

Summary
Branched Lipid II, required for the formation of indirectly crosslinked peptidoglycan, is generated by MurM, a protein essential for high-level penicillin resistance in the human pathogen Streptococcus pneumoniae. We have solved the X-ray crystal structure of Staphylococcus aureus FemX, an isofunctional homolog, and have used this as a template to generate a MurM homology model. Using this model, we perform molecular docking and molecular dynamics to examine the interaction of MurM with the phospholipid bilayer and the membrane-embedded Lipid II substrate. Our model suggests that MurM is associated with the major membrane phospholipid cardiolipin, and experimental evidence confirms that the activity of MurM is enhanced by this phospholipid and inhibited by its direct precursor phosphatidylglycerol. The spatial association of pneumococcal membrane phospholipids and their impact on MurM activity may therefore be critical to the final architecture of peptidoglycan and the expression of clinically relevant penicillin resistance in this pathogen.
Keywords: Streptococcus pneumoniae, MurM, penicillin resistance, peptidoglycan, indirect crosslinks, lipid bilayer, homology modeling, molecular dynamics, molecular docking
Graphical abstract
Highlights
-
•
S. aureus FemX structure generated an improved homology model of S. pneumoniae MurM
-
•
A Lipid II binding site in MurM was identified by docking and molecular dynamics
-
•
Modeling indicates cardiolipin is enriched at the MurM protein:membrane interface
-
•
Enzyme assays show that phospholipids influence MurM activity
With an improved homology model of MurM, York et al., use docking and molecular dynamics studies to identify a potential Lipid II binding site and simulate how MurM interacts with this membrane-embedded substrate. Cardiolipin, a membrane phospholipid, was also found to associate with MurM and upregulate its enzymatic activity.
Introduction
The peptidoglycan (PG) of the bacterial cell wall is a polymer consisting of alternating β-1,4-linked N-acetyl glucosamine (GlcNAc) and N-acetyl muramic acid (MurNAc) residues. Appended to the MurNAc sugar is a pentapeptide stem that can be crosslinked directly or indirectly to form a rigid mesh-like structure (Bugg et al., 2011). PG biosynthesis begins with the cytoplasmic formation of a Park nucleotide, which is subsequently converted into a lipid-linked PG precursor known as Lipid II. Lipid II is then transported across the membrane, where it is polymerized and crosslinked by the penicillin-binding proteins (PBPs) (Figure 1). PG is an essential component of the cell wall, involved in cell growth and division, maintaining structural integrity, and resisting high osmotic pressures. Inhibition of cell wall biosynthesis is a key mechanism for many antibiotics, including β-lactams, glycopeptides, and amino acid analogs (Schneider and Sahl, 2010).
Figure 1.

The PG biosynthesis pathway
(1) The cytoplasmic stage is characterized by the formation of UDP-MurNAc-pentapeptide (UDP-MurNAc-5P) by MurA-MurF. The pentapeptide stem peptide usually comprises L-Ala-γ-D-Glu-L-Lys-D-Ala-D-Ala in most Gram-positive organisms including the pneumococcus.
(2) At the internal face of the cytoplasmic membrane, MraY catalyzes the formation of Lipid I from UDP-MurNAc-5P to undecaprenyl-pyrophosphate, which is then converted to Lipid II by MurG. In S. pneumoniae, the second position D-glutamyl α-carboxyl is amidated to D-iso-glutamine (iGln) by the MurT/GatD complex, and in some cases a dipeptide branch of either L-Ser/L-Ala or L-Ala/L-Ala may be appended at the ε-amino group of the third-position lysine by MurM and MurN, respectively. The exact order of the cytoplasmic membrane steps remains uncertain, but here for clarity they appear as a linear sequence, with conversion to Lipid II occurring before peptide stem modifications, and amidation occurring before branching. Lipid II is translocated across the membrane by MurJ.
(3) At the external face of the cytoplasmic membrane, class A PBPs and complexes of SEDs-Class B PBPs form glycan chains by transglycosylation (TG), with the concomitant release of undecaprenyl-pyrophosphate, and form either direct or indirect crosslinks throughout the PG layer via transpeptidation (TP). Nucleotide sugars UDP-GlcNAc and UDP-MurNAc and the sugars GlcNAc and MurNAc are signified by blue, violet, dark-blue, and purple elongated hexagons, respectively. Figure created using BioRender.com.
In Streptococcus pneumoniae and other Gram-positive bacteria, the glutamate at the second position of the Lipid II pentapeptide is α-amidated to form iso-glutamine by the essential GatT/MurD complex (Figueiredo et al., 2012; Münch et al., 2012; Zapun et al., 2013; Morlot et al., 2018). In addition, branched Lipid II, capable of generating indirect crosslinks, can be formed by the non-essential aminoacyl-tRNA-dependent ligases, MurM and MurN. MurM and MurN are responsible for the sequential addition of amino acids to the third-position lysine of the pentapeptide stem of Lipid II (Filipe et al., 2000). MurM can append either L-serine or L-alanine at the first position of the dipeptide bridge, while MurN extends this modification by addition of an invariable L-alanyl moiety. Branched PG precursors are also found in several other Gram-positive bacterial pathogens, for example the glycyl-tRNAGly-dependent enzymes FemX, FemA, and FemB are responsible for the addition of a pentaglycyl bridge in Staphylococcus aureus (Schneider et al., 2004). In comparison with other Gram-positive organisms, the PG of S. pneumoniae is highly heterogeneous: the predominant C-terminal amino acid at position 1 of the dipeptide and the proportion of indirect crosslinks throughout the PG vary significantly between different strains (Severin and Tomasz, 1996; Garcia-Bustos et al., 1987; Garcia-Bustos and Tomasz, 1990). In vitro and in vivo studies indicate that MurM from the penicillin-resistant strain S. pneumoniae(159) preferentially incorporates L-alanine, while MurM from a penicillin-sensitive strain S. pneumoniae(Pn16) preferentially incorporates L-serine (Lloyd et al., 2008). In addition, penicillin-resistant strains demonstrated higher levels of indirect crosslinking in the PG compared with penicillin-susceptible isolates, although the overall degree of crosslinking remained constant (Garcia-Bustos and Tomasz, 1990).
Resistance to β-lactam antibiotics in S. pneumoniae is characterized by extensive interspecies recombination of PBP transpeptidase domains, which results in sequence heterogeneity within and around the mosaic PBP active site with a consequential lowering of β-lactam binding affinity (Smith et al., 1991). This mechanism of resistance contrasts with that of many other bacteria that have acquired genes for β-lactamase enzymes which inactivate the antibiotic before it binds to and inhibits the PBPs. Interestingly, deletion of the murM gene in S. pneumoniae eliminates indirect crosslinks from the PG and results in a complete loss of penicillin resistance (Filipe et al., 2001). It has been proposed that the changes to the PBP active site, which prevent β-lactam binding, may also alter the Lipid II substrate specificity such that the PBPs bind branched Lipid II more tightly than unbranched Lipid II. MurM is therefore necessary but not sufficient for resistance in clinical strains of S. pneumoniae, making it an interesting target for the development of new inhibitors of antimicrobial resistance (Filipe and Tomasz, 2000).
The cytoplasmic membrane of S. pneumoniae contains two phospholipids, phosphatidylglycerol and cardiolipin (Trombe et al., 1979; Pesakhov et al., 2007), where cardiolipin synthase is responsible for generating cardiolipin from two molecules of phosphatidylglycerol (Schlame, 2008). The proportion of cardiolipin and phosphatidylglycerol, as a percentage of the overall membrane lipids, varies in S. pneumoniae between anaerobic and aerobic growth conditions. Cardiolipin was found to decrease from 15.3% to 8.3% while phosphatidylglycerol increased from 12.7% to 16.3% in anaerobic conditions compared with aerobic conditions (Pesakhov et al., 2007). The peptidoglycan precursor, Lipid II, is tethered to the cell membrane by virtue of its C55 Lipid II tail. Therefore, MurM acts on its lipid substrate in close proximity to the cytoplasmic leaflet of the cell membrane and is potentially influenced by membrane phospholipid composition.
Previously, MurM inhibitors have been identified; however, none have shown growth inhibition or any effect on penicillin minimum inhibitory concentration, indicating that these compounds cannot effectively cross the cytoplasmic membrane of S. pneumoniae (Cressina et al., 2007, 2009). MurM has thus far resisted extensive crystallization in our laboratory, and consequently its X-ray-solved structure is not available. However, in a related study we were able to solve the X-ray structure of the isofunctional homolog of MurM from S. aureus (FemX), which we have used here as a template for homology modeling of MurM. Using this MurM homology model, we have successfully identified the Lipid II binding site and have used molecular dynamics (MD) simulations to investigate interactions between MurM and both membrane phospholipids and its Lipid II substrate (Witzke et al., 2016). We subsequently studied the effects of these membrane-embedded phospholipids, on the enzymatic activity of MurM in vitro, corroborating our in silico analysis. These studies provide insights into the structure and activity of MurM, providing a link between phospholipid membrane composition and peptidoglycan architecture. This may be useful for the development of chemical probes for these proteins and has important implications for future studies on penicillin resistance mechanisms in S. pneumoniae.
Results
X-ray crystallography and structure determination of S. aureus FemX
The crystal structure of S. aureus FemX was solved to a resolution of 1.62 Å, and the structure was deposited in the PDB with accession number PDB: 6SNR. A summary of the data collection and refinement statistics is given in Table 1.
Table 1.
Summary of crystallographic data collection and refinement statistics from the S. aureus FemX structure
| FemX | |
|---|---|
| Data collection | |
| Synchrotron radiation detector, wavelength (Å) | Pilatus 6M-F, 0.920 |
| Unit cell a, b, c (Å), α, β, γ (°) | 45.01, 83.62, 133.93, 90.0, 90.0, 90.0 |
| Space group | P212121 |
| Resolution (Å) | 52.27–1.62 [1.66–1.62] |
| Observations | 422,822 [29,596] |
| Unique reflections | 65,058 [4,782] |
| I/σ(I) | 15.7 [2.6] |
| Rsyma | 0.065 [0.567] |
| Rmeas | 0.078 [0.690] |
| Rp.i.m | 0.031 [0.273] |
| Completeness (%) | 99.7 [99.8] |
| Refinement | |
| Non-hydrogen atoms | 3,397 (including 177 waters) |
| Rcrystb | 0.221 [0.262] |
| Reflections used | 61,691 [4,531] |
| Rfreec | 0.262 [0.296] |
| Reflections used | 3,294 [244] |
| Rcryst (all data)b | 0.222 |
| Average temperature factor (Å2) | 26 |
| RMSDs from ideal values | |
| Bonds (Å) | 0.013 |
| Angles (°) | 1.5 |
| DPI coordinate error (Å)d | 0.098 |
| Ramachandran plote | |
| Favored (%) | 98.0 |
| Outliers (%) | 0.0 |
The highest-resolution bin of data is indicated by square brackets. Numbers in square brackets refer to values in the highest-resolution shell.
, where Ih,j is the is the jth observation of reflection h, and Ih is the mean intensity of that reflection.
, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively.
Rfree is equivalent to Rcryst for a 4% subset of reflections not used in the refinement (Brünger, 1992).
DPI refers to the diffraction component precision index (Cruickshank, 1999).
As calculated by MolProbity (Williams et al., 2008).
The final solved structure of S. aureus FemX contains a globular domain and a coiled-coil domain. Similar to FemA (Benson et al., 2002), the globular domain can be divided into two subdomains. Each subdomain contains a central five-stranded mixed-polarity β sheet surrounded by four α helices. Subdomain 1A comprises residues 1–145 and 384–421 while subdomain 1B comprises residues 146–234 and 298–383. Unfortunately, residues 403–421 were not present in the density. The coiled-coil domain consists of two antiparallel α helices, comprising residues 235–297. FemX and FemA can be superimposed onto each other with a root-mean-square deviation (RMSD) of ∼2.7 Å over 384 residues. Similarly to FemA, FemX has a deep L-shaped channel of about 20 × 40 Å located alongside the globular domain and mainly in subdomain 1B. This channel comprises a peptidoglycan precursor binding site which was previously identified in S. aureus FemA (Benson et al., 2002). The identity of this peptidoglycan precursor binding site has been further confirmed by crystallographic analysis of Weissella viridescens FemX complexed with substrates (Biarrotte-Sorin et al., 2004).
Homology modeling of S. pneumoniae MurM
The structures of two MurM homologs, S. aureus FemA and W. viridescens FemX, were solved previously by X-ray crystallography (Benson et al., 2002; Fonvielle et al., 2013; Biarrotte-Sorin et al., 2004). The S. aureus FemA structure was subsequently used as a template for homology modeling of MurM by Fiser et al. (2003). However, alignment of S. aureus FemA, S. aureus FemX, S. pneumoniae MurM, and W. viridescens FemX (Figure S1) showed that S. aureus FemX possesses the highest sequence identity to MurM and is also more functionally homologous to S. pneumoniae MurM, as it appends the first amino acid of the cross-bridge to the Lipid II precursor (Matsuhashi et al., 1967; Schneider et al., 2004). In contrast, S. aureus FemA appends the second and third amino acid residues of the cross-bridge to the α-amino group of a glycyl residue appended to the ε-amino group of the stem peptide L-lysyl residue of the Lipid II precursor. Therefore, given the difficulties in obtaining MurM crystals, we were motivated to solve the structure of its functional homolog (S. aureus FemX) by X-ray crystallography to access the structure of MurM in silico.
Using the structure of S. aureus FemX, we generated a homology model for MurM, which consists of a globular domain comprising two subdomains and a coiled-coil helical arm (Figure 2). Each subdomain comprises two twisted β-sheet cores surrounded by α helices; subdomain 1A is formed of residues 1–153 and 382–401, while subdomain 1B is made up of residues 154–241 and 294–381. The coiled-coil domain comprises residues 242–293. While the MurM homology model presented here is similar to the previous model (Fiser et al., 2003), the RMSD of the two models is 3.8 Å over 368 residues, indicating that there are some key structural differences, namely: loss of N-terminal β1 and antiparallel β6/β13 from the previous model; addition of α5 and β11/β12; and presence of an α-helical secondary structure proximal to the C-terminal end of this MurM model.
Figure 2.

Cartoon representation of MurM predicted structure
Fourteen α helices (red), 12 β sheets (yellow), and unstructured regions (green). Best model obtained based on SOAP and DOPE scores following homology modeling using MODELLER with S. aureus FemX as a template. The structure was rendered in PyMOL (version 2.2.0).
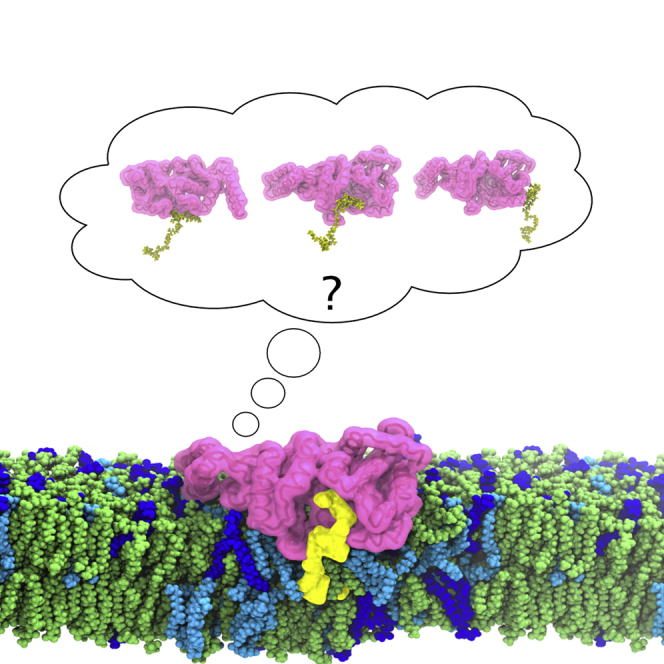
Identification of a possible Lipid II binding site of MurM
Our MurM model revealed a binding pocket that was not present in the previous model of MurM produced by Fiser et al. (2003). Structural comparison between W. viridescens FemX co-crystallized with its uridine diphosphate (UDP)-MurNAc-pentapeptide substrate and this MurM model allowed identification of a Lipid II binding site that corresponds to the peptidoglycan precursor binding sites identified previously in S. aureus FemA and W. viridescens FemX (Benson et al., 2002; Biarrotte-Sorin et al., 2004) and in our structure of S. aureus FemX. When this MurM model and the W. viridescens FemX were aligned and overlaid, the proposed MurM binding site appeared to easily accommodate the soluble UDP-MurNAc-pentapeptide substrate well (Figure 3A). The following eight residues—Tyr103, Lys36, Asn38, Trp39, Thr209, Arg211, Try215 and Tyr256—were independently proposed to be involved in substrate binding in the peptidoglycan precursor and tRNA liganded W. viridescens FemX structures (Biarrotte-Sorin et al., 2004; Fonvielle et al., 2013). The corresponding MurM residues, defined as having residues which have similar properties, and occupying a similar location and orientation in physical space with side chains facing the binding pocket, were identified in the MurM structure as Phe103, Lys35, Trp38, Arg215, and Tyr219; therefore, these residues may also be important for substrate binding in MurM.
Figure 3.

Surface representation of MurM binding site
(A) MurM159 model aligned and overlaid with the UDP-MurNAc-pentapeptide substrate, which was co-crystallized with W. viridescens FemX.
(B) MurM159 model with truncated Lipid II docked in the binding site, using AutoDock Vina.
(C) MurM159 model with Lipid II in the binding site, from membrane simulations. Figures were created with PyMOL (version 2.2.0) and Chimera (version 1.13.1).
See also Figures S2 and S3.
Next, molecular docking using AutoDock Vina (Trott and Olson, 2010) was conducted to independently investigate docking of the Lipid II substrate to the MurM model we generated. Lipid II is a large molecule that is, in general, unsuitable for molecular docking studies. In addition, the lipid tail is embedded in the membrane, and so is not itself available for binding to MurM. Therefore, a truncated Lipid II molecule, composed of a methyl capped diphospho GlcNAc-MurNAc-pentapeptide, was used for these docking experiments (Figure S2).
When AutoDock Vina was allowed to search the entire protein surface of MurM, all docking results returned were within the identified binding site, indicating that there are no other suitable binding sites on the protein. The search was then restricted to the binding site, and the top ten results were obtained. The top five results obtained all had identical binding affinities; however, two of these docking orientations, where the phosphates are located deep within the binding pocket, would be physically impossible for the natural substrate (Lipid II) in vivo, since the membrane-embedded prenyl lipid tail is appended via the phosphate. The remaining three docking orientations all orient the phosphates close to the opening of the binding site with the pentapeptide chain disappearing deep into the binding pocket. The exact orientation of the pentapeptide chain is variable, indicating that the binding site is spacious and that Lipid II may be accommodated in a number of different possible orientations. Figure 3B shows one conformation in which the docking of truncated Lipid II is similar to the orientation of the soluble UDP-MurNAc-pentapeptide from W. viridescens FemX overlaid with MurM (Figure 3A), the remaining four substrate orientations are shown in Figure S3. A key limitation of docking is that it considers the protein as rigid; therefore, multiple substrate orientations may indicate that conformational changes within the binding site may occur upon substrate binding or during catalysis.
Additional docking studies were performed to further explore the Lipid II binding pose in the putative MurM binding site and establish whether this binding site shows specific affinity for Lipid II or is indiscriminate between different lipid species. Therefore, Lipid II, cardiolipin, phosphatidylglycerol, and phosphatidylethanolamine lipids (full molecules) were docked into the putative binding site, where the binding site is defined by the residues Lys35, Trp38, Phe103, Arg215, and Tyr219. We found Lipid II bound into this site with atoms of the headgroup within 3 Å of the residues Lys35, Trp38, Phe215, and Arg219, and with the Lipid II tail outside of this site. In stark contrast, the cardiolipin and phosphatidylethanolamine headgroups did not show any preference for this site; instead the binding poses showed their tails inserted into the cavity. There was one binding pose for phosphatidylglycerol whereby the phospholipid headgroup was in the putative binding site, although a portion of the tail was also inside it; the remaining two phosphatidylglycerol poses had the tails inserted into this cavity. For each lipid type the top cluster of binding poses produced by HADDOCK2.4 (Van Zundert et al., 2016) were analyzed, and the top three binding poses for each lipid are shown in Figure S4.
Interactions between MurM and the lipid bilayer
Given the docking studies are performed in the absence of the membrane environment, a set of coarse-grained MD simulations was next performed to establish the likelihood of MurM being oriented on the membrane such that the putative binding site is available to Lipid II. MD simulations were used to model the interactions of MurM with the lipid bilayer. Six independent coarse-grained simulations were conducted for each of three membrane systems described in Table S1. In 16 of these simulation runs, MurM readily associated with the membrane in <3 μs and its orientation with respect to the membrane remained unchanged for the remaining 2 μs (Figure S5). In addition, in the same membrane leaflet as the peripheral MurM, more than 50% of Lipid II molecules were located within 2 nm of the protein. MurM associated with the membrane in a number of different orientations, which can be classified into two groups: those in which the binding site predicted from docking studies is available for lipid binding and those in which it is not. Table 2 shows that in 18 unbiased simulations; MurM adhered to the membrane in 16 of the simulations and of these, the binding site was available for Lipid II binding in 11 and unavailable in only 5. In 3 of these 18 unbiased simulations, MurM was oriented such that the Lipid II molecule was located in the putative binding site, which demonstrates that Lipid II is able to successfully enter this binding site even on the short timescale of an MD simulation (Figure 4). Back mapping of one of these systems to all-atom resolution allowed the binding site to be explored in more detail. Three independent atomistic simulations each of 250 ns duration found Lipid II located in the same binding site of MurM that was identified during molecular docking of the truncated Lipid II substrate, and in the co-crystal structure of UDP-MurNAc-pentapeptide with W. viridescens FemX (Figure 3A). In both the molecular docking and atomistic simulations, the Lipid II headgroup forms stable interactions with Lys35, Trp38, Arg215, and Tyr219.
Table 2.
Summary of MurM adherence to and orientation on the three different membrane systems
| Membrane | Available | Unavailable | Non-adherence | Lipid II in binding site |
|---|---|---|---|---|
| 1 (0% cardiolipin) | 4 | 1 | 1 | 1 |
| 2 (12% cardiolipin) | 4 | 1 | 1 | 2 |
| 3 (16% cardiolipin) | 3 | 3 | 0 | 0 |
Orientation of MurM was categorized such that the putative binding site was either available for Lipid II binding (facing/close to the membrane) or unavailable for Lipid II binding (facing away from the membrane).
Figure 4.

Lipid II binding to the putative MurM binding site
(A), (B), and (C) show the three simulations whereby Lipid II was found to bind in the putative MurM binding site. Each panel shows the MurM binding to Lipid II with respect to the membrane (top), and an enlarged image of the MurM binding to Lipid II, with the membrane removed (bottom). MurM binding site residues K35, W38, R215, and Y219 are depicted in yellow; the Lipid II headgroup and prenyl chain are rendered in red and blue, respectively.
See also Figure S1.
Similarly to the molecular docking findings, this MD simulation shows that the MurM binding site is flexible and allows the Lipid II molecule to adopt a wide variety of conformations (Figure 5). This may suggest that binding of a second substrate or a large conformational change may be required for catalysis.
Figure 5.

Different conformations of Lipid II inside MurM binding site
(A and B) MurM (gray) with Lipid II binding, colored on a blue to white to red scale with respect to simulation time, in system 5 (Table S2).
Interactions between MurM and membrane phospholipids
MD simulations were used to investigate the effects of membrane phospholipids (cardiolipin and phosphatidylglycerol) on MurM at the cytoplasmic membrane interface. To investigate local lipid enrichment/depletion, we calculated two-dimensional enrichment maps across the entire membrane and depletion-enrichment (D-E) indices within 1.1 nm of MurM, as described by Corradi et al. (2018) (see STAR Methods). A D-E index <0, or an enrichment percentage <0%, indicated that the specified lipid was depleted with respect to the bulk membrane composition. Figure 6 shows that upon association of MurM with the cytoplasmic membrane, there was no effect on the distribution of phosphatidylglycerol or phosphatidylethanolamine. However, in membranes containing 8% or 16% cardiolipin, cardiolipin was enriched at the MurM:membrane interface.
Figure 6.

Interactions between MurM and membrane phospholipids
(A–C) Depletion-enrichment (D–E) indices for phosphatidylethanolamine (PE), phosphatidylglycerol (PG), and cardiolipin (CL) occurring within a 1.1-nm perimeter of the MurM protein for (A) Systems 4 and 5 (molar ratio of 75% phosphatidylethanolamine and 25% phosphatidylglycerol), (B) Systems 6 and 7 (molar ratio of 76% phosphatidylethanolamine, 16% phosphatidylglycerol, and 8% cardiolipin), and (C) Systems 8 and 9 (molar ratio of 72% phosphatidylethanolamine, 12% phosphatidylglycerol, and 16% cardiolipin). The D-E index was determined from 150 to 250 ns in 50-ns blocks for all repeats for a total of eight values per plot.
(D) Example of a D-E map with MurM at the membrane. White dots represent the center of geometry of each protein amino acid residue, and the percentage enrichment of phospholipid is indicated by the color.
(E) Activation of MurM was calculated as the product of subtraction of MurM velocity in the absence of cardiolipin (v0(−C)) from MurM velocity in the presence of cardiolipin (v0(+C)) divided by v0(−C) and was plotted versus cardiolipin concentration.
(F) Inhibition of MurM was calculated as [(v0(-PhG)) − (v0(+PhG))]/v0(−PhG) × 100 (where PhG denotes phosphatidylglycerol) and was plotted versus phosphatidylglycerol concentration. Data were fitted as described in the text. GraphPad Prism (version 8.4.1) and Matplotlib (version 3.0.3) were used for data analysis and figure preparation.
See also Figure S2.
The importance of these observations was considered in vitro by measuring the enzymatic activity of MurM in the presence of varying concentrations of cardiolipin or phosphatidylglycerol. These enzymatic studies show that cardiolipin activates MurM while phosphatidylglycerol inhibits MurM in a concentration-dependent manner. Figure 6E shows the enzymatic activation of MurM with respect to cardiolipin concentration whereby a 9.1-fold activation of MurM was achieved, with 50% activation occurring at 0.4 mM cardiolipin. Figure 6F shows that the activity of MurM could be completely inhibited by phosphatidylglycerol, with an IC50 of 0.2 mM. Furthermore, Hill coefficients of 2.7 ± 0.3 and 2.8 ± 0.2 for cardiolipin and phosphatidylglycerol, respectively, indicate that both these phospholipids exhibit their effects on MurM in a cooperative manner. Phosphatidylethanolamine, used in the construction of the model pneumococcal membrane to which MurM bound, when tested at a concentration of 0.72 mM, only slightly activated MurM activity by 0.32-fold (duplicate determination with a difference of <10%). In comparison, 0.72 mM cardiolipin activated MurM by 8-fold (Figure 6E). Therefore, the impact of phosphatidylethanolamine on the disposition of MurM relative to its interaction with Lipid II and the phospholipid bilayer could be neglected.
Discussion
The crystal structure of S. aureus FemX has allowed us to generate an improved homology model of MurM leading to the identification of a putative Lipid II binding site. Fiser et al. (2003) proposed a different MurM model and speculated about an alternative binding site based on structural and functional analogy between MurM and N-myristoyltransferase (NMT) proteins. However, while the substrates of both NMT proteins and MurM are lipids, they are contextually very different. The NMT proteins are cytoplasmic proteins that contain a deep, narrow pocket which is highly specific for the myristoyl fatty acyl chain (Wright et al., 2010; Heuckeroth et al., 1988). In contrast, MurM binds the disaccharide headgroup and pentapeptide side chain of Lipid II, and the undecaprenyl C55 lipid tail is embedded in the membrane. Despite similarities with NMT proteins, the substrate binding site we propose more closely resembles those of W. viridescens FemX, S. aureus FemX, and FemA.
The orientations of truncated Lipid II in docking studies and Lipid II in MD simulations are strikingly similar to each other and also the orientation of UDP-MurNAc-pentapeptide substrate co-crystallized in W. viridescens FemX. This is consistent with the observation that, although inefficient compared with Lipid II, UDP-MurNAc pentapeptide is a MurM substrate (Lloyd et al., 2008). In all cases, the diphosphates are near the surface of the protein, and the protruding pentapeptide reaches into the binding pocket with the third-position lysine on the left-hand side of the binding pocket. In addition, previous studies suggest that the height of the Lipid II headgroup is 19 Å (Ganchev et al., 2006), and the binding pocket of this model was measured to be 15 Å. Since the Lipid II headgroup is flexible and the binding site provides enough room for the substrate to bend, these measurements are consistent. Together with our findings, this strongly supports the identification of this cavity as the Lipid II binding site and suggests that the Lipid II binds to MurM in an orientation similar to that of W. viridescens FemX binding to its substrate.
Alanyl-phosphatidylglycerol synthase (PDB: 4v34), similarly to MurM, also utilizes both lipid and alanyl-tRNAAla substrates (Hebecker et al., 2015). To successfully bring these two substrates together for catalysis it possesses two binding sites, located on opposite sides of the protein, which are connected by a channel. The protein itself provides a barrier between the hydrophobic lipid and the hydrophilic tRNA, such that they do not come into close proximity with each other. The negatively charged surface patch identified previously (Fiser et al., 2003) remains present on this homology model of MurM and is located on the opposite side of the protein with respect to the Lipid II binding site, which is located within a positively charged surface patch. This negative patch is unsuitable for the binding of negatively charged tRNA, so it is unlikely that MurM shares the same mechanism of action as alanyl-phosphatidylglycerol synthase. The negatively charged surface patch may, however, be important for protein-protein interactions occurring either at the cell surface or in the cytoplasm.
These modeling studies reveal that the stem peptide protrudes perpendicular to the surface of the membrane into the active site of MurM. Therefore, in order for alanyl-tRNAAla to simultaneously interact with MurM, while it is located over its lipid substrate the highly negatively charged hydrophilic tRNA would have to be brought into close proximity with the negatively charged phospholipid headgroups and/or the hydrophobic phospholipid tails below them. Given that this would be a highly unfavorable interaction, we propose an alternative “ping-pong” mechanism of action for MurM whereby MurM is initially aminoacylated by alanyl- or seryl-tRNA in the cytoplasm before translocation to the cell membrane for aminoacyl transfer to Lipid II. While the MurM is in the cytoplasm and not interacting with the membrane, the positively charged patch, of the proposed Lipid II binding site, may facilitate interaction with a polyanionic substrate such as tRNA. Once this has occurred, subsequent interaction of the aminoacyl-MurM with the surface of the membrane could accommodate the correct and catalytically productive interaction of aminoacylated-MurM with Lipid II. Although this proposed mechanism is at variance with the sequential mechanism of catalysis proposed for W. viridescens FemX (Hegde and Blanchard, 2003), here both substrates are highly hydrophilic nucleotide or polynucleotide derivatives in the same cellular subcompartment and are therefore without biophysical impediment with regard to their proximity during catalysis. Here, with regard to MurM, the chemical properties and location of both substrates indicate an advantage to a mechanism that avoids their simultaneous binding.
The pneumococcal peptidoglycan is heterogeneous with respect to its composition of directly and indirectly crosslinked stem peptides. It remains unclear as to whether the activity of MurM, and therefore the generation of indirect crosslinks, is distributed equally around the entire cell surface or whether it is localized to specific sites. Phospholipids are known to be involved in the spatial and temporal biochemistry of cells (Lin et al., 2019), and cardiolipin was shown to be enriched at the poles and septa of Escherichia coli and Bacillus subtilis, localizing specific membrane-associated proteins to these regions (Bramkamp and Lopez, 2015). Our simulations indicate that, while cardiolipin enrichment occurs within the membrane in the presence of MurM, this phospholipid is not essential for membrane association of MurM to occur. Therefore, it remains uncertain as to whether in vivo cardiolipin is highly concentrated in patches in the membrane and is used to recruit MurM to that location or whether association of MurM with the membrane drives the enrichment of cardiolipin in the membrane.
Despite this uncertainty, we show that cardiolipin stimulates the enzymatic activity of MurM, and while it is not clear if this increased activity is a result of a direct effect on the protein or the Lipid II substrate, or both, the spatial association of cardiolipin to the MurM protein suggests that at least some of this effect may be due to direct interactions with the MurM protein.
Cardiolipin has previously been found to bind to and activate a wide range of proteins including MurG (van den Brink-van der Laan et al., 2003), rat liver protein kinase N (Morrice et al., 1994; Peng et al., 1996), porcine heart AMP deaminase (Purzycka-Preis and Zydowo, 1987), rat liver multi-catalytic proteinase (Ruiz de Mena et al., 1993), E. coli glycerol-3-phosphate acyltransferase (Scheideler and Bell, 1989), E. coli dnaA (Sekimizu and Kornberg, 1988), and streptococcal hyaluronan synthases (Tlapak-Simmons et al., 1999a, 1999b, 2004; Weigel et al., 2006; Tlapak-Simmons et al., 1998). This further supports the contention that cardiolipin affects MurM activity by directly interacting with MurM. Similar cardiolipin-mediated sigmoidal stimulatory effects have been seen with other streptococcal membrane proteins such as the hyaluronan synthases from Streptococcus pyogenes and Streptococcus equisimilis (Tlapak-Simmons et al., 1999a, 1999b, 2004; Weigel et al., 2006). In these examples, up to 16 cardiolipin molecules are believed to associate with single hyaluronan synthase molecule (Tlapak-Simmons et al., 1998).
We also show that phosphatidylglycerol inhibits the catalytic activity of MurM and that the concentration of this lipid in the membrane environment surrounding the MurM changes very little. Therefore, the inhibitory effect of phosphatidylglycerol may be exerted by altering the presentation of the Lipid II substrate to MurM rather than by having a direct effect on the protein itself. It is possible that in S. pneumoniae, as in E. coli and B. subtilis, cardiolipin gathers in specific regions of the membrane, where in the pneumococcus it localizes and upregulates the activity of MurM, resulting in higher levels of indirect crosslinking in these regions.
While MurM alone is not sufficient for penicillin resistance, the enzyme is crucial together with mosaic S. pneumoniae PBPs for the generation of a highly resistant phenotype. Deletion of murM from resistant strains resulted in a virtual abolition of penicillin resistance that could not be restored by PBP DNA. Indeed, additional murM DNA from a resistant strain was required for full expression of donor-level penicillin resistance (Filipe and Tomasz, 2000; Smith and Klugman, 2001). Given the importance of MurM for penicillin resistance, the enrichment of cardiolipin at the MurM:membrane interface which activates MurM, and the inhibition of MurM activity by phosphatidylglycerol may regulate the penicillin resistance phenotype imparted by MurM activity, which may therefore be regulated by cardiolipin synthase activity. These findings have therefore revealed a crucial and hitherto unexplored facet of penicillin resistance suggesting the involvement of other areas of pneumococcal metabolism in the expression of clinical antibiotic resistance.
Conclusions
The MurM structural model presented in this work allowed identification of the Lipid II binding site and the contextual presentation of this substrate to MurM. Furthermore, this work characterized the impact of membrane phospholipids on MurM at the MurM:membrane interface and may have spatial mechanistic implications for the catalytic activity of this protein. MD enabled the in silico investigation into MurM-membrane interactions, which are often overlooked when studying enzymes that act at the cytoplasmic membrane interface. The subsequent in vitro experiments on the importance of phospholipids for MurM activity corroborate the in silico findings, supporting the role of phospholipids as an important contributor to the regulation of MurM at the membrane. These studies provide insights into the structure of MurM, which may guide future mutational studies and allow a more detailed analysis of the structure-function relationship of this protein. This research contributes important findings toward achieving a more complete understanding of the role of MurM in pneumococcal penicillin resistance mechanisms.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
|
E. coli BL21 Star (DE3): F- ompT hsdSB (rB- mB-) gal dcm rne131 (DE3) |
Invitrogen | Cat# C601003 |
|
E. coli B834 (DE3) - F- ompT hsdSB (rB- mB-) gal dcm met (DE3) |
Novagen | Cat# 69401 |
| E.coli Rosetta 2 (DE3). F- ompT hsdSB(rB- mB-) gal dcm (DE3) pRARE2 (CamR) | Novagen | Cat# 71405 |
| E. coli BL21 Star pRosetta2 (DE3) - F- ompT hsdSB (rB- mB-) gal dcm rne131 pRARE (DE3) | Lloyd et al. (2008) | N/A |
| E. coli C41 (DE3) (a derivative of BL21(DE3) [F_ ampT hsdS8 (r8m8)gal dcm DE3]) pRIL (CamR) | Lloyd et al. (2008) | N/A |
| E. coli B834 (DE3): pRosetta2 - F- ompT hsdSB (rB- mB-) gal dcm met pRARE2 (DE3) | Novagen | Cat# 69041 |
| M. flavus (M. luteus) (Stanley130.21) | NCIMB | Cat# 8166 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Poly(ethylene glycol) 8000 | Fluka | Cat# 81272 |
| L-Alanine, [2,3-3H] | Moravek | Cat# MT-886 |
| Lipid II (Lys) | BACWAN/Lloyd et al. (2008) | Cat# C55-LII-5K |
| Phosphatidylglycerol L-alpha-Phosphatidyl-DL-glycerol sodium salt from egg yolk lecithin, =99% (TLC), lyophilized powder |
MERCK LIFE SCIENCE UK LTD | Cat# P8318 |
| Cardiolipin Cardiolipin sodium salt from bovine heart, =98% (TLC), lyophilized powder |
MERCK LIFE SCIENCE UK LTD | Cat# C0563 |
| JCSG-plus™ crystallization screen | Molecular Dimensions | Cat# MD1-37 |
| PACT premier™ crystallization screen | Molecular Dimensions | Cat# MD1-29 |
| Morpheus® crystallization screen | Molecular Dimensions | Cat# MD1-46 |
| Cobalt TALON resin | Takara | Cat# 635502 |
| Nickel- Chelating Sepharose | Cytiva | Cat# 17526801 |
| Superdex 75 Size exclusion media | Cytiva | Cat# 17104404 |
| Sephacryl S200 size exclusion media | Cytiva | Cat# 17058401 |
| Selenomethionine | Acros Organics | Cat# 259960025 |
| Tomato etch virus (TEV) Protease | New England Biolabs | Cat# P8112 |
| BsaI | New England Biolabs | Cat# R0535S |
| Xho1 | New England Biolabs | Cat# R0146S |
| Hen egg white lysozyme | MERCK LIFE SCIENCE UK LTD | Cat# L6976 |
| Deposited Data | ||
| S. aureus FemX | This paper | PDB: 6SNR |
| Oligonucleotides | ||
| FemX Forward TTTGCGGGTGGTCTCCCATGGAAA AGATGCATATCACTAATCAGG |
IDT DNA | N/A |
|
S. aureus FemX Reverse TTTGCGCTCGAGGCCCTGAAAATACAG GTTTTCTTTTCGTTTTAATTTACGAGATA TTTTAATTTTAGC |
IDT DNA | N/A |
| Recombinant DNA | ||
| pRosetta 2 (purified from Escherichia coli Rosetta™ 2 (DE3)) | Novagen | Cat# 71405 |
| pET28a(+) | Novagen | Cat# 69864 |
| pET21b::MurM159 | Lloyd et al. (2008) | N/A |
| pET26b::alaRS | Lloyd et al. (2008) | N/A |
| pET28::FemX | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism Version 8.4.1. | GraphPad Soft-ware, San Diego, CA, USA. | www.graphpad.com |
| Xia2 | Winter (2010) | https://xia2.github.io/ |
| XDS package | Kabsch (2010) | http://xds.mpimf-heidelberg.mpg.de |
| SHELX suite | Sheldrick (2010) | https://www.shelxle.org/shelx/eingabe.php |
| ARP/wARP | Langer et al. (2008) | https://www.embl-hamburg.de/ARP/ |
| REFMAC | Vagin et al. (2004) | https://www2.mrc-lmb.cam.ac.uk/groups/murshudov/content/refmac/refmac.html |
| COOT | Emsley et al. (2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| MODELLER | Eswar et al. (2006); Martí-Renom et al., 2000; sali and Blundell (1993); Fiser et al. (2000) | https://salilab.org/modeller/ |
| Chimera (Version 1.13.1) | Pettersen et al., (2004) | https://www.cgl.ucsf.edu/chimera/ |
| Discrete Optimized Protein Energy (DOPE-HR) | Shen and sali (2006) | |
| Statistically Optimized Atomic Potentials (SOAP) | Dong et al. (2013) | https://github.com/salilab/SOAP |
| PyMOL (Version 2.2.0) | Schrodinger, LLC. 2010. The PyMOL Molecular Graphics System, Version 2.1.0 | https://pymol.org/2/#download |
| Avogadro2 software | Hanwell et al. (2012) | https://www.openchemistry.org/downloads/ |
| AutoDock Vina | Trott and Olson (2010) | http://vina.scripps.edu/download.html |
| HADDOCK web server | Van Zundert et al. (2016) | https://bianca.science.uu.nl/haddock2.4/ |
| GROMACS package (Version 2018) | Abraham et al. (2015) | http://www.gromacs.org/ |
| Auto-mated topology builder (ATB) web-interface | Malde et al. (2011) | https://atb.uq.edu.au/ |
| PyCGTOOL | Graham et al. (2017) | https://pypi.org/project/pycgtool/ |
| CHARMM-GUI web interface | Jo et al. (2017) | http://charmm-gui.org/ |
| backward script | Wassenaar et al. (2014) | http://www.cgmartini.nl/index.php/downloads/tools/240-backward |
| Particle mesh Ewald (PME) algorithm | Darden et al. (1993) | N/A |
| Visual Molecular Dynamics (VMD) | Humphrey et al. (1996) | https://www.ks.uiuc.edu/Research/vmd/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Roper (David.Roper@warwick.ac.uk).
Materials availability
This study did not generate any new unique reagents. Plasmids will be available by Materials Transfer Agreement (MTA) request in line with University of Warwick IP requirements.
Data and code availability
No novel code was generated during this work. Modelling scripts and raw data are available from the authors upon request.
Experimental model and subject details
E. coli B834 (DE3) - F- ompT hsdSB (rB- mB-) gal dcm met (DE3), referred to as E. coli B834 (DE3) in the method details section was transformed with pRosetta2 to yield E. coli B834 (DE3): pRosetta2 - F- ompT hsdSB (rB- mB-) gal dcm met (DE3) which was employed as the expression host for selenomethionine substituted S. aureus FemX. E. coli C41 (DE3) (a derivative of BL21(DE3) [F_ ampT hsdS8 (r8m8)gal dcm DE3]) was transformed with pRIL (CamR)). The resulting transformant referred to as E. coli C41 (DE3)/pRIL in the method details section was employed as the expression host for expression of S. pneumoniae (159) MurM. E. coli BL21 Star (DE3): F- ompT hsdSB (rB- mB-) gal dcm rne131 (DE3) was transformed with pRosetta2 to yield E. coli BL21 Star pRosetta2 (DE3) - F- ompT hsdSB (rB- mB-) gal dcm rne131 (DE3) referred to as E. coli BL21(DE3) star/pRosetta2 in the methods details section which was utilized as the expression host for expression of S. pneumoniae (Pn16) alanyl-tRNAAla synthetase (AlaRS). Expression of selenomethionine-labelled FemX was performed in M9 media supplemented with all 19 canonical amino acids and 40 mg mL−1 [LA1] L-selenomethionine in place in L-methionine. Expression of the remaining proteins were performed in luria broth. For all expression experiments freshly transformed expression hosts were cultured shaken at 180–200 rpm at 37°C in the presence of the appropriate antibiotics (30 mg mL−1 kanamycin, 30 mg mL−1 chloroamphenicol for FemX; 50 mg mL−1 carbenicillin, 30 mg mL−1 chloroamphenicol for MurM and 30 mg mL−1 kanamycin, 30 mg mL−1 chloroamphenicol for AlaRS) until mid-exponential phase (Optical density at 600 nm (OD600) = 0.4–0.6). At this point, cultures were supplemented with fresh antibiotics and protein expression was induced by 1 mM isopropyl-D-1-thiogalactopyranoside at 25°C for 4 h. Micrococcus flavus (Synonymous with M. luteus) was cultured shaken at 200 rpm at 37°C in tryptic soy broth to late exponential phase (OD600 of 4).
Method details
Cloning, overexpression and purification of S. aureus FemX
The S. aureus Mu50 FemX gene was amplified from chromosomal DNA using Oligonucleotides FemX forward: TTTGCGGGTGGTCTCCCATGGAAAAGATGCATATC ACTAATCAGG and FemX Reverse: TTTGCGCTCGAGGCCCTGAAAATACAGGTTTC TTTTCGTTTTAATTTACGAGATATTTTAATTTTAGC. The resulting PCR fragment was cleaved with BsaI and XhoI and cloned into pET28 between the NcoI and XhoI restriction sites to create pET28::FemX, containing a Tobacco Etch Virus (TEV) protease cleavable C-terminal hexa-histidine tag. E. coli B834 (DE3) harbouring plasmid pRosetta2 (which supplies seven rare tRNAs to support expression of genes in E. coli and derived from the Rosetta 2(DE3) strain) were transformed with pET28::FemX. Transformed E. coli B834 (DE3) pRosetta2 were used to inoculate M9 media supplemented with 30 mg mL−1 kanamycin, 30 mg mL−1 chloroamphenicol, and 40 mg mL−1 of each of the canonical amino acids [LA1] except L-methionine, which was replaced by 40 mg mL−1 (Doublié, 1997). Transformants were cultured at 37°C at 180 rpm until an optical density at 600 nm (OD600) of 0.4-0.6 was reached. Protein expression was induced by 1 mM isopropyl-D-1-thiogalactopyranoside at 25°C for 4 hours. Cells were harvested by centrifugation at 6,000 xg for 15 minutes and cell pellets containing 4-6 g of cells were resuspended in 20 mL of 50 mM sodium phosphate pH 7.0, 1M NaCl and 2.5 mg.mL-1 lysozyme to which one tablet of Pierce EDTA free Protease Inhibitor was added. The cell suspension was incubated with slow rotation for 30 minutes at 4°C before disruption using a Bandelin Sonopuls sonicator with 3 x 30 second bursts at 70% power. The lysate was clarified by centrifugation at 50,000 xg at 4°C for 30 minutes. FemX was then purified by immobilised metal affinity chromatography (IMAC) using a 5 mL gravity fed column of cobalt Talon resin equilibrated with 50 mL of 50 mM sodium phosphate pH 7.0, 500 mM NaCl, 20% (v/v) glycerol (equilibration buffer) supplemented with 10 mM imidazole. Once the 50,000 xg supernatant was loaded onto the column, it was eluted sequentially with 50 mL of equilibration buffer with 10 mM imidazole, 30 mL of equilibration buffer with 50 mM imidazole and 30 mL of equilibration buffer with 200 mM imidazole. 10 mL Fractions were analysed by SDS-PAGE and those containing FemX were pooled and concentrated, using a vivaspin 20 centrifugal concentrator (10,000 molecular weight cut off (MWCO), as required. Size exclusion chromatography in 50 mM sodium phosphate pH 7.0, 500 mM NaCl and 20% (v/v) glycerol was used to further purify FemX on a Superdex 75 10/300 column. The histidine tag was then removed from the FemX protein by digestion with histidine-tagged TEV protease at a molar ratio of 100:1 FemX : TEV protease at 4°C overnight. Cleaved and uncleaved protein were separated by a reverse IMAC following the procedure described above.
FemX crystallisation and data collection
FemX was exchanged into 50 mM ethanolamine pH 10.0, 100 mM NaCl and 20% (v/v) glycerol, concentrated to 15 mg.mL-1 using a vivaspin 20 centrifugal concentrator column with a 10,000 MWCO and screened for suitable crystallisation conditions using a honeybee 963 crystallisation robot against JCSG plus, PACT primer and Morpheus crystallisation screens. Crystals obtained from the Morpheus screen were used directly for data collection experiments, although crystallization conditions were further refined to 0.12 M ethylene glycol, 0.1 M MES/imidazole pH 6.3 and 28% (w/v) ethylene glycol-PEG 8000. Crystals were frozen directly for X-ray diffraction data experiments on the I04-1 beamline at the Diamond synchrotron (Didcot, UK) using a Pilatus 6M-F detector. Data were processed automatically using Xia2 (Winter, 2010) to 1.62 Å. Molecular replacement was not successful so selenomethionine containing FemX protein was produced and used to obtain FemX crystals (FemX-SeMet) in the same crystallisation conditions and the structure was solved by single anomalous diffraction (Leahy et al., 1992). X-ray data from the FemX-SeMet crystal were collected on the I02 beamline at the Diamond synchrotron (Didcot, UK) using a Pilatus 6M detector. All data were indexed, integrated and scaled using the XDS package (Kabsch, 2010). All 10 of expected selenium atoms in the asymmetric unit were located and refined by the SHELX suite (Sheldrick, 2010). These sites were used to obtain preliminary phases. The starting model was built by ARP/wARP (Langer et al., 2008). This model was used to refine the higher resolution data. The structure was refined using iterative cycles of REFMAC (Vagin et al., 2004) and model building/solvent addition with COOT (Emsley et al., 2010).
Cloning, Overexpression and Purification of S. pneumoniae(159) MurM
As described in Lloyd et al. (2008), the MurM allele of S. pneumoniae(159) was cloned with a C-terminal histidine tag into pET21b and over-expressed in E. coli C41 (DE3)/pRIL. Cells were harvested by centrifugation and treated with 2.5 mg.mL-1 hen egg white lysozyme prior sonication. MurM was solubilised with 1 M NaCl and fractionated between 25% and 50% of saturation ammonium sulfate followed by purification by size exclusion chromatography with Sephacryl S-200 and by immobilized metal affinity chromatography (IMAC) using cobalt Talon resin. The purity and identity of the final products of these purifications were assessed by SDS-PAGE.
Cloning, Overexpression and Purification of S. pneumoniae(Pn16) AlaRS
As described in Lloyd et al. (2008), the AlaRS allele of S. pneumoniae(Pn16) was cloned into pET26a and over-expressed in E. coli BL21(DE3) star/pRosetta2 The soluble protein was purified using nickel-chelated Chelating sepharose, desalted and further purified by anion exchange chromatography on a 0.98 ml MonoQ™, column. The purity and identity of the final products of these purifications were assessed by SDS-PAGE.
Preparation of MurM substrates
The substrates used for assays of MurM were prepared as follows:
Lipid II(Lys): The peptidoglycan intermediate Lipid II(Lys) (undecaprenyl pyrophosphoryl N-N-acetyl muramyl (N-acetyl glucosaminyl) L-alanyl-γ-D-glutamyl-L-lysyl-D-alanyl-D-alanine) was prepared by re-capitulation of the peptidoglycan synthesis pathway as described (Lloyd et al., 2008).
[3H]-Alanyl-tRNAAla: Micrococcus flavus tRNA was isolated from cell pellets of M. flavus cultures grown to late exponential phase by phenol extraction followed by isopropanol precipitation, anion exchange chromatography and ethanol precipitation as described by Zubay (1962) as adapted by Lloyd et al. (2008). tRNAs were renatured in 2 mM MgCl2 at 60°C and aminoacylated with [2,3-3H]-L-alanine S. pneumoniae (Pn16) AlaRS as described by Lloyd et al. (2008) and quantitated by liquid scintillation counting.
S. pneumoniae MurM enzymology
MurM was assayed as described by Lloyd et al. (2008) in duplicate in a final volume of 35 μl of 50 mM 3-(N-morpholino)-propane sulphonic acid adjusted to pH 6.8, 30 mM KCl, 10 mM MgCl2, 1.5% (w/v) CHAPS (Assay Buffer), 1 mM DTT, 1 mM L-alanine, 10 μM Lipid II-Lys and 24.3 nM MurM. Reactions were initiated by the addition of 0.45 M [3H]-alanyl-tRNAAla (1000 cpm.pmol-1) and were incubated at 37°C for two minutes, over which time frame, product accumulation was linear with respect to time. Where the impact of cardiolipin or phosphatidylglycerol on MurM activity was assessed, the required amounts of 10 mg/mL stocks of each phospholipid in ethanol or chloroform/methanol (49:1) were dried down in the reaction vials the assays were to be performed in, and solubilised by addition of assay buffer. Reactions were terminated by the addition of 35 μl of ice-cold 6 M pyridinium acetate pH 4.5 and 70 μl ice-cold n-butanol. The incubations were rapidly mixed and centrifuged for 5 minutes at 1°C at 13,000 xg, after which time the n-butanol phase was washed with 70 μl of water and then assayed for [3H]-Lipid II-L-Ala by liquid scintillation counting. Tritium counts accumulated in control reactions performed without Lipid II(Lys) were subtracted from corresponding data acquired in the presence of this substrate. MurM activities in the presence of phospholipid were related to the activity of the enzyme in the absence of phospholipid and plotted as fold activation or percentage inhibition vs phospholipid concentration. The data were then fitted using GraphPad Prism (Version 8.4.1) to either of equations 1 or 2 as appropriate:
| (Equation 1) |
| (Equation 2) |
Maximum activation and S0.5 (Activation) (Equation 1) corresponded to the degree of activation at infinite cardiolipin concentration and the cardiolipin concentration required to elicit half maximal activation respectively. IC50 (Equation 2) corresponds to the phosphatidylglycerol concentration that elicited half maximal inhibition. For both equations, h denoted the Hill coefficient.
Computational studies overview
A number of computational techniques were used in this study, to assist the reader in understanding the logistics of these methods, we have provided a summary flowchart (supplemental information: Figure S6).
Homology modelling of MurM
Due to the natural ability of streptococci to undergo homologous recombination, S. pneumoniae MurM genes are highly mosaic, and so, in line with the enzymology studies, the MurM sequence used for homology modelling was that of S. pneumoniae MurM159. S. pneumoniae MurM, S. aureus FemX (PDB ID: 6SNR) and FemA (PDB ID: 1LRZ), and Weissella viridescens FemX (PDB ID: 3GKR) were aligned (supplemental information: Figure S1) by multiple sequence alignment using Clustal Omega to determine sequence identity (Sievers et al., 2011).
The structure of S. aureus FemX was used as the template for homology modelling due to its high relatedness with MurM. S. aureus FemX and MurM159 sequences were aligned, and using MODELLER (Eswar et al., 2006; Martí-Renom et al., 2000; sali and Blundell, 1993; Fiser et al., 2000) a test model was generated to verify the validity of the template and the alignment. This model was evaluated by computing its energy profile according to the DOPE-HR (high-resolution version of the Discrete Optimized Protein Energy) (Shen and sali, 2006), smoothed via window averaging with a size of 15 residues. The profiles of template and model were compared (supplemental information: Figure S7), and further refinement was conducted in the region between Lys230 and Pro299, as well as in all loop regions. This optimisation was conducted by performing a very slow MD annealing on the selected regions, whilst maintaining the remaining structure. The scoring of the resulting conformations was obtained via a function built specifically to evaluate the geometry of loops. For this step, 64 different base models were created and their secondary structure was refined independently 16 times. The resulting 1024 models were evaluated and ranked using DOPE-HR as well as the SOAP (Statistically Optimized Atomic Potentials) (Dong et al., 2013). The 10 best scoring models for each score were selected and evaluated based on the number of physical constraint violations present.
The best model of MurM159 was aligned with the previously published MurM model (Fiser et al., 2003) or W. viridescens Femx homologues (Fonvielle et al., 2013; Biarrotte-Sorin et al., 2004) for visualisation and analysis in PyMOL (Version 2.1.0).
Molecular docking of truncated Lipid II to MurM
A truncated Lipid II substrate (supplemental information: Figure S2) was created for initial molecular docking simulations. The truncated Lipid II was drawn in ChemDraw Professional (Version 17.1) and converted to a pdb file using Avogadro (Version 1.2.0). To prepare the ligand file for docking, the protonation state in H2O at pH 7.4 was computed. Subsequently the equilibrium geometry minimizing the potential energy was computed using the general amber force field (GAFF) (Wang et al., 2004) from within the Avogadro2 software (Hanwell et al., 2012). Molecular docking was conducted using AutoDock Vina (Trott and Olson, 2010), for which pdbqt files were generated from the pdb files of receptor model and ligands using AutoDock Tools (Morris et al., 2009). Initially the location of the binding site was verified by providing the algorithm with a search space that included the entire protein. Docking was then repeated by restricting the search space to the identified binding site, in order to obtain the final docked conformation.
Molecular docking of Lipid II, Cardiolipin, Phosphatidylglycerol and Phosphatidylethanolamine to the putative MurM binding site.
Using the HADDOCK2.4 web server (Van Zundert et al., 2016), full length Lipid II, cardiolipin and phosphatidylglycerol and phosphatidylethanolamine were docked into the putative MurM binding site, where the binding site is defined by the residues Lys35, Trp38, Phe103, Arg215 and Tyr219.
Coarse-grained molecular dynamics simulations
All coarse-grained simulations were carried out with the GROMACS package (Version 2018) and the Martini (Version 2.2) forcefield (Abraham et al., 2015; de Jong et al., 2012). Simulations at the coarse-grained and atomstic resolutions were carried out at 313 K. For coarse-grained simulations, a stochastic velocity rescale thermostat with a coupling constant of 1:0 ps controlled the temperature.
The coordinates of the MurM homology model were used to generate a coarse-grained model using the ‘martinise.py’ script (de Jong et al., 2013). The protein was coarse grained to the ElNeDyn model (Periole et al., 2009) with an elastic network strength and cutoff of 500 kJmol-1nm-2 and 0:9 nm, respectively. The Lipid II model for inclusion in the membrane was parameterised using a united atom model (Gromos 53a6) generated by the Auto-mated topology builder (ATB) web-interface (Malde et al., 2011). Following this, the coarse-grained mapping was pursued iteratively, and the bonded terms were fitted with PyCGTOOL (Graham et al., 2017).
Since the pneumococcal membrane comprises a complex mixture of lipids, a simplified membrane composition was required for the simulations. In order to elucidate the effects of phosphatidylglycerol and cardiolipin on MurM, a non-pneumococcal lipid, phosphatidylethanolamine, was used as the majority lipid. Simulations were conducted with three different membrane systems (supplemental information: Table S1). System 1 comprised phosphatidylethanolamine and phosphatidylglycerol in a molar ratio of 75% and 25% respectively, system 2 contained phosphatidylethanolamine, phosphatidylglycerol and cardiolipin in a molar ratio of 76%, 16% and 8% respectively and system 3 comprised phosphatidylethanolamine, phosphatidylglycerol and cardiolipin at a molar ratio of 72%, 12% and 16% respectively. The membrane systems of size ∼16x16x11:5 nm were generated with the CHARMM-GUI web interface (Jo et al., 2017). Each system was relaxed with a series of minimisation and equilibration steps with timesteps of 5-20 fs, for up to 30 ns. The equilibration steps utilised a semi-isotropic Berendsen barostat, with a 4:0 ps coupling constant (Berendsen et al., 1984). Following equilibration, Lipid II molecules (10 in total) were added to each membrane. The systems were then minimised and equilbriated (for 10 ns), followed by a 2 μs production run to ensure sufficient mixing of all the lipid components. All production runs were carried out using a 10 fs timestep and a Parrinello-Rahman semi-isotropic barostat with a 12 ps coupling constant (Parrinello and Rahman, 1981). The Lennard-Jones potential was cutoff using the Potential shift Verlet scheme at long ranges. The reaction field method (Tironi et al., 1995) was used for electrostatics calculations, with dielectric constants of 15 and infinity for charge screening in the short- and long-range regimes, respectively. The short-range cutoff for non-bonded and electrostatic interactions was 1:2 nm. Once lipid mixing was ensured, the size of each system was increased to ∼32 nm in the dimension perpendicular to the membrane and MurM was added in a random orientation around 8 nm above each membrane. Biologically relevant salt concentrations (0.15 M NaCl) were added and 10% of the water molecules were changed to antifreeze particles to prevent localised freezing during simulations. After minimisation and 1 ns of equilibration, during which the protein backbone was restrained with 1000 kJmol-1nm-2 harmonic restraints, 6x5 μs production runs were generated per membrane composition (supplemental information: Table S1).
All-atom molecular dynamics simulations
Atomistic simulations were conducted using the CHARMM36m forcefield (Huang et al., 2017). The Lipid II model used here was also used in previous work (Witzke et al., 2016), while all other lipid models were obtained from the CHARMM-GUI membrane builder module (Jo et al., 2008). For each coarse-grained membrane system, two repeats were chosen where: 1) the last frame of the production run had a distinct orientation of MurM, relative to the membrane 2) MurM adhered to the membrane surface (supplemental information: Table S2). The last frame of the chosen coarse-grained repeats were then backmapped to the all-atom model, using the backward script (Wassenaar et al., 2014). Unfavourable ring conformers were corrected by carrying our minimisation and equilibration steps with dihedral restraints of 25000 kJmol-1rad-2 on key ring torsions. After the transformation was carried out, each system was cropped in the z dimension to a height of 16:5 nm, to remove unnecessary H2O molecules.
Each system was minimised and equilibrated for a total of 1 ns, while the backbone of the protein was restrained with 1000 kJmol-1nm-2 harmonic restraints. Two production runs of 250 ns were carried out for each system. During the production runs a timestep of 2 fs was used, and the pressure (1 bar) regulated with a semi-isotropic Parrinello-Rahman barostat, with a coupling constant of 5:0 ps. The Lennard-Jones potential was cutoff with the Force-switch modifier from 1:0 to 1:2 nm. The short range cutoff for the electrostatic interaction was also 1:2 nm and the Particle mesh Ewald (PME) algorithm (Darden et al., 1993) was used for the long-range regime.
Analysis was carried out over the final 100 ns of each simulation, unless stated otherwise. All simulations were visualised using Visual Molecular Dynamics (VMD) or PyMOL (Version 2.2.0) (Humphrey et al., 1996). Other analysis tools were written with a combination of GROMACS tools and in house scripts, that utilised the python module MDAnalysis (Gowers et al., 2016). The depletion/enrichment (D-E) indices were determined by first counting the number of lipids with a centre of geometry within 1.1 nm of the protein and then comparing this number to the number expected in the bulk of the membrane, using the procedure described by Corradi et al. (2018). The D-E index was obtained by dividing the lipid composition in the 1.1 nm shell around the protein by the bulk membrane composition. Thus a D-E index >1 indicates enrichment, while a D-E index <1 indicates depletion. The D-E index was determined for the last 100 ns of each simulation in 50 ns blocks for all repeats. For a given membrane composition, 8 D-E indices were obtained for each lipid, from which the average and standard errors were calculated. The enrichment maps were generated by first determining the 2D density map of the membrane using the GROMACS tool densmap. Following this, the enrichment percentage was determined using the procedure described by Corradi et al. (2018). An enrichment percentage <0% indicated that local membrane composition was depleted with respect to the bulk membrane composition. The code for the 2D enrichment maps and D-E indices was reported in Shearer et al. (2019).
Quatification and statistical analysis
Crystallographic statistics were calculated using software/programs as described in the methods, and values are reported in Table 1. Enzyme assays were performed in duplicate, generating data that differed by no more than 10%. Average values were then plotted. The standard errors of the fits of constants defining the relationships between the response of MurM to phospholipid and phospholipid concentration according to equations 1 and 2 were calculated by GraphPad Prism (Version 8.4.1).
Acknowledgments
This research was supported in part by the Midlands Integrative Biosciences Training Partnership (MIBTP) BBSRC grant BB/J014532/1, and the Center for Doctoral Training in Theory and Modeling in Chemical Sciences (TMCS DTC) EPSRC grant EP/L015722/1, as well as MRC grants G1100127, G0400848, MR/N002679/1 and BBSRC grant BB/N003241/1. The authors would like to acknowledge the help of the Media Preparation Facility in the School of Life Sciences at the University of Warwick. We would also like to thank Dr Allister Crow for his help and support with using PyMOL.
Author contributions
Conceptualization, A.Y., A.J.L., D.I.R., and S.K.; Investigation, K.F., K.J.H., V.F., A.J.L., A.Y., J.S., and C.I.d.G.; Methodology/software/formal analysis, C.I.d.G., J.S., A.Y., and S.K.; Writing – original draft, A.Y., J.S., and C.I.d.G.; Writing – review & editing, A.Y., A.J.L., S.K., D.I.R., and C.G.D.; Funding acquisition, A.Y., J.S., and C.G.D.; Supervision, A.J.L., D.I.R., S.K., and C.G.D.; Project administration, A.Y.; Visualization, A.Y., A.J.L., J.S., and S.K.
Declaration of interests
The authors declare no competing interests.
Published: March 18, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.str.2021.03.001.
Contributor Information
Syma Khalid, Email: s.khalid@soton.ac.uk.
David.I. Roper, Email: david.roper@warwick.ac.uk.
Supplemental information
References
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. [Google Scholar]
- Benson T.E., Prince D.B., Mutchler V.T., Curry K.A., Ho A.M., Sarver R.W., Hagadorn J.C., Choi G.H., Garlick R.L. X-ray crystal structure of Staphylococcus aureus FemA. Structure. 2002;10:1107–1115. doi: 10.1016/s0969-2126(02)00807-9. [DOI] [PubMed] [Google Scholar]
- Berendsen H.J., Postma J.V., van Gunsteren W.F., DiNola A., Haak J. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- Biarrotte-Sorin S., Maillard A.P., Delettré J., Sougakoff W., Arthur M., Mayer C. Crystal structures of Weissella viridescens FemX and its complex with UDP-MurNAc-pentapeptide: insights into FemABX family substrates recognition. Structure. 2004;12:257–267. doi: 10.1016/j.str.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Bramkamp M., Lopez D. Exploring the existence of lipid rafts in bacteria. Microbiol. Mol. Biol. Rev. 2015;79:81–100. doi: 10.1128/MMBR.00036-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Bugg T.D.H., Braddick D., Dowson C.G., Roper D.I. Bacterial cell wall assembly: still an attractive antibacterial target. Trends Biotechnol. 2011;29:167–173. doi: 10.1016/j.tibtech.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Corradi V., Mendez-Villuendas E., Ingólfsson H.I., Gu R.-X., Siuda I., Melo M.N., Moussatova A., DeGagné L.J., Sejdiu B.I., Singh G. Lipid-protein interactions are unique fingerprints for membrane proteins. ACS Cent. Sci. 2018;4:709–717. doi: 10.1021/acscentsci.8b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cressina E., Lloyd A.J., De Pascale G., Roper D.I., Dowson C.G., Bugg T.D.H. Adenosine phosphonate inhibitors of lipid II: alanyl tRNA ligase MurM from Streptococcus pneumoniae. Bioorg. Med. Chem. Lett. 2007;17:4654–4656. doi: 10.1016/j.bmcl.2007.05.071. [DOI] [PubMed] [Google Scholar]
- Cressina E., Lloyd A.J., De Pascale G., Mok B.J., Caddick S., Roper D.I., Dowson C.G., Bugg T.D.H. Inhibition of tRNA-dependent ligase MurM from Streptococcus pneumoniae by phosphonate and sulfonamide inhibitors. Bioorg. Med. Chem. 2009;17:3443–3455. doi: 10.1016/j.bmc.2009.03.028. [DOI] [PubMed] [Google Scholar]
- Cruickshank D. Remarks about protein structure precision. Acta Crystallogr. D Biol. Crystallogr. 1999;55:583–601. doi: 10.1107/s0907444998012645. [DOI] [PubMed] [Google Scholar]
- Darden T., York D., Pedersen L. Particle mesh Ewald: an N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- de Jong D.H., Singh G., Bennett W.D., Arnarez C., Wassenaar T.A., Schäfer L.V., Periole X., Tieleman D.P., Marrink S.J. Improved parameters for the martini coarse-grained protein force field. J. Chem. Theor. Comput. 2012;9:687–697. doi: 10.1021/ct300646g. [DOI] [PubMed] [Google Scholar]
- de Jong D.H., Singh G., Bennett W.F., Arnarez C., Wassenaar T.A., Schäfer L.V., Periole X., Tieleman D.P., Marrink S.J. Improved parameters for the martini coarse-grained protein force field. J Chem Theory Comput. 2013;9:687–697. doi: 10.1021/ct300646g. [DOI] [PubMed] [Google Scholar]
- Dong G.Q., Fan H., Schneidman-Duhovny D., Webb B., Šali A. Optimized atomic statistical potentials: assessment of protein interfaces and loops. Bioinformatics. 2013;29:3158–3166. doi: 10.1093/bioinformatics/btt560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublié S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M., Eramian D., Shen M.-Y., Pieper U., Šali A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics. 2006;15:5–6. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo T.A., Sobral R.G., Ludovice A.M., de Almeida J.M.F., Bui N.K., Vollmer W., de Lencastre H., Tomasz A. Identification of genetic determinants and enzymes involved with the amidation of glutamic acid residues in the peptidoglycan of Staphylococcus aureus. PLoS Pathog. 2012;8:e1002508. doi: 10.1371/journal.ppat.1002508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipe S.R., Tomasz A. Inhibition of the expression of penicillin resistance in Streptococcus pneumoniae by inactivation of cell wall muropeptide branching genes. Proc. Natl. Acad. Sci. U S A. 2000;97:4891–4896. doi: 10.1073/pnas.080067697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipe S.R., Pinho M.G., Tomasz A. Characterization of the murMN operon involved in the synthesis of branched peptidoglycan peptides in Streptococcus pneumoniae. J. Biol. Chem. 2000;275:27768–27774. doi: 10.1074/jbc.M004675200. [DOI] [PubMed] [Google Scholar]
- Filipe S.R., Severina E., Tomasz A. The role of murMN operon in penicillin resistance and antibiotic tolerance of Streptococcus pneumoniae. Microb. Drug Resist. 2001;7:303–316. doi: 10.1089/10766290152773310. [DOI] [PubMed] [Google Scholar]
- Fiser A., Do R.K.G., Šali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A., Filipe S.R., Tomasz A. Cell wall branches, penicillin resistance and the secrets of the MurM protein. Trends Microbiol. 2003;11:547–553. doi: 10.1016/j.tim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Fonvielle M., Li de La Sierra-Gallay I., El-Sagheer A.H., Lecerf M., Patin D., Mellal D., Mayer C., Blanot D., Gale N., Brown T. The structure of FemXWv in complex with a peptidyl-RNA conjugate: mechanism of aminoacyl transfer from Ala-tRNAAla to peptidoglycan precursors. Angew. Chem. Int. Ed. Engl. 2013;125:7419–7422. doi: 10.1002/anie.201301411. [DOI] [PubMed] [Google Scholar]
- Ganchev D., Hasper H., Breukink E., de Kruijff B. Size and orientation of the lipid II headgroup as revealed by AFM imaging. Biochemistry. 2006;45:6195–6202. doi: 10.1021/bi051913e. [DOI] [PubMed] [Google Scholar]
- Garcia-Bustos J., Tomasz A. A biological price of antibiotic resistance: major changes in the peptidoglycan structure of penicillin-resistant pneumococci. Proc. Natl. Acad. Sci. U S A. 1990;87:5415–5419. doi: 10.1073/pnas.87.14.5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bustos J., Chait B.T., Tomasz A. Structure of the peptide network of pneumococcal peptidoglycan. J. Biol. Chem. 1987;262:15400–15405. [PubMed] [Google Scholar]
- Gowers R.J., Linke M., Barnoud J., Reddy T.J., Melo M.N., Seyler S.L., Domański J., Dotson D.L., Buchoux S., Kenney I.M. MDAnalysis: a Python package for the rapid analysis of molecular dynamics simulations. In: Benthall S., Rostrup S., editors. SciPy; 2016. (Proceedings of the 15th Python in Science Conference (SCIPY 2016)). [DOI] [Google Scholar]
- Graham J.A., Essex J.W., Khalid S. PyCGTOOL: automated generation of coarse-grained molecular dynamics models from atomistic trajectories. J. Chem. Inf. Model. 2017;57:650–656. doi: 10.1021/acs.jcim.7b00096. [DOI] [PubMed] [Google Scholar]
- Hanwell M.D., Curtis D.E., Lnie D.C., Vandermeersch T., Zurek E., Hutchison G.R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012;4:17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebecker S., Krausze J., Hasenkampf T., Schneider J., Groenewold M., Reichelt J., Jahn D., Heinz D.W., Moser J. Structures of two bacterial resistance factors mediating tRNA-dependent aminoacylation of phosphatidylglycerol with lysine or alanine. Proc. Natl. Acad. Sci. U S A. 2015;112:10691–10696. doi: 10.1073/pnas.1511167112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde S.S., Blanchard J.S. Kinetic and mechanistic characterization of recombinant Lactobacillus viridescens FemX (UDP-N-acetylmuramoyl pentapeptide-lysine N6-alanyltransferase) J. Biol. Chem. 2003;278:22861–22867. doi: 10.1074/jbc.M301565200. [DOI] [PubMed] [Google Scholar]
- Heuckeroth R.O., Glaser L., Gordon J.I. Heteroatom-substituted fatty acid analogs as substrates for N-myristoyltransferase: an approach for studying both the enzymology and function of protein acylation. Proc. Natl. Acad. Sci. U S A. 1988;85:8795–8799. doi: 10.1073/pnas.85.23.8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B.L., Grubmüller H., MacKerell A.D., Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017;14:71–73. doi: 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jo S., Kim T., Iyer V.G., Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Jo S., Cheng X., Lee J., Kim S., Park S.J., Patel D.S., Beaven A.H., Lee K.I., Rui H., Park S. CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comput. Chem. 2017;38:1114–1124. doi: 10.1002/jcc.24660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G., Cohen S.X., Lamzin V.S., Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008;3:1171. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leahy D.J., Hendrickson W.A., Aukhil I., Erickson H.P. Structure of a fibronectin type III domain from tenascin phased by MAD analysis of the selenomethionyl protein. Science. 1992;258:987–991. doi: 10.1126/science.1279805. [DOI] [PubMed] [Google Scholar]
- Lin T.Y., Gross W.S., Auer G.K., Weibel D.B. Cardiolipin alters Rhodobacter sphaeroides cell shape by affecting peptidoglycan precursor biosynthesis. mBio. 2019;10:e02401–e02418. doi: 10.1128/mBio.02401-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd A.J., Gilbey A.M., Blewett A.M., De Pascale G., El Zoeiby A., Levesque R.C., Catherwood A.C., Tomasz A., Bugg T.D., Roper D.I. Characterization of tRNA-dependent peptide bond formation by MurM in the synthesis of Streptococcus pneumoniae peptidoglycan. J. Biol. Chem. 2008;283:6402–6417. doi: 10.1074/jbc.M708105200. [DOI] [PubMed] [Google Scholar]
- Malde A.K., Zuo L., Breeze M., Stroet M., Poger D., Nair P.C., Oostenbrink C., Mark A.E. An automated force field topology builder (ATB) and repository: version 1.0. J. Chem. Theor. Comput. 2011;7:4026–4037. doi: 10.1021/ct200196m. [DOI] [PubMed] [Google Scholar]
- Martí-Renom M.A., Stuart A.C., Fiser A., Sánchez R., Melo F., Šali A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Matsuhashi M., Dietrich C.P., Strominger J.L. Biosynthesis of the peptidoglycan of bacterial cell walls III. The role of soluble ribonucleic acid and of lipid intermediates in Glycine incorporation in Staphylococcus aureus. J. Biol. Chem. 1967;242:3191–3206. [PubMed] [Google Scholar]
- Morlot C., Straume D., Peters K., Hegnar O.A., Simon N., Villard A.M., Contreras-Martel C., Leisico F., Breukink E., Gravier-Pelletier C. Structure of the essential peptidoglycan amidotransferase MurT/GatD complex from Streptococcus pneumoniae. Nat. Commun. 2018;9:3180. doi: 10.1038/s41467-018-05602-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrice N.A., Gabrielli B., Kemp B.E., Wettenhall R. A cardiolipin-activated protein kinase from rat liver structurally distinct from the protein kinases C. J. Biol. Chem. 1994;269:20040–20046. [PubMed] [Google Scholar]
- Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;16:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch D., Roemer T., Lee S.H., Engeser M., Sahl H.G., Schneider T. Identification and in vitro analysis of the GatD/MurT enzyme-complex catalyzing lipid II amidation in Staphylococcus aureus. PLoS Pathog. 2012;8:e1002509. doi: 10.1371/journal.ppat.1002509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- Peng B., Morrice N.A., Groenen L.C., Wettenhall R.E. Phosphorylation events associated with different states of activation of a hepatic cardiolipin/protease-activated protein kinase structural identity to the protein kinase N-type protein kinases. J. Biol. Chem. 1996;271:32233–32240. doi: 10.1074/jbc.271.50.32233. [DOI] [PubMed] [Google Scholar]
- Periole X., Cavalli M., Marrink S.J., Ceruso M.A. Combining an elastic network with a coarse-grained molecular force field: structure, dynamics, and intermolecular recognition. J. Chem. Theor. Comput. 2009;5:2531–2543. doi: 10.1021/ct9002114. [DOI] [PubMed] [Google Scholar]
- Pesakhov S., Benisty R., Sikron N., Cohen Z., Gomelsky P., Khozin-Goldberg I., Dagan R., Porat N. Effect of hydrogen peroxide production and the Fenton reaction on membrane composition of Streptococcus pneumoniae. Biochim. Biophys. Acta. 2007;1768:590–597. doi: 10.1016/j.bbamem.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Purzycka-Preis J., Zydowo M. Regulatory effect of pig heart phospholipids on heart muscle AMP-deaminase. Int. J. Biochem. 1987;19:565–568. doi: 10.1016/0020-711x(87)90142-x. [DOI] [PubMed] [Google Scholar]
- Ruiz de Mena I., Mahillo E., Arribas J., Castano J. Kinetic mechanism of activation by cardiolipin (diphosphatidylglycerol) of the rat liver multicatalytic proteinase. Biochem. J. 1993;296:93–97. doi: 10.1042/bj2960093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Scheideler M., Bell R.M. Phospholipid dependence of homogeneous, reconstituted sn-glycerol-3-phosphate acyltransferase of Escherichia coli. J. Biol. Chem. 1989;264:12455–12461. [PubMed] [Google Scholar]
- Schlame M. Thematic Review Series: glycerolipids. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J. Lipid Res. 2008;49:1607–1620. doi: 10.1194/jlr.R700018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T., Sahl H.G. An oldie but a goodie—cell wall biosynthesis as antibiotic target pathway. Int. J. Med. Microbiol. 2010;300:161–169. doi: 10.1016/j.ijmm.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Schneider T., Senn M.M., Berger-Bächi B., Tossi A., Sahl H.G., Wiedemann I. In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of Staphylococcus aureus. Mol. Microbiol. 2004;53:675–685. doi: 10.1111/j.1365-2958.2004.04149.x. [DOI] [PubMed] [Google Scholar]
- Sekimizu K., Kornberg A. Cardiolipin activation of dnaA protein, the initiation protein of replication in Escherichia coli. J. Biol. Chem. 1988;263:7131–7135. [PubMed] [Google Scholar]
- Severin A., Tomasz A. Naturally occurring peptidoglycan variants of Streptococcus pneumoniae. J. Bacteriol. 1996;178:168–174. doi: 10.1128/jb.178.1.168-174.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer J., Jefferies D., Khalid S. Outer membrane proteins OmpA, FhuA, OmpF, EstA, BtuB, and OmpX have unique lipopolysaccharide fingerprints. J. Chem. Theor. Comput. 2019;15:2608–2619. doi: 10.1021/acs.jctc.8b01059. [DOI] [PubMed] [Google Scholar]
- Sheldrick G.M. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 2010;66:479–485. doi: 10.1107/S0907444909038360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M.Y., Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.M., Klugman K.P. Alterations in MurM, a cell wall muropeptide branching enzyme, increase high-level penicillin and cephalosporin resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2001;45:2393–2396. doi: 10.1128/AAC.45.8.2393-2396.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.M., Dowson C.G., Spratt B.G. Localized sex in bacteria. Nature. 1991;349:29–31. doi: 10.1038/349029a0. [DOI] [PubMed] [Google Scholar]
- Tironi I.G., Sperb R., Smith P.E., van Gunsteren W.F. A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys. 1995;102:5451–5459. [Google Scholar]
- Tlapak-Simmons V.L., Kempner E.S., Baggenstoss B.A., Weigel P.H. The active streptococcal hyaluronan synthases (HASs) contain a single HAS monomer and multiple cardiolipin molecules. J. Biol. Chem. 1998;273:26100–26109. doi: 10.1074/jbc.273.40.26100. [DOI] [PubMed] [Google Scholar]
- Tlapak-Simmons V.L., Baggenstoss B.A., Clyne T., Weigel P.H. Purification and lipid dependence of the recombinant hyaluronan synthases from Streptococcus pyogenes and Streptococcus equisimilis. J. Biol. Chem. 1999;274:4239–4245. doi: 10.1074/jbc.274.7.4239. [DOI] [PubMed] [Google Scholar]
- Tlapak-Simmons V.L., Baggenstoss B.A., Kumari K., Heldermon C., Weigel P.H. Kinetic characterization of the recombinant hyaluronan synthases from Streptococcus pyogenes and Streptococcus equisimilis. J. Biol. Chem. 1999;274:4246–4253. doi: 10.1074/jbc.274.7.4246. [DOI] [PubMed] [Google Scholar]
- Tlapak-Simmons V.L., Baron C.A., Weigel P.H. Characterization of the purified hyaluronan synthase from Streptococcus equisimilis. Biochemistry. 2004;43:9234–9242. doi: 10.1021/bi049468v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombe M.C., Lanéelle M.A., Lanéelle G. Lipid composition of aminopterin-resistant and sensitive strains of Streptococcus pneumoniae. Effect of aminopterin inhibition. Biochim. Biophys. Acta. 1979;574:290–300. doi: 10.1016/0005-2760(79)90010-9. [DOI] [PubMed] [Google Scholar]
- Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.A., Steiner R.A., Lebedev A.A., Potterton L., McNicholas S., Long F., Murshudov G.N. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- van den Brink-van der Laan E., Boots J.-W.P., Spelbrink R.E., Kool G.M., Breukink E., Killian J.A., de Kruijff B. Membrane interaction of the glycosyl-transferase MurG: a special role for cardiolipin. J. Bacteriol. 2003;185:3773–3779. doi: 10.1128/JB.185.13.3773-3779.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zundert G., Rodrigues J., Trellet M., Schmitz C., Kastritis P., Karaca E., Melquiond A., vanDijk M., De Vries S., Bonvin A. The haddock2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016;428:720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Wassenaar T.A., Pluhackova K., Bockmann R.A., Marrink S.J., Tieleman D.P. Going backward: a flexible geometric approach to reverse transformation from coarse grained to atomistic models. J. Chem. Theor. Comput. 2014;10:676–690. doi: 10.1021/ct400617g. [DOI] [PubMed] [Google Scholar]
- Weigel P.H., Kyossev Z., Torres L.C. Phospholipid dependence and liposome reconstitution of purified hyaluronan synthase. J. Biol. Chem. 2006;281:36542–36551. doi: 10.1074/jbc.M606529200. [DOI] [PubMed] [Google Scholar]
- Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2008;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Cryst. 2010;43:186–190. [Google Scholar]
- Witzke S., Petersen M., Carpenter T.S., Khalid S. Molecular dynamics simulations reveal the conformational flexibility of lipid II and its loose association with the defensin plectasin in the Staphylococcus aureus membrane. Biochemistry. 2016;55:3303–3314. doi: 10.1021/acs.biochem.5b01315. [DOI] [PubMed] [Google Scholar]
- Wright M.H., Heal W.P., Mann D.J., Tate E.W. Protein myristoylation in health and disease. J. Chem. Biol. 2010;3:19–35. doi: 10.1007/s12154-009-0032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapun A., Philippe J., Abrahams K.A., Signor L., Roper D.I., Breukink E., Vernet T. In vitro reconstitution of peptidoglycan assembly from the Gram-positive pathogen Streptococcus pneumoniae. ACS Chem. Biol. 2013;8:2688–2696. doi: 10.1021/cb400575t. [DOI] [PubMed] [Google Scholar]
- Zubay G. The isolation and fractionation of soluble ribonucleic acid. J. Mol. Biol. 1962;4:347–356. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No novel code was generated during this work. Modelling scripts and raw data are available from the authors upon request.