia triggers in heart failure: the smoking gun of [Ca2+]i dysregulation. Heart Rhythm. 2011;8:1804‐1808. [DOI] [PubMed] [Google Scholar]
- 57. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold‐inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829‐839. [DOI] [PubMed] [Google Scholar]
- 58. Martin OJ, Lai L, Soundarapandian MM, et al. A role for peroxisome proliferator‐activated receptor γ coactivator‐1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ Res. 2014;114:626‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Arany Z, He H, Lin J, et al. Transcriptional coactivator PGC‐1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259‐271. [DOI] [PubMed] [Google Scholar]
- 60. Fulco M, Schiltz RL, Iezzi S, et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12:51‐62. [DOI] [PubMed] [Google Scholar]
- 61. Krishnan J, Danzer C, Simka T, et al. Dietary obesity‐associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2‐NAD+ system. Genes Dev. 2012;26:259‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nadal‐Ginard B, Kajstura J, Anversa P, Leri A. A matter of life and death: cardiac myocyte apoptosis and regeneration. J Clin Invest. 2003;111:1457‐1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Crocco P, Montesanto A, Passarino G, Rose G. Polymorphisms falling within putative miRNA target sites in the 3'UTR region of SIRT2 and DRD2 genes are correlated with human longevity. J Gerontol A Biol Sci Med Sci. 2016;71:586‐592. [DOI] [PubMed] [Google Scholar]
- 64. North BJ, Rosenberg MA, Jeganathan KB, et al. SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. EMBO J. 2014;33:1438‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dahl A, Lund C, Russell D. Aterosklerose og hjerneinfarkt [Atherosclerosis and cerebral infarction]. Tidsskr Nor Laegeforen. 2007;127:892‐896. [PubMed] [Google Scholar]
- 66. Jover E, Marín F, Roldán V, Montoro‐García S, Valdés M, Lip GY. Atherosclerosis and thromboembolic risk in atrial fibrillation: focus on peripheral vascular disease. Ann Med. 2013;45:274‐290. [DOI] [PubMed] [Google Scholar]
- 67. Lacy M, Atzler D, Liu R, de Winther M, Weber C, Lutgens E. Interactions between dyslipidemia and the immune system and their relevance as putative therapeutic targets in atherosclerosis. Pharmacol Ther. 2019;193:50‐62. [DOI] [PubMed] [Google Scholar]
- 68. Milutinović A, Šuput D, Zorc‐Pleskovič R. Pathogenesis of atherosclerosis in the tunica intima, media, and adventitia of coronary arteries: an updated review. Bosn J Basic Med Sci. 2020;20(1):21‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gimbrone MA Jr, García‐Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Moschonas IC, Tselepis AD. The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis. 2019;288:9‐16. [DOI] [PubMed] [Google Scholar]
- 71. Soehnlein O, Swirski FK. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol Metab. 2013;24:129‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee HK, Shim EB. Extension of the mitochondria dysfunction hypothesis of metabolic syndrome to atherosclerosis with emphasis on the endocrine‐disrupting chemicals and biophysical laws. J Diabetes Investig. 2013;4:19‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Oalmann MC, Strong JP, Tracy RE, Malcom GT. Atherosclerosis in youth: are hypertension and other coronary heart disease risk factors already at work? Pediatr Nephrol. 1997;11:99‐107. [DOI] [PubMed] [Google Scholar]
- 74. Yahagi K, Kolodgie FD, Lutter C, et al. Pathology of human coronary and carotid artery atherosclerosis and vascular calcification in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2017;37:191‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sarrazy V, Sore S, Viaud M, et al. Maintenance of macrophage redox status by ChREBP limits inflammation and apoptosis and protects against advanced atherosclerotic lesion formation. Cell Rep. 2015;13:132‐144. [DOI] [PubMed] [Google Scholar]
- 76. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. MacParland SA, Tsoi KM, Ouyang B, et al. Phenotype determines nanoparticle uptake by human macrophages from liver and blood. ACS Nano. 2017;11:2428‐2443. [DOI] [PubMed] [Google Scholar]
- 78. Fang S, Xu Y, Zhang Y, et al. Irgm1 promotes M1 but not M2 macrophage polarization in atherosclerosis pathogenesis and development. Atherosclerosis. 2016;251:282‐290. [DOI] [PubMed] [Google Scholar]
- 79. Zhang B, Ma Y, Xiang C. SIRT2 decreases atherosclerotic plaque formation in low‐density lipoprotein receptor‐deficient mice by modulating macrophage polarization. Biomed Pharmacother. 2018;97:1238‐1242. [DOI] [PubMed] [Google Scholar]
- 80. Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. 2017;120:713‐735. [DOI] [PubMed] [Google Scholar]
- 81. Liu QQ, Ren K, Liu SH, Li WM, Huang CJ, Yang XH. MicroRNA‐140‐5p aggravates hypertension and oxidative stress of atherosclerosis via targeting Nrf2 and Sirt2. Int J Mol Med. 2019;43:839‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yu H, Pan W, Huang H, et al. Screening analysis of sirtuins family expression on anti‐inflammation of resveratrol in endothelial cells. Med Sci Monit. 2019;25:4137‐4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liang Y, Li J, Lin Q, et al. Research progress on signaling pathway‐associated oxidative stress in endothelial cells. Oxid Med Cell Longev. 2017;2017:7156941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lin P, Liu J, Ren M, et al. Idebenone protects against oxidized low density lipoprotein induced mitochondrial dysfunction in vascular endothelial cells via GSK3β/β‐catenin signalling pathways. Biochem Biophys Res Commun. 2015;465:548‐555. [DOI] [PubMed] [Google Scholar]
- 85. Zhang W, Liu D, Ren J, Zhou P, Han X. Overexpression of sirtuin2 prevents high glucose‐induced vascular endothelial cell injury by regulating the p53 and NF‐κB signaling pathways. Biotechnol Lett. 2018;40:271‐278. [DOI] [PubMed] [Google Scholar]
- 86. Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505‐514. [DOI] [PubMed] [Google Scholar]
- 87. Wang Y, Branicky R, Noë A, Hekimi S. Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol. 2018;217:1915‐1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ewings KE, Wiggins CM, Cook SJ. Bim and the pro‐survival Bcl‐2 proteins: opposites attract, ERK repels. Cell Cycle. 2007;6:2236‐2240. [DOI] [PubMed] [Google Scholar]
- 89. Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med. 1994;330:1431‐1438. [DOI] [PubMed] [Google Scholar]
- 90. Osol G, Mandala M. Maternal uterine vascular remodeling during pregnancy. Physiology. 2009;24:58‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Thompson AAR, Lawrie A. Targeting vascular remodeling to treat pulmonary arterial hypertension. Trends Mol Med. 2017;23:31‐45. [DOI] [PubMed] [Google Scholar]
- 92. Hashimoto‐Komatsu A, Hirase T, Asaka M, Node K. Angiotensin II induces microtubule reorganization mediated by a deacetylase SIRT2 in endothelial cells. Hypertens Res. 2011;34:949‐956. [DOI] [PubMed] [Google Scholar]
- 93. Wong MK, Gotlieb AI. The reorganization of microfilaments, centrosomes, and microtubules during in vitro small wound reendothelialization. J Cell Biol. 1988;107:1777‐1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ye X, Li M, Hou T, Gao T, Zhu WG, Yang Y. Sirtuins in glucose and lipid metabolism. Oncotarget. 2017;8:1845‐1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li X, Kumar A, Carmeliet P. Metabolic pathways fueling the endothelial cell drive. Annu Rev Physiol. 2019;81:483‐503. [DOI] [PubMed] [Google Scholar]
- 96. Gogiraju R, Bochenek ML, Schäfer K. Angiogenic endothelial cell signaling in cardiac hypertrophy and heart failure. Front Cardiovasc Med. 2019;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wang F, Tong Q. SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1's repressive interaction with PPARgamma. Mol Biol Cell. 2009;20:801‐808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rocha B, Rodrigues AR, Tomada I, et al. Energy restriction, exercise and atorvastatin treatment improve endothelial dysfunction and inhibit miRNA‐155 in the erectile tissue of the aged rat. Nutr Metab (Lond). 2018;15:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhang J, Wang C, Nie H, Wu D, Ying W. SIRT2 plays a significant role in maintaining the survival and energy metabolism of PIEC endothelial cells. Int J Physiol Pathophysiol Pharmacol. 2016;8:120‐127. [PMC free article] [PubMed] [Google Scholar]