

ABSTRACT
Streptococcus pneumoniae is an opportunistic pathogen that is a common cause of serious invasive diseases such as pneumonia, bacteremia, meningitis, and otitis media. Transmission of this bacterium has classically been thought to occur through inhalation of respiratory droplets and direct contact with nasal secretions. However, the demonstration that S. pneumoniae is desiccation tolerant and, therefore, environmentally stable for extended periods of time opens up the possibility that this pathogen is also transmitted via contaminated surfaces (fomites). To better understand the molecular mechanisms that enable S. pneumoniae to survive periods of desiccation, we performed a high-throughput transposon sequencing (Tn-seq) screen in search of genetic determinants of desiccation tolerance. We identified 42 genes whose disruption reduced desiccation tolerance and 45 genes that enhanced desiccation tolerance. The nucleotide excision repair pathway was the most enriched category in our Tn-seq results, and we found that additional DNA repair pathways are required for desiccation tolerance, demonstrating the importance of maintaining genome integrity after desiccation. Deletion of the nucleotide excision repair gene uvrA resulted in a delay in transmission between infant mice, indicating a correlation between desiccation tolerance and pneumococcal transmssion. Understanding the molecular mechanisms that enable pneumococcal persistence in the environment may enable targeting of these pathways to prevent fomite transmission, thereby preventing the establishment of new colonization and any resulting invasive disease.
KEYWORDS: Streptococcus pneumoniae, desiccation, xerotolerance, DNA repair, nucleotide excision repair, uvrA, fomite transmission, desiccation tolerance, fomite, pneumococci, transmission
INTRODUCTION
For pathogens with no environmental reservoir, transmission between hosts is necessary for the species to survive. In the case of Streptococcus pneumoniae (the pneumococcus), a common member of the human nasopharyngeal microbiome, most transmission events result in asymptomatic and transient colonization, which has been termed the carrier state (1–4). However, in susceptible individuals, such as children and the elderly, S. pneumoniae can be aspirated into the lungs, resulting in pneumonia and invasive diseases such as bacteremia and meningitis (5). Due to high carriage rates of S. pneumoniae within the population, invasive pneumococcal disease continues to be a leading cause of lower respiratory morbidity and mortality, as well as a significant socioeconomic burden (6–8). As colonization precedes invasive pneumococcal disease, developing ways to prevent colonization, such as limiting fomite transmission, would serve to reduce the incidence of invasive disease.
The prevailing model of pneumococcal transmission posits that transmission occurs via respiratory droplets and direct contact with nasal secretions. However, previous work has demonstrated that S. pneumoniae can survive long periods of desiccation (9, 10). Upon subsequent rehydration, a proportion of the bacteria were found to remain viable and capable of establishing colonization. Thus, environmentally stable bacteria desiccated on surfaces, also referred to as fomites, may serve as an alternate source of pneumococcal infection.
Surfaces contaminated with infectious microbes are an important mode of transmission for a number of pathogens (11–18). In particular, fomites have been demonstrated to be a frequent source of nosocomial infections (19–24). Therefore, the demonstration that S. pneumoniae can be isolated from surfaces in a daycare provides evidence that fomite reservoirs of the bacterium exist in the community (10, 25). As S. pneumoniae is desiccation tolerant for an extended period of time, it is likely that the bacterium uses fomite transmission as one of multiple strategies to reach new hosts. Furthermore, increased desiccation tolerance of a pyruvate oxidase mutant has been shown to correlate with improved transmission between infant mice in a murine model of pneumococcal transmission, providing support to the hypothesis of pneumococcal fomite transmission (26).
Although fomites may constitute an important mode of transmission for S. pneumoniae, little is known about the molecular mechanisms that enable S. pneumoniae to remain stable in the environment as the bacteria desiccate and are left without access to nutrients. Desiccation is theorized to impose an enormous amount of stress on an organism. Some of these stresses include DNA damage, protein damage, osmotic shock, oxidative damage, protein denaturation, and cross-linking, and reduced membrane fluidity (27). These challenges are so great that the majority of bacteria are unable to survive extended periods of desiccation (28). Therefore, the pneumococcus must have evolved mechanisms to cope with the challenges imposed by desiccation. In this study, we used a high-throughput mutant-screening approach to identify genes that are involved in the desiccation tolerance response of S. pneumoniae in order to better characterize environmental persistence of the bacterium.
RESULTS
Tn-seq screen to identify genes involved in S. pneumoniae desiccation tolerance.
To uncover which S. pneumoniae factors are required for desiccation tolerance, we employed a high-throughput transposon sequencing (Tn-seq) approach (29). In vitro transposition of a minitransposon and subsequent transformation of the transposed DNA into bacteria produced a library of ∼64,000 unique insertion mutants in the serotype 2 strain D39. This high-complexity library was then screened for sensitivity to desiccation using a previously described desiccation assay (9). To perform the desiccation, bacteria were grown to near confluence on blood agar (BA) and then collected, spread thinly on polystyrene petri plate lids, and left in the dark to desiccate for 48 h. In order to isolate survivors, desiccated bacteria were resuspended and plated on blood agar and then grown overnight.
To prepare the libraries for sequencing, genomic DNA was isolated from the pooled desiccation survivors, as well as the input library. The genomic junctions of all transposon insertion mutants were amplified by homopolymer tail-mediated ligation (HTML)-PCR as described previously (29), and each sample was uniquely barcoded. The location of each transposon insertion was then identified using massively parallel sequencing on the Illumina platform, and the relative frequency of each mutant within the library was then determined using normalized read counts. The frequencies of each unique insertion mutant before and after desiccation were compared, and this was used to calculate a fitness (W) value for each insertion. The mean fitness of a gene was then calculated by averaging the fitness of all transposon insertions within a gene.
As expected, the majority of genes disrupted by the transposon had a neutral impact on bacterial fitness during desiccation, resulting in a fitness of ∼1 (Fig. 1; Table S1 in the supplemental material). All genes showing a 20% or greater change in fitness (W) with a P value below 2.33 × 10−4 [−log(Pvalue) > 3.633] were considered to have a significant deviation from the wild type. Both genes that contributed to desiccation tolerance (desiccation sensitive) and ones that hindered it (desiccation resistant) were identified (Fig. 1A and B, respectively). Reproducibility was high between the two biological replicates (Pearson’s correlation, R = 0.801), providing confidence in the results of the screen (Fig. 1C). In total, this screen identified 42 genes whose disruption by transposon insertion rendered the bacterium desiccation sensitive and 45 genes that resulted in improved survival (Table 1).
FIG 1.

Tn-seq desiccation tolerance screen results. (A, B) Volcano plots of Tn-seq screen results display statistical significance against fitness for both desiccation-sensitive transposon mutants (A) and desiccation-resistant mutants (B). Mutants with a 20% or greater change in fitness that are above the −log(Pvalue) cutoff of 3.633 are highlighted in black. The three components of the highly enriched nucleotide excision repair complex UvrABC are highlighted in green. (C) Reproducibility of the two biological replicates is demonstrated by a Pearson’s correlation of R = 0.801. (D) Significant hits from the screen were categorized by function using annotations and GO terms from the KEGG genome database and UniProt. The number of genes within each category is quantified on the x axis.
TABLE 1.
Putative desiccation tolerance genes from the Tn-seq screen
| Desiccation tolerance, functional category, D39 locus | Gene | Fitnessa |
|---|---|---|
| Desiccation sensitivity | ||
| Capsule synthesis | ||
| SPD_0320 | cps2T | 0.14 |
| Cell redox homeostasis | ||
| SPD_1298 | 0.55 | |
| Cell wall and cell division | ||
| SPD_0336 | pbp1A | 0.46 |
| SPD_0342 | mapZ | 0.57 |
| SPD_1821 | pbp2A | 0.58 |
| SPD_0535 | murM (fibA) | 0.72 |
| DNA replication recombination and repair | ||
| SPD_1096 | uvrB | 0.20 |
| SPD_0538 | uvrC | 0.26 |
| SPD_0176 | uvrA | 0.35 |
| SPD_1778 | rmuC | 0.39 |
| SPD_1104 | 0.44 | |
| SPD_0366 | recD2 | 0.69 |
| Lipid metabolism or envelope biogenesis | ||
| SPD_0185 | 0.12 | |
| SPD_0646 | 0.40 | |
| SPD_0996 | 0.48 | |
| Membrane-associated | ||
| SPD_0268 | 0.50 | |
| SPD_1867 | 0.66 | |
| SPD_2068 | htrA | 0.67 |
| Metabolism | ||
| SPD_0330 | rfbB | 0.26 |
| SPD_1779 | 0.31 | |
| SPD_0953 | ppc | 0.64 |
| SPD_0068 | gadE | 0.71 |
| Nucleotide metabolism | ||
| SPD_2055 | guaB | 0.18 |
| SPD_0980 | prs2 | 0.25 |
| SPD_1274 | guaA | 0.75 |
| Regulatory/signal transduction | ||
| SPD_1542 | stkP | 0.05 |
| SPD_2000 | adcR | 0.09 |
| SPD_1084 | vicK | 0.13 |
| SPD_1797 | ccpA | 0.17 |
| SPD_2032 | pde1 | 0.35 |
| RNA processing, modification, and catabolism | ||
| SPD_1549 | rny | 0.40 |
| SPD_0129 | gidA | 0.26 |
| Transport | ||
| SPD_1998 | adcB | 0.04 |
| SPD_1099 | 0.06 | |
| SPD_1098 | 0.09 | |
| SPD_1622 | 0.62 | |
| SPD_1621 | 0.71 | |
| Unknown function | ||
| SPD_1740 | cinA | 0.26 |
| SPD_1295 | Hemolysin III | 0.33 |
| SPD_0010 | 0.35 | |
| SPD_1121 | 0.38 | |
| SPD_1333 | 0.56 | |
| Desiccation resistance | ||
| Amino acid metabolism | ||
| SPD_0700 | pepN | 1.21 |
| SPD_1418 | 1.21 | |
| SPD_0542 | pepV | 1.22 |
| SPD_0787 | pepX | 1.26 |
| SPD_0513 | cysE | 1.45 |
| Cell redox homeostasis | ||
| SPD_0685 | gor | 1.24 |
| SPD_1375 | 1.41 | |
| Lipid metabolism or envelope biogenesis | ||
| SPD_1165 | 1.28 | |
| SPD_0819 | lspA | 1.32 |
| Membrane associated | ||
| SPD_0670 | 1.21 | |
| SPD_0437 | ribU | 1.26 |
| Metabolism | ||
| SPD_0641 | manA | 1.23 |
| SPD_1971 | 1.25 | |
| SPD_1634 | galK | 1.27 |
| SPD_1633 | galT-2 | 1.28 |
| SPD_0065 | bgaC | 1.32 |
| SPD_1678 | agaN | 1.47 |
| SPD_0636 | spxB | 1.67 |
| Nucleotide metabolism | ||
| SPD_1068 | udk | 1.23 |
| Regulatory/signal transduction | ||
| SPD_1635 | galR | 1.25 |
| SPD_1450 | mntR | 1.26 |
| SPD_1487 | 1.26 | |
| SPD_1487 | 1.27 | |
| SPD_0064 | cpsR | 1.30 |
| SPD_0818 | cmbR | 1.55 |
| RNA processing, modification, and catabolism | ||
| SPD_0820 | rluD | 1.24 |
| Transport | ||
| SPD_1677 | rafE | 1.21 |
| SPD_1676 | rafF | 1.22 |
| SPD_1667 | amiF | 1.24 |
| SPD_1669 | amiD | 1.25 |
| SPD_1409 | msmK | 1.26 |
| SPD_1668 | amiE | 1.26 |
| SPD_1670 | amiC | 1.26 |
| SPD_1671 | amiA | 1.28 |
| SPD_2046 | cbiQ | 1.32 |
| SPD_2047 | cbiO1 | 1.33 |
| SPD_2048 | cbiO2 | 1.33 |
| SPD_1170 | appA | 1.40 |
| SPD_1169 | appB | 1.42 |
| SPD_1167 | appD | 1.42 |
| SPD_1168 | appC | 1.45 |
| SPD_0150 | gshT | 1.53 |
| Unknown function | ||
| SPD_1491 | 1.22 | |
| SPD_1166 | 1.23 | |
| SPD_1171 | 1.28 | |
Average fitness between the two biological replicates. In cases where the gene did not meet analytical cutoffs for read counts and Tn insertions in one biological replicate, only the fitness of the significant replicate is displayed.
These genes were categorized by function using annotations and Gene Ontology (GO) terms from the KEGG genome database and UniProt (Fig. 1D). Multiple categories of gene disruption rendered the bacteria desiccation sensitive. In particular, genes required for DNA repair and replication (6) and nucleotide metabolism (3) were abundant among the sensitive mutants. To further support the significance of this category of genes, a Gene Ontology (GO) enrichment for cellular components revealed that the excision endonuclease (excinuclease) repair complex UvrABC, which carries out nucleotide excision repair, was enriched 23-fold among our hits. This was the only category identified to be enriched in our GO analysis for cellular components. This emphasizes the importance of repairing DNA damage after desiccation and suggests that there is substantial DNA damage that occurs. This is well supported by work in other bacteria that demonstrates the necessity of DNA repair for successful desiccation resistance (30–32). Other functional categories that render the bacterium sensitive to desiccation pertain to the composition of the membrane and cell wall. These include the penicillin-binding proteins (PBPs) pbp1A and pbp2A, which are responsible for modifying the cell wall, as well as cardiolipin synthetase, which produces the lipid cardiolipin that increases membrane fluidity. Functional categories that result in desiccation resistance include 12 metabolic genes, 5 of which are involved in amino acid metabolism, and 16 different transporters, 3 of which encode sugar transporters. These categories of gene indicate a role for metabolism in desiccation tolerance.
Validation of putative desiccation tolerance genes.
In order to validate the results of our screen, we used allelic replacement to produce deletion mutants of 28 genes. Genes were selected for validation if they had a substantial fitness change and are not pleiotropic in other conditions (33). Four of these genes (SPD_1092, SPD_1093, SPD_1095, and tig [SPD_0365]), were selected because they had P values near the statistical cutoff and share genetic loci with other screen hits. Genes of known and unknown function were chosen. These deletion mutants were then tested in desiccation tolerance competitions with wild-type bacteria. Each mutant was mixed at a 1:1 ratio with the wild type and then plated for overnight growth. The plate-grown bacteria were then challenged with a 4-day desiccation, and a competitive index (CI) was calculated as the ratio of mutant/wild type in the output divided by the ratio from the input. Similar to the fitness values, a CI of less than 1 represents a defect in desiccation tolerance, while a CI greater than 1 represents improved survival. We found that 22 genes were validated with competitive indices that were significantly different than that of a neutral gene deletion, SPD_0022 (Fig. 2; Table S2). The majority of genes were validated in desiccation competition assays, demonstrating the robustness of our screen.
FIG 2.

Competitive indices of desiccation tolerance gene mutants. Twenty-eight putative desiccation tolerance genes identified in the Tn-seq were deleted, and the mutants tested in a 4-day desiccation competition assay against the wild type. Strains in this figure are the 22 deletion mutants that were validated and a mutant with deletion of a neutral gene, SPD_0022. Competitive index was calculated as the ratio of mutant to wild type after desiccation divided by the ratio before desiccation. The median for each mutant is represented with a bar. Statistical analyses were performed using a nonparametric Mann-Whitney U two-sample rank test, comparing each mutant against the neutral-gene SPD_0022 mutant (***, P ≤ 0.001; **, P ≤ 0.002; *, P ≤ 0.033). The median for each mutant is represented with a bar. Error bars show the 95% confidence interval.
We found that both of the nucleotide excision repair genes tested, uvrA and uvrB, had a significant defect in desiccation tolerance, resulting in median competitive indices of 0.47 and 0.35 (Fig. 2). In addition, the homologous recombination helicase recD and nucleotide biosynthetic gene prs2 also displayed significant fitness defects. Due to the significant enrichment of the excinuclease DNA repair complex and the validation of other DNA repair and maintenance genes, we chose to further characterize the impact of DNA repair on desiccation tolerance.
DNA repair pathways involved in desiccation tolerance.
Previous work has demonstrated that the drying of bacteria results in extensive DNA damage (34–36). However, although the types of DNA damage that occur in desiccating bacteria have been theorized, there is little direct evidence. To genetically dissect the specific types of DNA damage that occur during desiccation, we chose to delete a variety of DNA repair genes, including some not identified in our Tn-seq screen because they were above the P value cutoff. Because specific DNA repair pathways are required for resolving particular DNA lesions, increased desiccation sensitivity resulting from disruption of a DNA repair pathway would suggest that a particular type of damage is occurring.
Due to the general essentially of DNA repair for bacterial viability, deletion of many DNA repair genes is lethal. For this reason, we selected genes that function in specific DNA repair pathways but are not essential. We tested multiple genes in the nucleotide excision repair (NER) pathway, including two that are part of the core NER complex (uvrA and uvrB), as well as a gene that is only involved in transcription-coupled NER (mfd). Deletion of uvrA and uvrB resulted in a significant competitive disadvantage in desiccation survival, while deletion of mfd had a neutral effect on desiccation tolerance (Fig. 3). This suggests that the global genome repair pathway of NER is important for desiccation tolerance, but transcription-coupled repair is dispensable. We were able to complement uvrA at a neutral locus in the chromosome, demonstrating that the uvrA deletion was indeed responsible for the observed desiccation sensitivity (Fig. 3). To query the significance of homologous recombination (HR), we deleted the HR helicase recD and found that this results in a significant loss of viability after desiccation, suggesting that homologous recombination is necessary for desiccation tolerance. Next, we deleted two glycosylases (mutM and mutY) involved in base excision repair (BER) and found that both glycosylases have a competitive disadvantage, although the competitive index of the ΔmutM mutant is significantly lower than that of the ΔmutY mutant, suggesting that deletion of mutM has a greater impact on repairing DNA damage resulting from desiccation (Fig. 3). Finally, we tested three factors involved in mismatch repair (MMR) (xseA, mutL, and mutS1). MutS1 and MutL act in a stepwise fashion with MutS, first recognizing the nucleotide mismatch, followed by binding of MutL, which then recruits an endonuclease to the complex. Neither of these genes displayed a competitive disadvantage in desiccation, suggesting that mismatches are not the primary type of DNA damage occurring during desiccation (Fig. 3). The desiccation sensitivity displayed by the ΔxseA mutant (Fig. 3), a bidirectional single-stranded DNA exonuclease (ExoVII) that hydrolyzes single-stranded DNA, can be explained by the fact that this protein is involved in three different DNA repair pathways: mismatch repair, single-strand break repair, and homologous recombination. Based on the neutral impact of mutL and mutS deletion, we suggest that XseA is likely required for repairing single- and double-strand breaks after desiccation, and not mismatched nucleotides. This makes sense, as a desiccated bacterium is likely dormant and not actively replicating its genome, which is where replication errors usually occur.
FIG 3.

Multiple DNA repair pathways are required for desiccation tolerance. Four-day desiccations were performed on mutants representing a number of DNA repair pathways: nucleotide excision repair (NER), homologous recombination (HR), base excision repair (BER), and mismatch repair (MMR). ΔuvrA/uvrA represent the uvrA deletion mutant with the full gene and native promoter complemented on the chromosome at neutral-gene locus SPD_0022. Competitive index was calculated as the ratio of mutant to wild type after desiccation over the input ratio. Statistical analyses were performed using a nonparametric Mann-Whitney U two-sample rank test, comparing each mutant against the neutral gene SPD_0022 (***, P ≤ 0.001; **, P ≤ 0.002; *, P ≤ 0.033). Error bars show standard deviations.
Having identified BER pathway genes mutM and mutY, which have both been characterized as repairing oxidatively damaged guanines (8-oxoG) (37), we wanted to see if endogenous hydrogen peroxide production was responsible for oxidative damage that may be repaired by BER. S. pneumoniae is well known to produce hydrogen peroxide without a detoxification mechanism. The primary producer of hydrogen peroxide is pyruvate oxidase (SpxB) (38), which when deleted resulted in improved desiccation resistance in our screen (Table 1). This desiccation resistance was recapitulated in competition against the wild type (Fig. 4). To probe the impact of hydrogen peroxide production on DNA damage during desiccation, we performed desiccation competitions where we removed the majority of hydrogen peroxide from the system by deleting spxB in both the wild-type background and our DNA repair mutants. If the DNA repair mutant were responsible for repairing oxidative damage to the DNA caused by endogenous hydrogen peroxide, we would expect to see an abrogation of the fitness defect when spxB was deleted. Deletion of spxB caused slight increases in the competitive indexes of the ΔmutM and ΔuvrA mutants, but the differences were not significant (Fig. 4), suggesting that endogenous hydrogen peroxide production by SpxB is not responsible for the majority of DNA damage that is repaired by either of these DNA repair pathways. This suggests that the improved desiccation tolerance of the ΔspxB mutant may have more to do with the metabolic role of SpxB in carbon utilization, as opposed to its production of hydrogen peroxide as a metabolic by-product.
FIG 4.

Hydrogen peroxide produced by SpxB is not a primary cause of DNA damage during desiccation. Pyruvate oxidase (SpxB) is responsible for the majority of hydrogen peroxide produced by S. pneumoniae. In order to determine if endogenous hydrogen peroxide results in oxidative DNA damage that is repaired by MutM or UvrA, we deleted spxB in both the wild-type (WT) and DNA repair mutant backgrounds. Competitive indices were calculated as the ratio of mutant to wild type (or double mutant to single mutant) after desiccation compared to the input. Statistical analyses were performed using a nonparametric Mann-Whitney U two-sample rank test (***, P ≤ 0.001; ns, nonsignificant). The median for each mutant is represented with a bar. Error bars show the 95% confidence interval.
In order to confirm that the UvrABC complex performs a function similar to that of its well characterized homolog in Escherichia coli, we challenged the ΔuvrA mutant with UV irradiation. A deletion in any one of the three components of the NER complex should successfully abrogate its function, as all three are required to make a functional complex (39). The uvrA deletion mutant was significantly more susceptible to UV treatment, resulting in a 3-log reduction in survival below that of the wild type (Fig. 5). We were able to rescue this phenotype by complementing the uvrA gene back at a neutral gene locus, resulting in a wild-type level of survival (Fig. 5). Having confirmed that uvrA has a significant impact on desiccation survival and that its behavior mimics that of its homologs in other bacteria, we next investigated the impact of the uvrA deletion mutation and other desiccation mutations on transmission.
FIG 5.
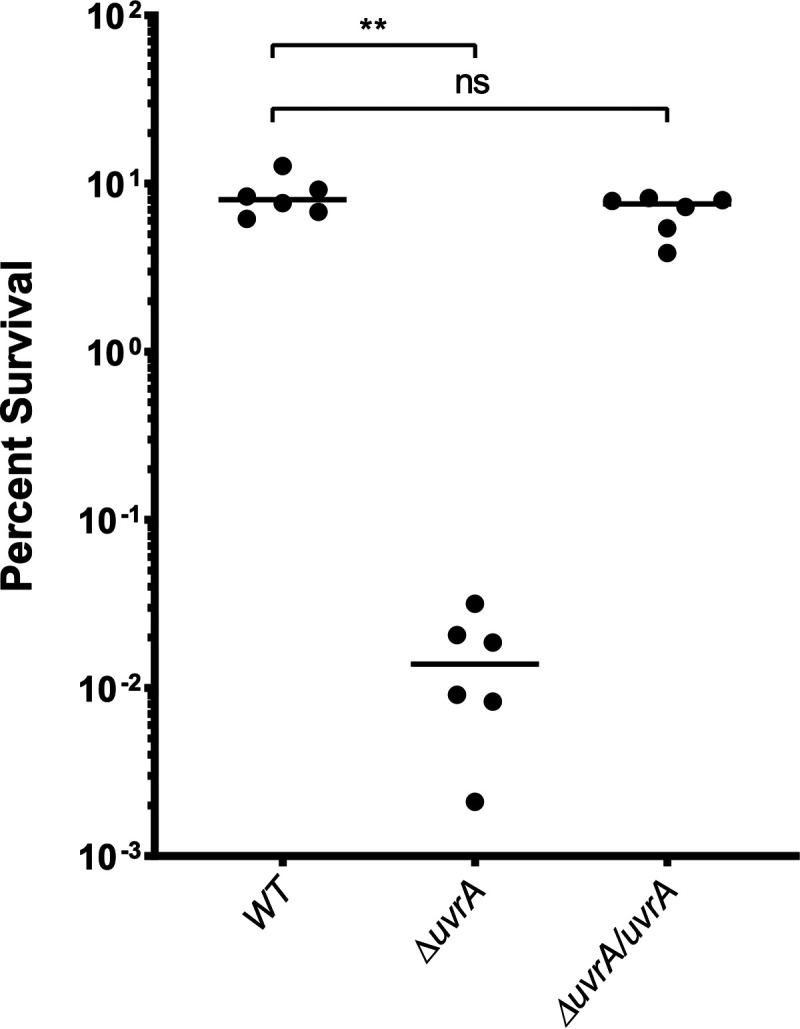
Bacterial survival after UV irradiation. Exponentially growing cultures of S. pneumoniae strains were washed and resuspended in PBS and then challenged with 15 mJ of UV light. Percent survival was quantified by plating bacteria for CFU before and after UV exposure. The uvrA deletion mutant (ΔuvrA) was complemented (ΔuvrA/uvrA) by placing the full gene and native promoter at neutral-gene locus SPD_0022. WT, wild type. Statistical analyses were performed using a nonparametric Mann-Whitney U two-sample rank test (**, P ≤ 0.002; ns, nonsignificant).
Transmission efficiency of selected desiccation mutants.
As we hypothesize that fomite transmission of S. pneumoniae is likely an important method of reaching new hosts, we wanted to see if our desiccation tolerance mutations would impact how efficiently bacteria are passed between mice in a murine model of transmission. Previous work has shown a correlation between transmission efficiency and desiccation tolerance using a ΔspxB mutant; spxB deletion results in both improved desiccation tolerance and increased transmission efficiency (26). Four hits from our screen were selected to be tested in the transmission assay: uvrA, bgaC, SPD_1622, and SPD_0996.
The transmission assay was performed by colonizing half of a mouse litter with serotype 2 S. pneumoniae (D39), the strain background which was used to develop the pneumococcal transmission assay (40). These colonized mice were the pneumococcal donors, while the uncolonized littermates were the contact mice. All mice were then returned to their cage with the dam, and transmission was tracked over the next 10 days by tapping the nares of the mice against a plate. Detection of colonies on two subsequent days was considered a colonization event. In this murine model of transmission, bacterial spread can occur through either of two routes: fomite transmission or direct contact. We found that transmission of the ΔuvrA mutant was significantly delayed compared to that of the wild type (D39), with a significant difference observed on days three and four (P = 0.0348) (Fig. 6). By day 6, transmission of the ΔuvrA mutant caught up to that of the wild type (P = 0.0654), resulting in equal transmission percentages at the conclusion of the experiment. The delayed transmission of the ΔuvrA mutant was not due to lower levels of colonization from the donor mice, as colonization levels were assessed at the end of the experiment and there was no significant difference between the wild type and the ΔuvrA nutant. Altogether, this demonstrates a correlation between decreased desiccation tolerance and delayed transmission.
FIG 6.

Transmission efficiency of a uvrA mutant. Litters of 4-day-old, C57BL/6 mice were split into two groups. The first group was colonized with a wild type (n = 11) or mutant (n = 14) strain of serotype 2 S. pneumoniae (D39), while the second half was left uncolonized. All mice were then returned to the cage, and uncolonized mice were surveyed daily for transmission by tapping the nares of each mouse against a blood agar plate. A colonization event was defined as detectable CFU on two subsequent days. (A) Transmission of the wild type (D39) and the ΔuvrA mutant was tracked over the course of 10 days. (B) Colonization levels of all donor mice were assessed at the end of the experiment. Statistics were performed with Mantel-Cox log-rank test for the transmission assay and Mann-Whitney U two-sample rank test for the colonization assay (*, P = 0.0348; ns, nonsignificant).
DISCUSSION
S. pneumoniae has been demonstrated to be desiccation tolerant, surviving in a dehydrated state for up to 30 days (9, 10). However, little is known about the mechanisms that enable the bacterium to persist in this state. Here, we have used transposon insertion sequencing (Tn-seq) to investigate the genetic factors that influence the desiccation tolerance of S. pneumoniae. We screened approximately 64,000 unique transposon insertion mutants using a 2-day desiccation assay on a plastic surface. After stringent analysis of the Tn-seq screen results, we identified 42 genes that resulted in reduced fitness and 45 genes that led to improved fitness. Within these hits were a number of functional categories that impacted desiccation tolerance.
A major category was that of DNA repair and replication. DNA damage is likely to be one of the most significant stresses of desiccation, as many parts of the cell can be remade, but the genome is the template for all necessary cellular components, and therefore, genome integrity is of the utmost importance. This is supported by the observation that in our screen, disruption of DNA repair and replication genes only resulted in sensitization to desiccation. Of particular interest was the nucleotide excision repair (NER) complex, composed of UvrA, UvrB, and UvrC. This complex was highly enriched in our data set based on a Gene Ontology (GO) enrichment for cellular components, and we found that deletion of any of these genes resulted in a significant fitness defect. UvrABC is best known to repair thymine dimers that are the result of UV damage (41, 42); however, our desiccations were performed in the dark, making UV an unlikely source of significant DNA damage. UvrABC has also been characterized as repairing other DNA lesions, including proteins that have been fused to DNA (43, 44). This may occur as the loss of water results in molecular crowding and loss of hydration shells surrounding proteins and DNA within the cell, causing various cellular components to interact more than they would in a normally hydrated cell (28). Study of DNA damage in desiccated Bacillus subtilis spores has previously demonstrated that significant DNA-protein cross-linking occurs during desiccation (45), suggesting that this type of DNA damage likely also occurs in desiccating S. pneumoniae cells.
Single- and double-stranded breaks have been shown to occur as a result of desiccation (35, 36), and oxidative damage is hypothesized to result from either desiccation or subsequent rehydration of bacteria (46). These other forms of DNA damage would require different repair pathways to resolve specific DNA lesions. When tested, we found that DNA repair pathways that are capable of repairing these types of damage were also required for desiccation tolerance. These pathways include homologous recombination (HR), which would repair double-strand breaks, and base excision repair (BER), which is capable of repairing modified nucleotides, such as oxidatively damaged bases. In addition, we found that nucleotide biosynthesis genes (prs2, guaA, and guaB) involved in maintaining the pool of available nucleotides required for DNA repair and replication also had decreased fitness in desiccation. Mismatch repair (MMR) was found to have little impact on desiccation tolerance, which can be explained by the fact that MMR generally repairs errors that occur during DNA replication. As we assume the bacteria are metabolically dormant, active DNA replication is unlikely to occur during desiccation. Our finding that deletion of additional DNA repair pathways results in a fitness defect suggests that multiple types of DNA damage are occurring during desiccation and a full complement of DNA repair systems is required for the bacteria to survive after desiccation.
A second category that emerged from our screen was genes that impact the structural integrity of the cell. These include genes involved in cell wall and cell division, as well as lipid metabolism and envelope biogenesis. During desiccation, the volume of the cell decreases while the membrane and cell wall remain their original size (28). This results in dense packing of phospholipids, resulting in decreased membrane fluidity and distortion of the membrane, which can eventually result in membrane rupture. We found that production of the phospholipid cardiolipin by cardiolipin synthetase (SPD_0185) significantly improves desiccation survival. Cardiolipin is known to increase membrane fluidity, which decreases packing of the membrane (47), and thus, may be instrumental during desiccation. Additionally, a previous study identified the cardiolipin synthetase gene as important for survival of Listeria monocytogenes in both desiccation and osmotic stress (48), suggesting that it may also be important for surviving either hyperosmotic shock that may occur during desiccation or subsequent hypoosmotic shock that occurs during rehydration. A structurally sound cell wall also likely helps avoid membrane rupture throughout desiccation, as well as during the osmotic shock of rehydration. We found that two class A penicillin-binding proteins (PBPs), Pbp1A and Pbp1B, were both important for wild-type levels of desiccation tolerance. The function of these two proteins is still not fully understood; however, they are known to be required for maturation of the cell wall, as opposed to the construction of nascent peptidoglycan (49). In addition, these genes are synthetically lethal, suggesting they share some functional redundancy in an essential process (50). Loss of type A PBPs has been characterized as leading to decreased cell wall stiffness and fewer peptidoglycan cross-links in E. coli and B. subtilis (51, 52). Improvements in cell wall integrity by Pbp1A and Pbp2A may increase the bacterium’s resistance to osmotic shock, resulting in improved desiccation survival. It is clear that the condition of the bacterial membrane and cell wall has a large impact on pneumococcal desiccation survival.
Another category of interest from our screen includes metabolic genes and transporters. Previous work has demonstrated that starvation and metal sequestration result in improved desiccation tolerance of S. pneumoniae (26). We found multiple sugar transporters, carbohydrate catabolic genes, and a putative metal transporter whose disruption resulted in increased desiccation resistance, which is in agreement with this previous finding. However, the exact mechanism of this improved desiccation tolerance of carbohydrate- and metal-starved bacteria is unknown. Slower growth could result in smaller cells that would undergo less shrinkage and membrane stress as they desiccated (53). Additionally, slow growth in Vibrio cholerae has been shown to improve resistance to osmotic shock (54). More work should be done to understand the impact of a decreased growth rate on desiccation tolerance in S. pneumoniae.
In order to demonstrate the impact of decreased desiccation tolerance on transmission, the desiccation-sensitive ΔuvrA mutant was tested in an infant mouse model of transmission. We found that deletion of uvrA results in delayed transmission between infant mice. It is known that pneumococcal shedding has a large impact on transmission efficiency (55); therefore, it was important to demonstrate that the ΔuvrA mutant did not have a colonization defect that could result in decreased shedding. We found that the bacterial load of the ΔuvrA mutant in the nasopharynx was the same as that of the wild type, suggesting that colonization density is not the cause of the transmission defect. We suggest that the transmission delay is due to the desiccation sensitivity of our mutant; however, we do not have direct evidence that transmission occurs from desiccated bacteria in our murine model. The possibility remains that transmission occurs by direct contact between mice. However, we hypothesize that some of the shed S. pneumoniae cells become desiccated on surfaces in the cage, as well as the skin of the pups and the dam. This is supported by the observation that desiccated S. pneumoniae cells remain capable of colonizing a new host (9). Additionally, an association between desiccation tolerance and transmission efficiency has been observed in a pyruvate oxidase mutant, which is not only more desiccation resistant but also has increased transmission rates in the infant mouse model (26). While these results do not directly demonstrate fomite transmission, they do exhibit a strong correlation between desiccation tolerance and transmission efficiency.
This work has highlighted a number of genetic factors that influence the desiccation tolerance of S. pneumoniae. In particular, the ability to repair damaged DNA appears to be a key factor that enables bacterial survival and transmission between hosts. The use of DNA-damaging agents may be an effective strategy to eliminate bacteria from surfaces. For example, far-UVC light (222 nm) has been demonstrated to effectively kill infectious bacteria while leaving mammalian skin undamaged (56–58). Utilization of such sterilizing techniques that cause additional DNA damage may prove to be an effective method to decrease the bacterial load on surfaces, thereby reducing pathogen transmission.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All experiments were performed with S. pneumoniae serotype 2 strain D39 and isogenic mutants. Bacteria were cultivated in a 37°C incubator with 5% CO2. Liquid cultures were grown on Todd-Hewitt broth (BD Biosciences) supplemented with 5% yeast extract (Fisher Scientific) and 300 U/ml catalase (Worthington Biochemicals) (THY broth). Overnight growth was performed on blood agar (BA) plates consisting of tryptic soy agar (Sigma-Aldrich) with 5% sheep’s blood (Northeast Laboratory Services). Antibiotics were used at the following concentrations: chloramphenicol at 4 μg/ml and spectinomycin at 200 μg/ml.
Strain construction.
Marked deletion strains were constructed by transforming competent S. pneumoniae bacteria with PCR products carrying the desired deletion. Allelic-exchange PCR products were made using splicing by overlap extension (SOE) PCR as described previously (59), where the chloramphenicol cassette was spliced to a minimum of 1 kb of sequencing flanking each side of the gene to be deleted. The flanking sequences allow allelic replacement by double-crossover homologous recombination. Complementation was performed by placing the promoter region, coding sequence, and spectinomycin cassette into a neutral gene locus (SPD_0022). All mutations were confirmed by Sanger sequencing or whole-genome sequencing. Strains used in this study are listed in Table 2.
TABLE 2.
Bacterial strains used in this study
| Strain | Description | Reference or source |
|---|---|---|
| S. pneumoniae strains | ||
| D39 | S. pneumoniae, serotype 2 | Laboratory stock |
| AC6529 | D39 SPD_0185::Cmr | This study |
| AC6539 | D39 SPD_2032 (pde1)::Cmr | This study |
| AC6547 | D39 SPD_0366 (recD)::Cmr | This study |
| AC6532 | D39 SPD_1096 (uvrB)::Cmr | This study |
| AC6541 | D39 SPD_0996::Cmr | This study |
| AC6533 | D39 SPD_0980 (prs2)::Cmr | This study |
| AC6540 | D39 SPD_0336 (pbp1A)::Cmr | This study |
| AC6538 | D39 SPD_0176 (uvrA)::Cmr | This study |
| AC6543 | D39 SPD_1622::Cmr | This study |
| AC6546 | D39 SPD_1621::Cmr | This study |
| AC6549 | D39 SPD_1093::Cmr | This study |
| AC6534 | D39 SPD_1740 (cinA)::Cmr | This study |
| AC6553 | D39 SPD_0820 (rluD)::Cmr | This study |
| AC6548 | D39 SPD_1092::Cmr | This study |
| AC6545 | D39 SPD_1867::Cmr | This study |
| AC6537 | D39 SPD_2000 (adcR)::Cmr | This study |
| AC6528 | D39 SPD_1098::Cmr | This study |
| AC6542 | D39 SPD_1821 (pbp1A)::Cmr | This study |
| AC6551 | D39 SPD_1095::Cmr | This study |
| AC6550 | D39 SPD_0128::Cmr | This study |
| AC6554 | D39 SPD_1450 (mntR)::Cmr | This study |
| AC6535 | D39 SPD_1999 (adcC)::Cmr | This study |
| AC6561 | D39 SPD_0022::Cmr | This study |
| AC6530 | D39 SPD_1099::Cmr | This study |
| AC6552 | D39 SPD_1094::Cmr::Cmr | This study |
| AC6531 | D39 SPD_2055 (guaB)::Cmr | This study |
| AC6544 | D39 SPD_0365 (tig)::Cmr | This study |
| AC6555 | D39 SPD_0064::Cmr | This study |
| AC6556 | D39 SPD_0065 (bgaC)::Cmr | This study |
| AC6674 | D39 SPD_0006 (mfd)::Cmr | This study |
| AC6675 | D39 SPD_1135 (mutM)::Cmr | This study |
| AC6676 | D39 SPD_1086 (mutY)::Cmr | This study |
| AC6677 | D39 SPD_1067 (xseA)::Cmr | This study |
| AC6678 | D39 SPD_0165 (mutL)::Cmr | This study |
| AC6679 | D39 SPD_0371 (mutS1)::Cmr | This study |
| AC6680 | AC6538 SPD_0022::SPD_0176 | This study |
| AC6681 | D39 SPD_0636 (spxB)::Specr | This study |
| AC6682 | AC6538 SPD_0636 (spxB)::Specr | This study |
| AC6683 | AC6675 SPD_0636 (spxB)::Specr | This study |
| E. coli strains | ||
| AC1304 | E. coli(pMalC9); Apr | 29 |
| AC3687 | E. coli(pMagellan6); Apr Spr | 29 |
Desiccation protocol.
S. pneumoniae cells were struck from a frozen glycerol stock onto blood agar plates and grown overnight. Colonies were subsequently resuspended into THY broth and diluted to an optical density at 600 nm (OD600) of 0.1. Fifty microliters of a 10-fold dilution was then spread onto a blood agar plate and allowed to grow for 16 h. The resulting semiconfluent colonies were pooled, scraped off a plate using a plastic wedge (gel releaser, product number 1653320; Bio-Rad), and split into equal sections. Each section was spread very thinly on a polystyrene petri plate lid using the wedge. Input CFU were quantified by immediate resuspension of the bacteria from several lids in THY and plating of 10-fold dilutions on blood agar. The remaining bacteria were then allowed to desiccate on lids for 2 or 4 days depending on the experiment, after which bacteria were resuspended in THY and plated for viable CFU counts. Bacterial counts were used to calculate percent survival.
Competitions were performed as described above using a 1:1 mixture of unmarked wild type and a chloramphenicol-resistant mutant to plate the bacterial lawn. Dilutions of bacteria collected from lids on day zero and day 4 were plated on blood agar and incubated for 16 h. The resulting colonies were replica plated onto blood agar containing 4 μg/ml chloramphenicol to assess the ratio of mutant to wild type. All were done with 5 to 10 biological replicates.
Transposon library construction.
The transposon library was constructed as previously described (60). Briefly, in vitro transposition was performed using purified transposase MarC9, genomic DNA, and the minitransposon Magellan6, which contains a spectinomycin resistance gene. Transposed DNA was then transformed into competent S. pneumoniae cells, and bacteria carrying a transposon were selected for by plating on blood agar supplemented with 200 μg/ml spectinomycin. This pool of mutants was grown up in THY broth and then collected and frozen down in 20% glycerol (final concentration) for further experimentation.
Tn-seq desiccation tolerance screen.
Libraries that were previously frozen were plated on blood agar and grown for 16 h. Each biological replicate consisted of 10 150-mm-diameter blood agar plates. The following day, the bacterial lawns were collected, mixed together, and desiccated as described above. Three input samples were collected immediately after spreading on lids and plated on blood agar. Five output samples were collected after 2 days of desiccation and plated on blood agar. After overnight growth, bacteria were collected and frozen as glycerol stocks for future isolation of genomic DNA.
Sequencing and analysis pipeline.
Genomic DNA was isolated using the DNeasy blood and tissue kit (catalog number 69504; Qiagen). Samples were prepared for sequencing using the HTML-PCR method (29). Briefly, genomic DNA was sheared via sonication in a cup horn sonicator and poly(C) tails were added to the 3′ ends of all fragments using terminal deoxynucleotidyl transferase. Transposon junctions were amplified through PCR amplification using primers specific for the Magellan6 transposon and the poly(C) tail. A subsequent nested PCR was performed to add a unique barcode to each sample. Sequencing was performed as 50-bp single-end reads on an Illumina HiSeq 2500 at the Tufts University Core Facility.
Fitness was calculated as previously described (60). Briefly, reads were mapped to the D39 genome using Bowtie (61). Transposon insertions at each gene locus were quantified for all input and output samples, and the data were normalized to the total number of reads in each sample to account for slight variations in read depth. Fitness for each unique insertion was calculated as previously described (29). No change is quantified as a fitness of 1, representing a neutral gene. Increased presence in the output results in a fitness greater than 1, while decreased presence in the output produces a fitness less than 1. Fitness values were then normalized against a list of neutral genes from D39 to artificially set those genes’ fitness to 1, and the same factor was used to normalize all other fitness values. Mean fitness of a gene was calculated by averaging all unique insertions across a gene. A minimum cutoff of 4 unique transposon insertions per gene was applied, in addition to a read cutoff of 15 reads per transposon insertion. Next a fitness cutoff was applied to remove all genes with less than a 20% fitness change from the neutral fitness of 1. Finally, statistical significance was determined using a sample t test with Bonferroni correction for multiple comparisons.
UV irradiation challenge.
Strains were grown up in THY broth to mid-log phase. Cultures were washed and resuspended in phosphate-buffered saline (PBS), and then 50 μl of each was spotted onto parafilm. Bacteria were exposed to 15 mJ of UV light (254 nm) using a Stratagene UV cross-linker. Bacteria from before and after UV treatment were plated on blood agar to quantify CFU, and this was used to calculate percent survival. All were done with six biological replicates over two separate days.
Transmission assay.
This assay was performed as previously described (26). Briefly, litters of 4-day-old C57BL/6 infant mice were split into two equal groups. The first group was colonized with 5 × 103 CFU of either wild-type or mutant serotype 2 strain D39, termed the donor mice. The other group was left uncolonized and referred to as the contact mice. All mice from the litter were then placed back in the cage with the dam. Transmission was tracked over the course of 10 days by tapping the nares of the contact mice against a blood agar plate. Twenty taps were collected from donor and contact mice each day for 10 days. Detection of bacteria on two subsequent days was defined as a transmission event. At the conclusion of the transmission experiment, all mice were sacrificed, and the level of nasopharyngeal colonization was quantified to ensure that varied transmission levels were not the result of increased or decreased shedding from the donor mice. All experiments involving animals were performed with prior approval of and in accordance with guidelines of the St. Jude Institutional Animal Care and Use Committee. The St. Jude laboratory animal facilities have been fully accredited by the American Association for Accreditation of Laboratory Animal Care. Laboratory animals are maintained in accordance with the applicable portions of the Animal Welfare Act and the guidelines prescribed in the DHHS publication, Guide for the Care and Use of Laboratory Animals.
ACKNOWLEDGMENT
This research was supported by National Institutes of Health training grant number GM139772 (A.J.M.).
Footnotes
Supplemental material is available online only.
Contributor Information
Andrew Camilli, Email: andrew.camilli@tufts.edu.
Nancy E. Freitag, University of Illinois at Chicago
REFERENCES
- 1.Bogaert D, de Groot R, Hermans P. 2004. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis 4:144–154. 10.1016/S1473-3099(04)00938-7. [DOI] [PubMed] [Google Scholar]
- 2.Sleeman KL, Griffiths D, Shackley F, Diggle L, Gupta S, Maiden MC, Moxon ER, Crook DW, Peto TEA. 2006. Capsular serotype–specific attack rates and duration of carriage of Streptococcus pneumoniae in a population of children. J Infect Dis 194:682–688. 10.1086/505710. [DOI] [PubMed] [Google Scholar]
- 3.Mosser JF, Grant LR, Millar EV, Weatherholtz RC, Jackson DM, Beall B, Craig MJ, Reid R, Santosham M, O’Brien KL. 2014. Nasopharyngeal carriage and transmission of Streptococcus pneumoniae in American Indian households after a decade of pneumococcal conjugate vaccine use. PLoS One 9:e79578. 10.1371/journal.pone.0079578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Austrian R. 1986. Some aspects of the pneumococcal carrier state. J Antimicrob Chemother 18:35–45. 10.1093/jac/18.Supplement_A.35. [DOI] [PubMed] [Google Scholar]
- 5.van der Poll T, Opal SM. 2009. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374:1543–1556. 10.1016/S0140-6736(09)61114-4. [DOI] [PubMed] [Google Scholar]
- 6.Troeger C, Blacker B, Khalil IA, Rao PC, Cao J, Zimsen SRM, Albertson SB, Deshpande A, Farag T, Abebe Z, Adetifa IMO, Adhikari TB, Akibu M, Al Lami FH, Al-Eyadhy A, Alvis-Guzman N, Amare AT, Amoako YA, Antonio CAT, Aremu O, Asfaw ET, Asgedom SW, Atey TM, Attia EF, Avokpaho EFGA, Ayele HT, Ayuk TB, Balakrishnan K, Barac A, Bassat Q, Behzadifar M, Behzadifar M, Bhaumik S, Bhutta ZA, Bijani A, Brauer M, Brown A, Camargos PAM, Castañeda-Orjuela CA, Colombara D, Conti S, Dadi AF, Dandona L, Dandona R, Do HP, Dubljanin E, Edessa D, Elkout H, Endries AY, Fijabi DO, et al. 2018. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect Dis 18:1191–1210. 10.1016/S1473-3099(18)30310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wahl B, O’Brien KL, Greenbaum A, Majumder A, Liu L, Chu Y, Lukšić I, Nair H, McAllister DA, Campbell H, Rudan I, Black R, Knoll MD. 2018. Burden of Streptococcus pneumoniae and Haemophilus influenzae type b disease in children in the era of conjugate vaccines: global, regional, and national estimates for 2000–15. Lancet Global Health 6:e744–e757. 10.1016/S2214-109X(18)30247-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang D, Petigara T, Yang X. 2018. Clinical and economic burden of pneumococcal disease in US adults aged 19–64 years with chronic or immunocompromising diseases: an observational database study. BMC Infect Dis 18:436. 10.1186/s12879-018-3326-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsh RL, Camilli A. 2011. Streptococcus pneumoniae is desiccation tolerant and infectious upon rehydration. mBio 2:e00092-11. 10.1128/mBio.00092-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marks LR, Reddinger RM, Hakansson AP. 2014. Biofilm formation enhances fomite survival of Streptococcus pneumoniae and Streptococcus pyogenes. Infect Immun 82:1141–1146. 10.1128/IAI.01310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kraay ANM, Hayashi MAL, Hernandez-Ceron N, Spicknall IH, Eisenberg MC, Meza R, Eisenberg JNS. 2018. Fomite-mediated transmission as a sufficient pathway: a comparative analysis across three viral pathogens. BMC Infect Dis 18:540. 10.1186/s12879-018-3425-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guerrero DM, Nerandzic MM, Jury LA, Jinno S, Chang S, Donskey CJ. 2012. Acquisition of spores on gloved hands after contact with the skin of patients with Clostridium difficile infection and with environmental surfaces in their rooms. Am J Infect Control 40:556–558. 10.1016/j.ajic.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Williams C, Davis DL. 2009. Methicillin-resistant Staphylococcus aureus fomite survival. Clin Lab Sci 22:34–38. [PubMed] [Google Scholar]
- 14.Lopez GU, Gerba CP, Tamimi AH, Kitajima M, Maxwell SL, Rose JB. 2013. Transfer efficiency of bacteria and viruses from porous and nonporous fomites to fingers under different relative humidity conditions. Appl Environ Microbiol 79:5728–5734. 10.1128/AEM.01030-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicas M, Sun G. 2006. An integrated model of infection risk in a health-care environment. Risk Anal 26:1085–1096. 10.1111/j.1539-6924.2006.00802.x. [DOI] [PubMed] [Google Scholar]
- 16.Hall CB, Douglas RG, Geiman JM. 1980. Possible transmission by fomites of respiratory syncytial virus. J Infect Dis 141:98–102. 10.1093/infdis/141.1.98. [DOI] [PubMed] [Google Scholar]
- 17.Boone SA, Gerba CP. 2007. Significance of fomites in the spread of respiratory and enteric viral disease. Appl Environ Microbiol 73:1687–1696. 10.1128/AEM.02051-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fekety R, Kim K-H, Brown D, Batts DH, Cudmore M, Silva J. 1981. Epidemiology of antibiotic-associated colitis. Am J Med 70:906–908. 10.1016/0002-9343(81)90553-2. [DOI] [PubMed] [Google Scholar]
- 19.Han JH, Sullivan N, Leas BF, Pegues DA, Kaczmarek JL, Umscheid CA. 2015. Cleaning hospital room surfaces to prevent health care-associated infections: a technical brief. Ann Intern Med 163:598–607. 10.7326/M15-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carling PC, Bartley JM. 2010. Evaluating hygienic cleaning in health care settings: what you do not know can harm your patients. Am J Infect Control 38:S41–S50. 10.1016/j.ajic.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Otter JA, Yezli S, Salkeld JAG, French GL. 2013. Evidence that contaminated surfaces contribute to the transmission of hospital pathogens and an overview of strategies to address contaminated surfaces in hospital settings. Am J Infect Control 41:S6–S11. 10.1016/j.ajic.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Hartmann B, Benson M, Junger A, Quinzio L, Röhrig R, Fengler B, Färber UW, Wille B, Hempelmann G. 2003. Computer keyboard and mouse as a reservoir of pathogens in an intensive care unit. J Clin Monit Comput 18:7–12. 10.1023/B:JOCM.0000025279.27084.39. [DOI] [PubMed] [Google Scholar]
- 23.Cobrado L, Silva-Dias A, Azevedo MM, Rodrigues AG. 2017. High-touch surfaces: microbial neighbours at hand. Eur J Clin Microbiol Infect Dis 36:2053–2062. 10.1007/s10096-017-3042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kramer A, Schwebke I, Kampf G. 2006. How long do nosocomial pathogens persist on inanimate surfaces? A systematic review. BMC Infect Dis 6:130. 10.1186/1471-2334-6-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee L, Tin S, Kelley ST. 2007. Culture-independent analysis of bacterial diversity in a child-care facility. BMC Microbiol 7:27. 10.1186/1471-2180-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rowe HM, Karlsson E, Echlin H, Chang T-C, Wang L, van Opijnen T, Pounds SB, Schultz-Cherry S, Rosch JW. 2019. Bacterial factors required for transmission of Streptococcus pneumoniae in mammalian hosts. Cell Host Microbe 25:884–891.e6. 10.1016/j.chom.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.García AH. 2011. Anhydrobiosis in bacteria: from physiology to applications. J Biosci 36:939–950. 10.1007/s12038-011-9107-0. [DOI] [PubMed] [Google Scholar]
- 28.Greffe VRG, Michiels J. 2020. Desiccation-induced cell damage in bacteria and the relevance for inoculant production. Appl Microbiol Biotechnol 104:3757–3770. 10.1007/s00253-020-10501-6. [DOI] [PubMed] [Google Scholar]
- 29.van Opijnen T, Lazinski DW, Camilli A. 2014. Genome-wide fitness and genetic interactions determined by Tn-seq, a high-throughput massively parallel sequencing method for microorganisms. Curr Protoc Mol Biol 106:7.16.1–7.16.24. 10.1002/0471142727.mb0716s106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mattimore V, Battista JR. 1996. Radioresistance of Deinococcus radiodurans: functions necessary to survive ionizing radiation are also necessary to survive prolonged desiccation. J Bacteriol 178:633–637. 10.1128/jb.178.3.633-637.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humann JL, Ziemkiewicz HT, Yurgel SN, Kahn ML. 2009. Regulatory and DNA repair genes contribute to the desiccation resistance of Sinorhizobium meliloti Rm1021. Appl Environ Microbiol 75:446–453. 10.1128/AEM.02207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pitcher RS, Green AJ, Brzostek A, Korycka-Machala M, Dziadek J, Doherty AJ. 2007. NHEJ protects mycobacteria in stationary phase against the harmful effects of desiccation. DNA Repair (Amst) 6:1271–1276. 10.1016/j.dnarep.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 33.van Opijnen T, Camilli A. 2012. A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res 22:2541–2551. 10.1101/gr.137430.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asada S, Takano M, Shibasaki I. 1979. Deoxyribonucleic acid strand breaks during drying of Escherichia coli on a hydrophobic filter membrane. Appl Environ Microbiol 37:266–273. 10.1128/AEM.37.2.266-273.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dose K, Bieger-Dose A, Labusch M, Gill M. 1992. Survival in extreme dryness and DNA-single-strand breaks. Adv Space Res 12:221–229. 10.1016/0273-1177(92)90176-X. [DOI] [PubMed] [Google Scholar]
- 36.Dose K, Bieger-Dose A, Kerz O, Gill M. 1991. DNA-strand breaks limit survival in extreme dryness. Orig Life Evol Biosph 21:177–187. 10.1007/BF01809446. [DOI] [PubMed] [Google Scholar]
- 37.Hazra TK, Hill JW, Izumi T, Mitra S. 2001. Multiple DNA glycosylases for repair of 8-oxoguanine and their potential in vivo functions. Prog Nucleic Acids Res Mol Biol 68:193–205. 10.1016/s0079-6603(01)68100-5. [DOI] [PubMed] [Google Scholar]
- 38.Lisher JP, Tsui H-CT, Ramos-Montañez S, Hentchel KL, Martin JE, Trinidad JC, Winkler ME, Giedroc DP. 2017. Biological and chemical adaptation to endogenous hydrogen peroxide production in Streptococcus pneumoniae D39. mSphere 2:e00291-16. 10.1128/mSphere.00291-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeung AT, Mattes WB, Oh EY, Grossman L. 1983. Enzymatic properties of purified Escherichia coli UvrABC proteins. Proc Natl Acad Sci U S A 80:6157–6161. 10.1073/pnas.80.20.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diavatopoulos DA, Short KR, Price JT, Wilksch JJ, Brown LE, Briles DE, Strugnell RA, Wijburg OL. 2010. Influenza A virus facilitates Streptococcus pneumoniae transmission and disease. FASEB J 24:1789–1798. 10.1096/fj.09-146779. [DOI] [PubMed] [Google Scholar]
- 41.Kisker C, Kuper J, Van Houten B. 2013. Prokaryotic nucleotide excision repair. Cold Spring Harb Perspect Biol 5:a012591. 10.1101/cshperspect.a012591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sidler C. 2016. Genomic instability and aging: causes and consequences, p 511–525. In Kovalchuk I, Kovalchuk O (ed), Genome stability: from virus to human application. Elsevier/Academic Press, London, United Kingdom. 10.1016/B978-0-12-803309-8.00029-X. [DOI] [Google Scholar]
- 43.Minko IG, Kurtz AJ, Croteau DL, Van Houten B, Harris TM, Lloyd RS. 2005. Initiation of repair of DNA−polypeptide cross-links by the UvrABC nuclease. Biochemistry 44:3000–3009. 10.1021/bi0478805. [DOI] [PubMed] [Google Scholar]
- 44.Snowden A, Kow YW, Van Houten B. 1990. Damage repertoire of the Escherichia coli UvrABC nuclease complex includes abasic sites, base-damage analogues, and lesions containing adjacent 5′ or 3′ nicks. Biochemistry 29:7251–7259. 10.1021/bi00483a013. [DOI] [PubMed] [Google Scholar]
- 45.Bieger-Dose A, Dose K, Meffert R, Mehler M, Risi S. 1992. Extreme dryness and DNA-protein cross-links. Adv Space Res 12:265–270. 10.1016/0273-1177(92)90181-V. [DOI] [PubMed] [Google Scholar]
- 46.Potts M. 1994. Desiccation tolerance of prokaryotes. Microbiol Rev 58:755–805. 10.1128/MMBR.58.4.755-805.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Unsay JD, Cosentino K, Subburaj Y, García-Sáez AJ. 2013. Cardiolipin effects on membrane structure and dynamics. Langmuir 29:15878–15887. 10.1021/la402669z. [DOI] [PubMed] [Google Scholar]
- 48.Hingston PA, Piercey MJ, Truelstrup Hansen L. 2015. Genes associated with desiccation and osmotic stress in Listeria monocytogenes as revealed by insertional mutagenesis. Appl Environ Microbiol 81:5350–5362. 10.1128/AEM.01134-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Straume D, Piechowiak KW, Olsen S, Stamsås GA, Berg KH, Kjos M, Heggenhougen MV, Alcorlo M, Hermoso JA, Håvarstein LS. 2020. Class A PBPs have a distinct and unique role in the construction of the pneumococcal cell wall. Proc Natl Acad Sci U S A 117:6129–6138. 10.1073/pnas.1917820117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoskins J, Matsushima P, Mullen DL, Tang J, Zhao G, Meier TI, Nicas TI, Jaskunas SR. 1999. Gene disruption studies of penicillin-binding proteins 1a, 1b, and 2a in Streptococcus pneumoniae. J Bacteriol 181:6552–6555. 10.1128/JB.181.20.6552-6555.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McPherson DC, Popham DL. 2003. Peptidoglycan synthesis in the absence of class A penicillin-binding proteins in Bacillus subtilis. J Bacteriol 185:1423–1431. 10.1128/jb.185.4.1423-1431.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vigouroux A, Cordier B, Aristov A, Alvarez L, Özbaykal G, Chaze T, Oldewurtel ER, Matondo M, Cava F, Bikard D, van Teeffelen S. 2020. Class-A penicillin binding proteins do not contribute to cell shape but repair cell-wall defects. Elife 9. 10.7554/eLife.51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alpert P. 2006. Constraints of tolerance: why are desiccation-tolerant organisms so small or rare? J Exp Biol 209:1575–1584. 10.1242/jeb.02179. [DOI] [PubMed] [Google Scholar]
- 54.Silva-Valenzuela CA, Lazinski DW, Kahne SC, Nguyen Y, Molina-Quiroz RC, Camilli A. 2017. Growth arrest and a persister state enable resistance to osmotic shock and facilitate dissemination of Vibrio cholerae. ISME J 11:2718–2728. 10.1038/ismej.2017.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zafar MA, Wang Y, Hamaguchi S, Weiser JN. 2017. Host-to-host transmission of Streptococcus pneumoniae is driven by its inflammatory toxin, pneumolysin. Cell Host Microbe 21:73–83. 10.1016/j.chom.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buonanno M, Ponnaiya B, Welch D, Stanislauskas M, Randers-Pehrson G, Smilenov L, Lowy FD, Owens DM, Brenner DJ. 2017. Germicidal efficacy and mammalian skin safety of 222-nm UV light. Radiation Res 187:493–501. 10.1667/RR0010CC.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matafonova GG, Batoev VB, Astakhova SA, Gómez M, Christofi N. 2008. Efficiency of KrCl excilamp (222 nm) for inactivation of bacteria in suspension. Lett Appl Microbiol 47:508–513. 10.1111/j.1472-765X.2008.02461.x. [DOI] [PubMed] [Google Scholar]
- 58.Welch D, Buonanno M, Grilj V, Shuryak I, Crickmore C, Bigelow AW, Randers-Pehrson G, Johnson GW, Brenner DJ. 2018. Far-UVC light: a new tool to control the spread of airborne-mediated microbial diseases. Sci Rep 8:2752. 10.1038/s41598-018-21058-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535. [PubMed] [Google Scholar]
- 60.van Opijnen T, Bodi KL, Camilli A. 2009. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 6:767–772. 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download IAI00713-20_Supp_1_seq10.xlsx, XLSX file, 0.2 MB (177.3KB, xlsx)
Table S2. Download IAI00713-20_Supp_2_seq11.xlsx, XLSX file, 0.01 MB (11.7KB, xlsx)