
Fig. 2. Performance of SARGE on SGDP and archaic hominin dataset.
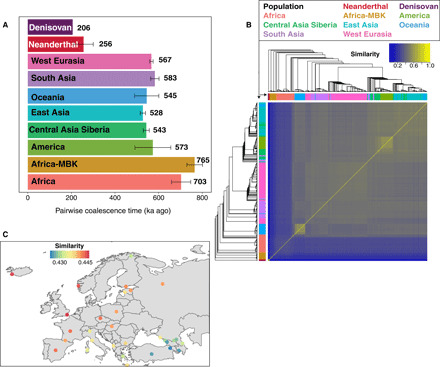
(A) Pairwise coalescence times for randomly sampled sets of up to 10 pairs of phased genome haplotypes per population (every possible pair was considered for archaic hominins, since fewer genomes were available). Values are calibrated using a 13-Ma human-chimp divergence time (see Supplementary Methods) and averaged across every variable site in the dataset, error bars show one SD, and branch shortening values for archaic samples were incorporated into calculations using mean values reported in (1). The lower value for humans comes from removing archaic-admixed clades from trees. (B) UPGMA (unweighted pair group method with arithmetic mean) trees computed using nucleotide diversity from SNP data (top and left) against similarity matrix from shared recombination events inferred by SARGE. Light yellow boxes (similar groups) are Native Americans and Papuans. (C) Average similarity between Orcadian haplotypes in the SGDP panel and other European haplotypes calculated on the basis of the number of shared ancestral recombination events. The best matches are in England, Iceland, and Norway, as expected.