
Fig. 1. Data structure and performance of SARGE on simulated data.
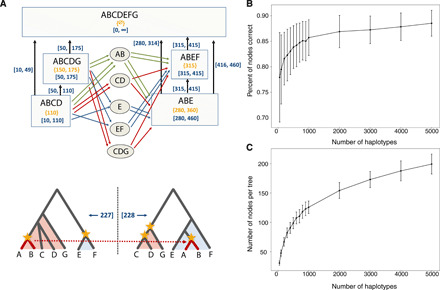
(A) Schematic of data structure. Top: Rectangles are “tree nodes” representing clades in trees. Each clade has member haplotypes (shown with letters A to G) and a start and end coordinate (blue numbers in brackets) determined by coordinates of single-nucleotide polymorphism (SNP) sites tagging the clade (yellow numbers in braces), along with a propagation distance parameter (100 in this example). Parent/child edges (vertical arrows) also have start and end coordinates determined by the nodes. Ovals are candidates for clades sharing an ancestral recombination event that can explain four-gamete test failures; colored edges indicate potential paths between tree nodes through candidate nodes that could explain four-gamete test failures (colors indicate types of paths). The candidate node with the most edges (here, AB) is eventually chosen as the most parsimonious branch movement, allowing for the inference of new nodes. The two trees at the bottom show the “solved” ancestral recombination event with the branch movement marked in red and all clades inferred without SNP data marked with yellow stars (haplotypes A and B share an ancestral recombination event; their ancestry is shared with haplotypes C, D, and G upstream of the recombination event and haplotype E downstream of it). The coordinates of the recombination event (blue numbers in brackets) are taken to be midway between the highest-coordinate upstream site (left side) and the lowest-coordinate downstream site (right side) involved in recombination. For a more detailed overview of the data structure, see figs. S3 to S5. (B) Accuracy of SARGE on simulated data (defined as percent of all clades correct according to the true ARG in the simulation), with increasing numbers of human-like haplotypes from an unstructured population. Error bars are one SD across five replicates. (C) Number of nodes per tree with increasing number of haplotypes in simulated data.